
Abstract
Solvents employed for perovskite film fabrication not only play important roles in dissolving the precursors but also participate in crystallization process. High boiling point aprotic solvents with O-donor ligands have been extensively studied, but the formation of a highly uniform halide perovskite film still requires the participation of additives or an additional step to accelerate the nucleation rate. The volatile aliphatic methylamine with both coordinating ligands and hydrogen protons as solvent or post-healing gas facilitates the process of methylamine-based perovskite films with high crystallinity, few defects, and easy large-scale fabrication as well. However, the attempt in formamidinium-containing perovskites is challenged heretofore. Here, we reveal that the degradation of formamidinium-containing perovskites in aliphatic amines environment results from the transimination reaction of formamidinium cation and aliphatic amines along with the formation of ammonia. Based on this mechanism, ammonia is selected as a post-healing gas for a highly uniform, compact formamidinium-based perovskite films. In particular, low temperature is proved to be crucial to enable formamidinium-based perovskite materials to absorb enough ammonia molecules and form a liquid intermediate state which is the key to eliminating voids in raw films. As a result, the champion perovskite solar cell based on ammonia post-healing achieves a power conversion efficiency of 23.21% with excellent reproducibility. Especially the module power conversion efficiency with 14 cm2 active area is over 20%. This ammonia post-healing treatment potentially makes it easier to upscale fabrication of highly efficient formamidinium-based devices.
Similar content being viewed by others


Introduction
Halide perovskite materials with the general formula ABX3, where A refers to a monovalent cation such as methylammonium (CH3NH3+, MA+), formamidinium (HC(NH2)2+, FA+), Cs+, B represents a divalent cation such as Pb2+, Sn2+, Ge2+, and X represents a halide ion such as I-, Br-, Cl-, have emerged as a class of promising light-harvesting materials in photovoltaics since 20091. The highest certified power conversion efficiency (PCE) of perovskite solar cells (PSCs) has so far been up to 25.7% 2, which is comparable to that of widely commercialized Si-based solar cells. The crystal structure of halide perovskites can be regarded as a [MX6]4− octahedron in a three-dimensional (3D) space with a common apex angle connecting. The A site ions fill up the gap of the octahedral and the structure is stabilized by Van der Waals forces3. These special structural characteristics provide a variety of solution processing methods to prepare perovskite films4,5,6.
The high boiling point aprotic N,N-dimethylformamide (DMF), γ-butyrolactone (GBL), dimethyl sulfoxide (DMSO) and N,N-dimethylacetamide (DMA) are the common solvents employed to dissolve the perovskite precursors with the formation of solvated iodoplumbate complexes because of the strong coordination between O-donor ligands (OLs) and Pb(II)4,7,8,9. The Lewis basicity of solvents is thought to correlate with the “coordinating ability” with lead halide salts, which can be predicted by their Gutmann’s Donor Number (DN) with a trend of DMSO > DMA > DMF > GBL10,11. High DN solvents with strong coordination with the Pb(II) center also result in the formation of intermediate phase OL-PbI2-RAI (RA refers to MA or FA, etc.) prior to perovskite phase during the film fabrication process. The anisotropic growth nature of the intermediate phase leads to rough films with one-dimensional (1D) fiber-like structures and a large proportion of void area12. In this regard, stronger coordinating additives (such as thiourea13 and pyridine14) and/or the fast nucleation strategies (such as anti-solvent extraction, gas-quenching, and vacuum-assisted drying) have been introduced to regulate the growth process of the intermediate phase to achieve highly uniform perovskite films with good crystallinity7,15,16.
Among them, methylamine (MA0) featured with the presence of hydrogen bonding and low boiling point has become an impressive coordination agent for the MAPbI3 material. In this case, the formed intermediate phase is a metastable (PbI2-MAI)·xMA0 complex, instead of simple coordinate bond-dominated OL-PbI2-MAI adducts. Easy formation of highly uniform (PbI2-MAI)·xMA0 films and following conversion to highly uniform perovskite films are closely related to the self-leveling behavior of (PbI2-MAI)·xMA0 liquid intermediate phase and the ultrafast evaporation of MA0 gas. MA0 employed as a post-healing gas to eliminate the voids in the MAPbI3 perovskite film is firstly reported by Zhou et al. in 201517, and the adoption of MA0 as a volatile solvent system has become a commercially viable technology for MAPbI3 devices with excellent device reproducibility18,19,20,21,22,23. In comparison with MAPbI3, α-FAPbI3 has higher theoretical efficiency and thermal stability. The undesired phase transition to non-perovskite phase δ-FAPbI3 has been well dissolved by alloying a small amount of Cs+, MA+ to tailor the tolerance factor24,25,26,27,28,29. We have attempted to introduce MA0 gas-related methods for the fabrication of FA-containing perovskite layers, unfortunately, resulting in degradation of the 3D perovskite phase18. The underlying reason for the irreversible transformation of FAPbI3 with MA0 treatment is still not fully clear, let alone make efforts to solve this problem.
Here, we have systematically studied the underlying chemical reactions between aliphatic amines/formamidine (FA0) gases and FAI salt, and elucidated the addition-elimination reaction between amine compound and the imine band of FA0 with the formation of ammonia (NH3), also named transimination reaction. Based on this mechanism, NH3 is selected as a post-healing gas to avoid the degradation of the FA-based perovskite phase during the post treatment. It is demonstrated that decreasing processing temperature is crucial for FA-based perovskite layer to enhance the absorption of NH3 molecules, leading to the formation of a desired liquid intermediate state. Self-leveling behavior of the liquid intermediate state can quickly heal the voids in the rough perovskite film and finally form a highly uniform and compact film with the evaporation of NH3. At last, the PCE of FA-based PSC based on this NH3 post-healing strategy is more than 23% with a certified PCE of 22.22%. Especially, the PSC module achieves a PCE of 20.61% which is comparable with the highest reports in PSC modules. These results demonstrate the large advantage of the NH3 gas post-healing technology in upscaling fabrication of highly efficient PSCs.
Results
The degradation evidence of formamidinium-containing perovskites in aliphatic amines environment
Nuclear magnetic resonance (NMR) spectroscopy is a particularly practical tool for quantifying the relative amounts of the organic cations and related chemical reactions in the perovskite precursor30,31,32,33. We started with the study of the underlying chemical reactions between FA0/R-NH2(RA0) gases and FAI salt (Table 1). The gases involved include FA0, MA0, ethylamine (EA0), n-propylamine (PA0), n-butylamine (BA0), and NH3. The detailed preparation procedures of the relevant gases are provided in the methods section.
The synthesis of FA0 is tricky because of its instability. Mixed FACl and NaOH powders are expected to form FA0 by the neutralization as shown in Supplementary Scheme 1a. To study their product, deuterated DMSO (DMSO-d6) solvent is used to collect released gas from the mixed powders at room temperature (RT) by a simple equipment (Supplementary Fig. 1a) for 1H nuclear magnetic resonance (1H NMR) measurement. Only NH3 signal is detected in the 1H NMR (Table 1 (No. 1) and Supplementary Fig. 4). We think that the high boiling point of FA0 likely makes FA0 molecules difficult to diffuse into DMSO-d6 solvent. The formation of NH3 is indicative of the existence of some chemical reactions in the process of gas preparation (Supplementary Fig. 2). When the temperature of the reactor and the gas pipeline is increased to 150 °C and DMSO-d6 solvent is still kept at RT, four main compounds are detected: s-triazine, formamide, FA0, and NH3 (Table 1 (No. 2) and Supplementary Fig. 5). The formation of s-triazine is because of the transimination reaction of three FA0 molecules along with the formation of NH3 (Supplementary Fig. 2b)30,31. The s-triazine has also been detected when the FAI powder is heated at 150 °C32. Formamide is attributed to the hydrolyzation product of FA0 (Supplementary Fig. 2c) in the presence of H2O which is generated from the neutralization between FACl and NaOH. Considering the long-time interval from gas collection to 1H NMR measurement, we then employ acetic acid (HOAc) to capture FA0 so as to limit these chemical reactions in DMSO-d6 solvent by the quick formation of FAAc. As expected, 1H NMR spectrum verifies that the reaction product at 150 °C only exists FAAc rather than s-triazine, formamide, or NH4Ac (Table 1 (No. 3) and Supplementary Fig. 6). The above results strongly indicate the formation and instability of FA0. When FAI powder is exposed to FA0, there is hardly any change in chemical compositions except the formation of a tiny amount of formamide as the presence of H2O (Table 1 (No. 4), Supplementary Fig. 7). These spontaneous reactions and instability of FA0 seriously limit its practical application in post-healing FA-based perovskite films.
In comparison with FA0, amines are more stable. When FAI powder is treated with MA0, EA0, PA0, or BA0, the powder quickly transforms into a liquid state. After the liquid is held at 60 °C for 6 h and then vacuumed (Supplementary Fig. 1b), it is unexpected that there are no FAI signals in 1H NMR spectrum of the final powder samples (Table 1 (No. 5–8), Supplementary Figs. 8–11).
Taking MA0 as an example (Table 1 (No. 5)), we find that the main signals in 1H NMR spectrum belong to N,N’-dimethyl formamidinium iodide (DMFAI) (Supplementary Figs. 3 and 8)33. The related chemical reactions are illustrated in Fig. 1 (i, ii). The lone-pair electrons of N atom in nucleophilic MA0 molecule can attack electrophilic imine bond in FAI, which leads to the formation of N-methyl formamidinium iodide (MFAI) by a transimination process (Fig. 1(i), Supplementary Fig. 3)34. The imine bond in the formed MFAI can carry out the second transimination reaction with MA0 to form DMFAI (Fig. 1(ii), Supplementary Fig. 3). With excessive MA0 and holding for enough time, FAI can fully transform into DMFAI. Similarly, when FAI powders are treated with EA0, PA0 and BA0 gases, the transimination reaction also occur with the formation of DEFAI, DPFAI and DBFAI, respectively (Table 1 (No. 6–8), Supplementary Figs. 9–11, Fig. 1(i, ii)).

The transimination reactions and the ion exchange reactions between FAI and RA0 molecules (R refers to H, Me, Et, n-Pr, or n-Bu etc.), and the hydrolysis reactions of FA0, RFA0, and DRFA0.
When FAI powder is treated by RA0 (R is referred to H, Me, Et etc.) gas with some H2O vapor evaporated from their aqueous solution (Supplementary Fig. 1c), the final product is RAI rather than FAI, RFAI or DRFAI (Table 1 (No. 9–11), Supplementary Figs. 12–14). Considering there is no reaction between FAI salt and H2O (Table 1 (No. 12), Supplementary Fig. 15), the formation of RAI mentioned above is due to the reversible ion exchange reactions between FAI, RFAI or DRFAI and RA0 with the formation of RAI and FA0, RFA0, or DRFA0. The hydrolysis reactions of FA0, RFA0, and DRFA0 lead to the formation of volatile HCOOH and RA0, which in turn promotes the ion exchange reactions towards the formation of RAI (Fig. 1 (iii–x))35.
Supplementary Fig. 16 shows the optical photos and XRD patterns of α-FAPbI3 films before and after MA0 post-healing treatment. δ-FAPbI3, MFAPbI3, and DMFAPbI3 films are also measured for comparison. The formation of the non-perovskite phase after MA0 post-healing the α-FAPbI3 film is because of the conversion from FA+ to MFA+ and DMFA+. This is why aliphatic amines cannot be employed as solvents or post-healing gases for the fabrication of FA-containing perovskite materials. Inspired by the above transimination reaction, NH3 is selected to avoid the change of composition after the gas post-healing treatment. The 1H NMR spectrum well verifies no composition change of FAI powder after being treated with NH3 gas (Table 1 (No. 13), Supplementary Fig. 17).
Feasibility analysis of ammonia as the healing gas
The reversible absorption-desorption process of gas is crucial for the post-healing strategy. MAI and FAI samples are weighted during MA0/NH3 absorption and desorption as shown in Fig. 2a. The whole process can be divided into gas absorption at RT (Stage I), desorption at RT (Stage II), and desorption at 80 °C (Stage III). When exposed to MA0, MAI quickly absorbs MA0 and reaches a saturated state with the x value of about 2.5 in MAI·xMA0 (the x value is very sensitive to temperature and pressure). The liquid MAI·xMA0 complex removed from MA0 atmosphere desorbs MA0 gas spontaneously until it transforms into a relative stable semi-solid-state with the x value of about 1.1, which is denoted as stage II. After further heating at 80 °C for 10 min (Stage III), the sample returns to a white powder with the same weight as the initial MAI, which means the complete reversibility of absorption-desorption process in MAI-MA0 pair. In FAI-MA0 pair, the Stage I and Stage II are similar to these of MAI-MA0 pair, but the weight of the final powder in the Stage III cannot return back to the initial value of FAI because of the formation of MFAI and DMFAI as presented in Fig. 2a.

a Absorption and desorption behavior of MAI-MA0, FAI-MA0, and FAI-NH3. b The solution 15N NMR spectra of 15NH3, FAI(15N), FAI·x15NH3, PbI2·x15NH3, and FAPbI3·x15NH3. Signals: i, 15N signal in 15NH3; ii, 15N signal in FAI (15N); iii, 15N signal in FAI hydrogen bonded with 15NH3; iv, 15N signal of 15NH3 coordinated with Pb(II). c Reaction mechanism of transimination reactions between NH3 and FAI. d The photographs of PbI2/FAI powder in NH3 atmosphere under different temperatures. e The calculated x value of FAPbI3·xNH3 complex in an open condition under different temperatures.
FAI powder exposed to NH3 gas atmosphere rapidly achieves a liquid phase FAI·xNH3 (Supplementary Fig. 18) with the x value of ∼3.0 (Stage I). The liquid phase transforms into an ice-like transparent solid-state FAI·xNH3 with x ∼ 1.3 as NH3 desorption in Stage II and the XRD pattern of the ice-like complex is shown in Supplementary Fig. 17. The following thermal annealing at 80 °C fully removes the absorbed NH3 molecules and leads to the formation of a white powder with the same weight as the initial FAI (Stage III). This phenomenon, similar to that of MAI-MA0 pair, preliminarily indicates the feasibility of the NH3 as a post-healing gas for FA-based perovskite films.
To make a profound study, the chemical reaction and intermolecular interactions between FAPbI3 precursors and NH3 molecules, the solution 15N NMR spectra are measured as shown in Fig. 2b and Supplementary Figs. 19–24. The measured isotopically labeled 15N samples are 15NH3, FAI treated with 15NH3, and vacuumed (FAI(15N)), FAI absorbing 15NH3 molecules (FAI·x15NH3), PbI2 absorbing 15NH3 molecules (PbI2·x15NH3), and FAPbI3 absorbing 15NH3 molecules (FAPbI3·x15NH3). The 15N chemical shift of 15NH3 in DMSO-d6 is −377.46 ppm. The 15N NMR spectrum of FAI after treatment with 15NH3 (FAI (15N)) shows the 15N signal at −356.33 ppm, which indicates the existence of transimination reaction between NH3 and FAI (Supplementary Fig. 3). The reaction process between the NH3 and FAI is similar to the transimination reaction involving the aliphatic amine. The lone-pair electrons of N atom in NH3 can attack electrophilic imine bond in FAI, leading to the exchange of 15N in NH3 and 14N in FAI and formation of FAI(15N), as shown in Fig. 2c. Besides, the 15N chemical shift of FAI in FAI ·x15NH3 is at −265.22 ppm, which moves towards the low field about 91 ppm in comparison with FAI(15N). It suggests the presence of hydrogen bond interaction between NH3 and FAI. Furthermore, the 15N chemical shift of PbI2·x15NH3 at −357.51 ppm moves towards the low field about 20 ppm compared to that of 15NH3, which is due to the coordination of 15NH3 to Pb(II). The 15N chemical shifts of FAPbI3·x15NH3 at −265.02 ppm and −357.53 ppm correspond to that of FA+(15N) with hydrogen bond interaction with NH3 and that of 15NH3 with coordination bond interaction with Pb(II), respectively. The coexistence of hydrogen bond and coordination bond in the intermediate phase FAPbI3·x15NH3 is also supported by the solid-state 15N NMR result in Supplementary Fig. 25.
NH3, employed as a post-healing gas, should also have good solubility for the FA-based perovskite precursors, including PbI2 and FAI powders. The color of mixed PbI2/FAI powder exposed in NH3 gas atmosphere turns into light yellow from the yellow of PbI2 at 20 °C36. The mass difference before and after absorption shows that the absorbed NH3 molecule number per FAPbI3 is ∼4. While FAPbI3·4NH3 is still a solid-state rather than an expected liquid state, which is very different from liquid MAPbI3·xMA with x of only 317. One possible reason is the much smaller size of NH3 than MA0. We have found that the absorbed amount of gas is highly dependent on processing temperature23. Lowering the processing temperature to −5 °C leads to the formation of a white powder with x value of ∼6. At −15 °C, a flowable slurry with the x value of ∼12 is formed, as shown in Fig. 2d.
The kinetic process of NH3 desorption from the flowable FAPbI3·xNH3 complex is measured by monitoring its weight change with temperature increased step-by-step from 20 °C to 120 °C under an open condition. The absorbing NH3 number per FAPbI3 as the function of time illustrated in Fig. 2e shows that the weight of FAPbI3·xNH3 slurry drops rapidly at the initial stage and then decreases slowly in 30 min at 20 °C. Then the sample was annealed at 40 °C for 30 min and 60 °C for 30 min showing continuously slow weight loss by weighing at regular intervals. At 80 °C, the sample quickly reaches a relatively stable state then almost no weight loss in the maintained 30 min and the NH3 molecular number per FAPbI3 is about 1.0. Finally, the x value quickly decreases to 0 when the sample is annealed at 120 C.
Preparation and characterization of thin films
Then the NH3 post-healing strategy is employed to prepare FA-based perovskite films based on a homemade chamber with a semiconductor chilling plate controlling chamber temperature (Supplementary Fig. 26). As shown in Supplementary Fig. 27a, b, the raw FAPbI3 film with voids by the traditional one-step spin-coating method can transform into a uniform film after NH3 post-healing at −15 °C. The NH3 post-healing FAPbI3 is denoted as NH3-FAPbI3. The XRD spectra show that the NH3-FAPbI3 film has better crystallinity and less undesired δ phase than the raw film (Supplementary Fig. 27c). This leads to the increasing PCE of the NH3-FAPbI3 device compared to the raw-FAPbI3 device (Supplementary Fig. 27d). While the PCE of NH3-FAPbI3 device is still limited due to the existence of undesired δ-phase in the perovskite film. To prepare highly efficient PSCs, cesium (Cs) doped FAPbI3 material system FA0.9Cs0.1PbI3 (FACsPbI3) is selected. The NH3 post-healing process of FACsPbI3 film is schematically illustrated in Fig. 3a. Due to the thin thickness of perovskite films, the NH3 gas absorption and desorption are much easier and faster than those of powder samples. The SEM images show that the raw FACsPbI3 film could be well healed by the NH3 post-healing method in the temperature range from −15 °C to 0 °C, and −15 °C was used for NH3 gas post-healing in the following study (Supplementary Fig. 28).

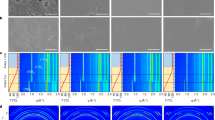
a Schematic illustration of NH3 post-healing FACsPbI3 perovskite thin film. b, c Scanning electron microscope (SEM) images of (b) raw FACsPbI3 and (c)NH3-FACsPbI3 perovskite films. d, e X-ray diffraction (XRD) patterns, f Ultraviolet-visible (UV-Vis) spectra and steady photoluminescence (PL) spectra of raw FACsPbI3 and NH3-FACsPbI3 perovskite films, respectively.
The scanning electron microscope (SEM) image (Fig. 3b) shows that there are some micrometer-scale voids in the raw FACsPbI3 film. While the NH3-FACsPbI3 film is dense and smooth (Fig. 3c), resulting from the self-leveling of the liquid intermediate state and the quick desorbing process of the NH3. This gas post-healing treatment with the self-leveling behavior is essentially different from the traditional post-treatment using DMF37, MASCN vapor25, etc. The corresponding atomic force microscope (AFM) images (Supplementary Fig. 29) show that the root mean square (RMS) roughness of NH3-FACsPbI3 film is only 9 nm over a 20 × 20 µm2 area, which is even lower than that of the film prepared by the traditional antisolvent method (24 nm) (denoted as anti-FACsPbI3).
X-ray diffraction (XRD) patterns in Fig. 3d and e show that the NH3 post-healing strategy can improve the orientation of the perovskite film, and the diffraction intensity of (110) peak of the NH3-FACsPbI3 perovskite film is about 100 times stronger than that of the raw FACsPbI3 perovskite film. Besides, NH3 post-healing decreases the full-width half-maximum (FWHM) of the (110) peak from 0.203o to 0.114o (Fig. 3e). As shown in ultraviolet-visible (UV-Vis) optical absorption spectra (Fig. 3f) and their Tauc plot curves (Supplementary Fig. 30), the raw and NH3-FACsPbI3 perovskite films present almost the same absorption edge and band gap of 1.54 eV. While the absorbance of NH3-FACsPbI3 perovskite film exhibits an obvious increase compared to that of raw perovskite film, resulting from the removal of voids. Photoluminescence (PL) peak of the NH3-FACsPbI3 film is similar to that of the anti-FACsPbI3 film (Supplementary Fig. 31), but obviously blue shift and narrow in comparison with the raw FACsPbI3 film, which indicates a low trap density of NH3-FACsPbI3 and anti-FACsPbI3 films in comparison with the raw film38. The reduced defect density is also evidenced by the charge-limited current (SCLC) and time-resolved photoluminescence (TRPL) results in Supplementary Fig. 32. The calculated defect density from the SCLC curves decreases from 3.97 × 1016 to 9.62 × 1015 cm−3. And the TRPL results show that the NH3-FACsPbI3 film has a much longer trap-assisted nonradiative lifetime (τ = 5.6 μs) than the Raw FACsPbI3 film (τ = 3.2 μs).
Device performance evaluation
The planar devices with a configuration of FTO/TiO2-SnO2/perovskite/2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenyl-amine)−9,9′-spirobifluorene (Spiro-OMeTAD)/Au are fabricated. The champion solar cell (Fig. 4a) based on NH3-FACsPbI3 film displays a PCE of 23.21%, with open-circuit voltage (VOC) of 1.16 V, short-circuit current density (JSC) of 24.65 mA/cm2, fill factor (FF) of 81.20%, demonstrating obvious improvement in comparison with the PCE of 10.92% of the control device based on raw FACsPbI3 film and comparable performance compared to the devices (PCE = 22.99%) based on the anti-FACsPbI3 film in our lab (Supplementary Fig. 33). The certified PCE of NH3-FACsPbI3 PSC is 22.22% (Supplementary Fig. 34). Meanwhile, there is less hysteresis for the NH3-FACsPbI3 device (Supplementary Fig. 35). The stabilized power output (SPO) of NH3-FACsPbI3 champion cells is 22.93%, shown in Supplementary Fig. 36. The JSC of NH3-FACsPbI3 PSC is comparable with the integrated JSC from EQE results (24.41 mA/cm2) in Fig. 4b. The distribution histogram of PCE based on 50 NH3-FACsPbI3 devices at reverse scan direction (Fig. 4c, Supplementary Table 1) shows excellent reproducibility of high-performance NH3 post-healing devices. The cross-sectional SEM image of NH3-FACsPbI3 PSC shows the uniform thickness and good interfacial contact of perovskite film with transport layers (Supplementary Fig. 37). Combined NH3 gas healing and the doctor-blading technology (Suzhou GCL Nano Co. Ltd.), a prototype PSC module consisting 10 cells in series connection achieves a PCE of 20.61% (Fig. 4d) and the certified PCE of the module is 19.38% (Supplementary Fig. 38) with an estimated active area of 14.00 cm2, which is comparable with the reported highest efficiencies in perovskite modules39,40,41.

a Current density–voltage (J-V) curve of the champion PSCs based on NH3-FACsPbI3. b External quantum efficiency (EQE, red) and the integrated short-circuit current density (blue) of the champion NH3-FACsPbI3 device. c Histogram of the power conversion efficiency for 50 NH3-FACsPbI3 PSCs. d J-V curve of NH3-FACsPbI3 PSC module. e Normalized power conversion efficiency (PCE) of unencapsulated NH3-FACsPbI3, anti-FACsPbI3, anti-FMCsPbI3, and anti-MAPbI3 devices with the storage time of 320 days under 10–30% relative humidity at room temperature. f Evolution of the normalized PCE over time measured by maximum power point tracking of unencapsulated NH3-FACsPbI3, anti-FACsPbI3, anti-FMCsPbI3, and anti-MAPbI3 devices under light soaking with full solar intensity. Standard deviation (error bar) was calculated from three individual devices in the same batch.
To further evaluate the device stability based on NH3 post-healing treatment, the high-efficiency NH3-FACsPbI3, anti-FACsPbI3, anti-FA0.90Cs0.05MA0.05PbI3 (anti-FMCsPbI3), and anti-MAPbI3 PSCs are employed. As the low PCE, raw FACsPbI3 device has not been chosen to study its stability. The shelf stability of unencapsulated devices in Fig. 4e shows that NH3-FACsPbI3 device has negligible performance loss after 320 days of storage, which is similar to the anti-FACsPbI3 devices and much better than anti-FMCsPbI3 and anti-MAPbI3. The XRD and UV-Vis spectra of these aged perovskite films in 320-day-aged PSCs (Supplementary Figs. 39–41) show that NH3-FACsPbI3 film keeps its strong light absorption, and has few PbI2. Meanwhile, NH3-FACsPbI3 device still maintains decent cross-sectional morphology with clear grain boundaries (Supplementary Fig. 42). What’s more, the maximum power point (MPP) of the unencapsulated NH3-FACsPbI3, anti-FACsPbI3, anti-FMCsPbI3, and anti-MAPbI3 PSCs is tracked under white light emitting diode (LED) irradiation (Fig. 4f, Supplementary Fig. 43) with an intensity equivalent to 1 sun in N2 atmosphere. After 500 h of continuous light soaking, the NH3-FACsPbI3 device maintains 80% of its initial efficiency, and the stability trend of MPP tracking is consistent with that of shelf stability in these devices.
Discussion
We have systematically studied the transimination reactions between FA/RA gases and FAI salt, and further developed an NH3 gas post-healing strategy for upscale fabrication of high-quality FA-based perovskite films. The low operating temperature during NH3 post-healing process is vital to enable FA-based perovskite materials to absorb enough NH3 molecules and transform into a flowable intermediate state. Based on this strategy, the champion device achieves a PCE of 23.21% (Certified PCE of 22.22%), and the PCE of the perovskite module is up to 20.61%. Meanwhile, the device stability based on this strategy is also comparable with that of the state-of-the-art anti-solvent method. This NH3 gas post-healing technology is compatible with established commercial technologies to efficiently remove voids in the raw FA-based films and therefore opens a direction for fabrication of large-scale high-efficient FA-based PSCs.
Methods
Materials
N,N-dimethylformamide (DMF, anhydrous 99.8%), dimethyl sulfoxide (DMSO, anhydrous 99.9%) were procured from Sigma-Aldrich. Formamidine hydrochloride (FACl, 96%), Ethylamine Hydrochloride (EACl, 98%), n-Propylamine, (PA, 99%), n-Butylamine (BA, 98%), and Lead iodide (PbI2, >98.0%) was purchased from TCI. Formamidinium iodide (FAI, 99.5%), Methanaminium iodine (MAI, 99.5%), Cesium iodide (CsI, 99.99%), and Spiro-OMeTAD (99.8%) were purchased from Xi’an Polymer Light Technology Corp. (PLT). Ammonium chloride (15NH4Cl, 10 atom% 15N labelled, ≥98.5%) was purchased from Macklin. Ammonium chloride (15NH4Cl, 99 atom% 15N labelled, >98%) and DMSO-d6 (99 atom% D labelled) were purchased from Cambridge Isotope Laboratories, Inc. (CIL). Sodium hydroxide (NaOH, 96%), Methylamine alcohol solution (30–33 wt. % in anhydrous ethanol), and Ammonium Hydroxide (NH3 ∙ H2O, 25∼28%) were purchased from Sinopharm Chemical Reagent Co., Ltd. The above chemicals were used as received without any purification.
Material synthesis
For the synthesis of N-methyl formamidinium iodide (MFAI), Methylamine alcohol solution (30–33 wt. % in anhydrous ethanol, 425 μl, about 2.8 mmol) was added dropwise to a solution of formamidinium iodide (FAI, 516 mg, 3 mmol) in 30 ml anhydrous ethanol under nitrogen protection, and the mixture was stirred at 0 °C for 2 h. Subsequently, the solvent was evaporated under reduced pressure and the residue was recrystallized in the mix solvent of isopropanol and ethyl acetate in glove box to obtain the MFAI powder. The synthesis of N, N’-dimethyl formamidinium iodide (DMFAI) was similar to that of MFAI, but the amount of the methylamine alcohol solution was increased to 1 ml.
NMR sample fabrication
For solution 1H NMR characterization of the sample in Table 1 (No.1), the mixture of 5.0 g FACl and 5.0 g NaOH was reacted at RT in a conical flask, and the generated gas was collected by 0.5 ml DMSO-d6. For samples in Table 1 (No. 2–4), the mixture of 5.0 g FACl and 5.0 g NaOH was reacted at 150 °C, and the generated gas was collected by 0.5 ml DMSO-d6, 1 ml HOAc or 0.2 g FAI powder at RT. Samples No. 3 and 4 were vacuumed before dissolving in DMSO-d6. For samples in Table 1 (No. 5–12), the gas sources of R-NH2 were heated at 60 °C, and the generated gases were collected by 0.2 g FAI powder. The gas sources of R-NH2 were methylamine alcohol solution, the mixture of EACl and NaOH, PA, BA, NH3·H2O, MA aqueous solution, EA aqueous solution, or H2O, respectively. The samples (No. 5–12) were kept at 60 °C for 6 h, and then vacuum treated to remove the low boiling point components. For the sample in Table 1 (No. 13), the NH3 gas was collected with 0.2 g FAI powder. The sample was kept at RT for 6 h and then vacuumed. The obtained solid powders in Table 1 (No. 3–13) were dissolved in DMSO-d6 for 1H NMR characterization.
For solution 15N NMR characterization, 15NH3 was produced from the reaction between 5.0 g 15NH4Cl (10% 15N labelled) and 5.0 g NaOH at 60 °C, and the generated gas was collected by 0.5 ml DMSO-d6. FAI (15N) was produced based on the reaction between 0.2 g FAI and excess 15NH3 at RT for 6 h, followed by vacuum treatment. FAI·x15NH3, PbI2·x15NH3, and FAPbI3·x15NH3 were generated between the reaction between 0.2 g FAI, 0.2 g PbI2, or 0.2 g FAPbI3 precursor powders and excess 15NH3 at RT for 6 h, but without vacuum treatment. The above samples were directly dissolved in DMSO-d6 for 15N NMR measurement.
The NMR sample fabrication of FAPbI3·x15NH3 for the solid-state NMR spectrum was similar to that of the solution 15N NMR spectrum, but 15N isotope source for preparing 15NH3 was used 15NH4Cl (99% 15N labelled) as the requirement of high 15N concentration for solid-state NMR.
NMR measurements
The solution 1H NMR spectra were measured with a Bruker AVANCE III 600 instrument operating at 600.13 MHz at 298 K. Chemical shifts d (ppm) were referenced to the internal solvent signals.
The solution 1H-decoupled 15N NMR spectra (pules program zgig) were acquired using a Bruker Avance III 600 instrument operating at 600.13 MHz for 1H and 60.82 MHz for 15N. The 15N chemical shifts were determined from 1 M urea in DMSO (−303.20 ppm, 10% 15N labelled) as external standard reference 42. The relaxation delay was 10 s, the acquisition time was 1.3 s, and 128 scans were accumulated.
The solid-state NMR experiment was performed on a Bruker AVNCE 400 spectrometer operating at 399.95 MHz for 1H and 40.54 MHz for 15N. The obtained sample was packed into a 7 mm ZrO2 rotor. The 15N chemical shifts were determined from the 15NH4Cl (−341.17 ppm, 10% 15N labelled)43. The HPEDC pulse sequence was used. The acquisition time was 50 ms, the spin rate was 3 kHz, and the recycle delay was 5 s. The two-pulse phase-modulation (TPPM) decoupling was used during the acquisition.
Perovskite film fabrication
Raw FACsPbI3 films: The perovskite precursor was prepared by mixing PbI2: FAI: CsI (1:0.9:0.1 molar ratio) in DMF solvent (1.40 M). Raw FAPbI3 films: The perovskite precursor was prepared by mixing PbI2: FAI (1:1 molar ratio) in DMF solvent (1.40 M). The raw FACsPbI3 and FAPbI3 films were simply fabricated by one-step spin-coating at 4000 rpm for 30 s and then heated at 140 °C for 20 min.
NH3-FACsPbI3 film: After spin-coating, the raw FACsPbI3 film was annealed at 140 °C for 5 min, and then transferred into a homemade chamber with a temperature from 20 to −15 °C. N2 gas was used to remove the moisture-laden air in the chamber. Subsequently, NH3 gas was quickly introduced into the chamber and maintained for ∼5 s. Then the NH3 gas is removed from the chamber and the film was further annealed at 140 °C for 20 min. For NH3-FAPbI3 film, the film treatment process is the same as above, and the temperature of the homemade chamber is −15 °C for NH3 post-healing process.
For anti-FACsPbI3, anti-FMCsPbI3, and anti-MAPbI3 films, the perovskite precursors were prepared in DMF: DMSO (3:7, 1.40 M) by stoichiometric ratios. The perovskite solution was spin-coated in a two-step at 1000 rpm and 4000 rpm for 10 s and 30 s, respectively. During the second step, 300 μl anisole was drop-casted quickly at the tenth second of the second step. The anti-MAPbI3 perovskite film was then heated at 100 °C for 20 min, and the other films were heated at 140 °C for 20 min.
The α-FAPbI3, δ-FAPbI3, MFAPbI3, and DMFAPbI3 films were fabricated from their DMF solution with a concentration of 1.40 M using chlorobenzene as the antisolvent. The α-FAPbI3, MFAPbI3, and DMFAPbI3 films were obtained after heating treatment at 140 °C for 20 min. The δ-FAPbI3 film was achieved by exposing α-FAPbI3 film in the air (RH > 50%) until fully transformed into yellow. The MA-FAPbI3 film was prepared by the MA0 gas post-healing the α-FAPbI3 film and then heated at 140 °C for 20 min.
The annealing process of all thin films is in ambient air conditions (30–40% humidity).
Device fabrication
The methods described are similar to that reference 44. Fluorine doped tin oxide (FTO)-coated glass (2.20 mm, 7Ω sq−1) was used as the substrate for the devices. The compact TiO2 layer (∼10 nm) was deposited by atomic layer deposition (ALD) for 200 cycles and annealed at 500 °C for 30 min in ambient air. For ALD TiO2 deposition, titanium (IV) isopropoxide (TTIP) and H2O as Ti and O sources, respectively. The TTIP precursor was held at 75 °C. Pulse/exposure/purge times of 1 s/8 s/25 s were used for the TTIP and 0.1 s/8 s/25 s for H2O precursor, and the deposition temperature was set to 120 °C. On top of the c-TiO2 layer, SnOx-Cl layer was deposited by spin-coating at a speed of 3000 rpm for 30 s from an aged SnCl4 aqueous solution (1:75 with deionized water by volume), followed by a sintering heat-treatment of 200 °C for 30 min in air and then transferred to the glove box for device fabrication. The perovskite layers were fabricated as described above. For the surface passivation, 1 mg ml−1 of Phenyltrimethylammonium tribromide (PTAB) solution in isopropanol was spin-coated on these perovskite films at 4000 rpm for 30 s.
The spiro-OMeTAD chlorobenzene solution (72.3 mg ml−1) with 28.8 μl 4-tert-butylpyridine (96%, Aldrich-Sigma) and 17.5 μl lithium bis(trifluoro-methanesulfonyl) imide (Li-TSFI, Aldrich-Sigma) solution (520 mg Li-TSFI (98%) in 1 ml acetonitrile (99.8%, Aldrich-Sigma)) was spin-coated on top of the perovskite film at 3000 rpm for 30 s. The devices were put into a dry-air box (RH < 3%) for 12 h. Finally, 80 nm thick Au electrode was thermally evaporated.
For fabricating modules, P1 etching process was pre-patterned on FTO glass (5 cm × 5 cm) with a 1064 nm fiber laser (Han’s laser). The laser power ratio, laser duty cycle, and laser frequency were 30%, 5%, and 50 kHz, respectively. Then, patterned FTO substrates were cleaned and treated by UV Ozone Cleaner (Ossila) for 15 min. The TiO2/SnO2, PTAB, and Spiro-OMeTAD layers were prepared with the same procedure as presented above. The large-size raw FACsPbI3 perovskite film was prepared through the doctor-blading method (provided by Suzhou GCL Nano Co. Ltd.). The NH3 post-healing strategy was carried out on it to form a large-scale NH3-FACsPbI3 film. For P2 etching process, the laser used was a 532 nm laser with a laser power ratio of 65%, a laser duty cycle of 5%, and a laser frequency of 100 kHz. 80 nm thick Au electrodes were thermally evaporated under vacuum to complete the modules fabrication. Finally, P3 etching used the same laser with P2 with a laser power ratio of 50%, a laser duty cycle of 5%, and a laser frequency of 100 kHz. P4 is an etching procedure for cleaning the edge of the modules, the laser used in P4 is the same with P1 with a laser power ratio of 40%, the laser duty cycle of 10%, and a laser frequency of 100 kHz.
Perovskite film and device characterization
The characterization described are similar to that reference 44. XRD spectra were measured by Ultima IV of Rigaku with Cu Kα radiation (1.5406 Å). The UV-Vis absorbance spectra were measured by QE Pro (Ocean Optics). Top view, cross-section SEM images were obtained with a field-emission SEM (S-4800, Hitachi). Steady PL spectra were recorded on QE Pro excited at 460 nm. AFM measurements were performed in contact mode (5400, Agilent). EQE measurement was calculated using certified incident photon to current conversion efficiency equipment from Enlitech (QE-R). Time-resolved photoluminescence (TRPL) experiments were performed by Steady State and Transient State Fluorescence Spectrometer (Edinburgh FLS980). The testing conditions as the films were photoexcited at 483.6 nm pulse width ∼ 118.6 ps, 5 mW/pulse, and emission were collected on the surface side of the film (perovskite/ glass substrate).
J-V curves of the as-fabricated PSCs were measured using a SourceMeter (Keithley 2400) under simulated one-sun AM 1.5 G 100 mW cm−2 intensity (Oriel Sol3A Class AAA, Newport) with a scan rate of 200 mV/s (the voltage step is 20 mV with no delay time) from reverse and forward two scanning directions in air condition around 25 °C. The typical active area of PSCs is 0.09 cm2 defined by a metal mask. The area of the mask (0.08713 cm2) used for certification was certified by the National Institute of Metrology, China, No. CDjc2021-10891. The intensity of one-sun AM 1.5 G illumination was calibrated using a Si-reference cell certified by the National Renewable Energy Laboratory. SPO is measured by tracking the current under a fixed voltage which is decided by the voltage of maximum power point at the J-V curve. Here, the fixed voltage of NH3-FACsPbI3 device is 1.002 V.
For the stability tests, all PSCs were without encapsulation. For shelf-life stability, the devices were stored in a dark environment with a humidity of 10–30%, and the photovoltaic performance of PSCs was measured every ten days. The operational stability was performed using a stability setup (LC Auto-Test 24, Shenzhen Lancheng Technology Co., Ltd.), tested under continuous light illumination and maximum power point tracking (controlled and monitored to be 15 °C). The light source consisted of an array of white LEDs powered by a constant current. The LED type is MG-A200A-AE with an emission spectrum of 400–750 nm (Supplementary Fig. 40). Equivalent sun intensities were calibrated using a calibrated Si-reference cell. During aging, the device is connected with a 100 Ohm load resistance. The PSCs were masked and placed inside a sample holder purged with continuous N2 flow. J-V curves with reverse voltage scans were recorded every 12 h during the whole operational test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data that support the findings of this study are available in Supplementary Data Files in the Supplementary Information section. Source data are provided with this paper.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Research-Cell Efficiency Records NREL, https://www.nrel.gov/pv/cell-efficiency.html (Accessed: June 7, 2021).
Green, M. A., Ho-Baillie, A. & Snaith, H. J. The emergence of perovskite solar cells. Nat. Photonics 8, 506–514 (2014).
Jeon, N. J. et al. Solvent engineering for high-performance inorganic-organic hybrid perovskite solar cells. Nat. Mater. 13, 897–903 (2014).
Burschka, J. et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499, 316–319 (2013).
Jiang, Q. et al. Surface passivation of perovskite film for efficient solar cells. Nat. Photonics 13, 460–466 (2019).
Liu, C., Cheng, Y. B. & Ge, Z. Understanding of perovskite crystal growth and film formation in scalable deposition processes. Chem. Soc. Rev. 49, 1653–1687 (2020).
Li, L. et al. The additive coordination effect on hybrids perovskite crystallization and high-performance solar cell. Adv. Mater. 28, 9862–9868 (2016).
Yang, W. S. et al. High-performance photovoltaic perovskite layers fabricated through intramolecular exchange. Science 348, 1234–1237 (2015).
Hamill, J. C., Schwartz, J. & Loo, Y.-L. Influence of solvent coordination on hybrid organic-inorganic perovskite formation. ACS Energy Lett. 3, 92–97 (2017).
Radicchi, E., Mosconi, E., Elisei, F., Nunzi, F. & De Angelis, F. Understanding the solution chemistry of lead halide perovskites precursors. ACS Appl. Energy Mater. 2, 3400–3409 (2019).
Li, Y. et al. Investigation on crystallization of CH3NH3PbI3 perovskite and Its intermediate phase from polar aprotic solvents. Cryst. Growth Des. 19, 959–965 (2018).
Fei, C. B. et al. Highly efficient and stable perovskite solar cells based on monolithically grained CH3NH3PbI3 film. Adv. Energy Mater. 7, 1602017 (2017).
Zhang, H. et al. Toward all room-temperature, solution-processed, high-performance planar perovskite solar cells: a new scheme of pyridine-promoted perovskite formation. Adv. Mater. 29, 1604695 (2017).
Li, B., Binks, D., Cao, G. & Tian, J. Engineering halide perovskite crystals through precursor chemistry. Small 15, e1903613 (2019).
Lee, J. W., Lee, D. K., Jeong, D. N. & Park, N. G. Control of crystal growth toward scalable fabrication of perovskite solar cells. Adv. Funct. Mater. 29, 1807047 (2018).
Zhou, Z. M. et al. Methylamine-gas-induced defect-healing behavior of CH3NH3PbI3 thin films for perovskite solar cells.Angew. Chem. Int. Ed. 54, 9705–9709 (2015).
Zhang, Y. et al. Trash into treasure: δ-FAPbI3 polymorph stabilized MAPbI3 perovskite with power conversion efficiency beyond 21%. Adv. Mater. 30, e1707143 (2018).
Wang, K. et al. A nonionic and low-entropic MA(MMA)nPbI3-ink for fast crystallization of perovskite thin films. Joule 4, 615–630 (2020).
Long, M. et al. Nonstoichiometric acid-base reaction as reliable synthetic route to highly stable CH3NH3PbI3 perovskite film. Nat. Commun. 7, 13503 (2016).
Liu, Z. et al. Gas-solid reaction based over one-micrometer thick stable perovskite films for efficient solar cells and modules. Nat. Commun. 9, 3880 (2018).
Noel, N. K. et al. A low viscosity, low boiling point, clean solvent system for the rapid crystallisation of highly specular perovskite films. Energy Environ. Sci. 10, 145–152 (2017).
Pang, S. et al. Transformative evolution of organolead triiodide perovskite thin films from strong room-temperature solid-gas interaction between HPbI3-CH3NH2 precursor pair. J. Am. Chem. Soc. 138, 750–753 (2016).
Lee, J. W. et al. 2D perovskite stabilized phase-pure formamidinium perovskite solar cells. Nat. Commun. 9, 3021 (2018).
Lu, H. et al. Vapor-assisted deposition of highly efficient, stable black-phase FAPbI3 perovskite solar cells. Science 370, 74 (2020).
Saliba, M. et al. Cesium-containing triple cation perovskite solar cells: improved stability, reproducibility and high efficiency. Energy Environ. Sci. 9, 1989–1997 (2016).
Yi, C. et al. Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells. Energy Environ. Sci. 9, 656–662 (2016).
Bi, D. et al. Multifunctional molecular modulators for perovskite solar cells with over 20% efficiency and high operational stability. Nat. Commun. 9, 4482 (2018).
Lee, J.-W. et al. Formamidinium and cesium hybridization for photo- and moisture-stable perovskite solar cell. Adv. Energy Mater. 5, 1501310 (2015).
Schaefer, F. C., Hechenbleikner, I., Peters, G. A. & Wystrach, V. P. Synthesis of the sym-triazine system. I. trimerization and cotrimerization of amidines. J. Am. Chem. Soc. 81, 1466–1470 (2002).
Dahlman, C. J., Kubicki, D. J. & Reddy, G. N. M. Interfaces in metal halide perovskites probed by solid-state NMR spectroscopy. J. Mater. Chem. A. 9, 19206–19244 (2021).
Kubicki, D. J., Stranks, S. D., Grey, C. P. & Emsley, L. NMR spectroscopy probes microstructure, dynamics and doping of metal halide perovskites. Nat. Rev. Chem. 5, 624–645 (2021).
Wang, X. et al. Perovskite solution aging: what happened and how to inhibit? Chem 6, 1369–1378 (2020).
Ciaccia, M., Cacciapaglia, R., Mencarelli, P., Mandolini, L. & Di Stefano, S. Fast transimination in organic solvents in the absence of proton and metal catalysts. A key to imine metathesis catalyzed by primary amines under mild conditions. Chem. Sci. 4, 2253–2261 (2013).
Shriner, R. L. & Neumann, F. W. The chemistry of the amidines. Chem. Rev. 35, 351–425 (2002).
Preda, N. et al. Films and crystalline powder of PbI2 intercalated with ammonia and pyridine. J. Mater. Sci.: Mater. Electron. 20, 465–470 (2008).
Xiao, Z. et al. Solvent annealing of perovskite-induced crystal growth for photovoltaic-device efficiency enhancement. Adv. Mater. 26, 6503–6509 (2014).
Dong, Q. et al. Electron-hole diffusion lengths >175 μm in solution-grown CH3NH3PbI3 single crystals. Science 347, 967–970 (2015).
Bu, T. et al. Lead halide-templated crystallization of methylamine-free perovskite for efficient photovoltaic modules. Science 372, 1327–1332 (2021).
Chen, S. et al. Stabilizing perovskite-substrate interfaces for high-performance perovskite modules. Science 373, 902–907 (2021).
Huang, H.-H. et al. A simple one-step method with wide processing window for high-quality perovskite mini-module fabrication. Joule 5, 958–974 (2021).
Andersson, H., Carlsson, A.-C. C., Nekoueishahraki, B., Brath, U. & Erdélyi, M. Solvent effects on nitrogen chemical shifts. Annu. Rep. NMR Spectrosc. 86, 73–210 (2015).
Hayashi, S. & Hayamizu, K. Chemical shift standards in high-resolution solid-state NMR (2) 15N nuclei. Bull. Chem. Soc. Jpn. 64, 688–690 (1991).
Li, Z. et al. Inhibiting ion migration by guanidinium cation doping for efficient perovskite solar cells with enhanced operational stability. Sol. RRL 6, 220003 (2022).
Acknowledgements
S.P.P. acknowledges funding from the Youth Innovation Promotion Association CAS (Y201944), Funding of Young Taishan Scholars (tsqn201812110), Natural Science Foundation of Shandong Province (ZR2020KB001), and funding from Shandong Energy Institute (SEI I202129), X.W. thanks the Natural Science Foundation of Shandong Province (ZR2020KB001). Z.P.S. thanks the National Natural Science Foundation of China (51902324). We thank Suzhou GCL Nano Co. Ltd. for the fabrication of large-sized perovskite films and the Qingdao Key Lab of Solar Energy Utilization & Energy Storage Technology.
Author information
Authors and Affiliations
Contributions
L.W., G.L.C., and S.P.P. conceived and planned the experiments with additional input from Z.W.W. and Z.P.S. Z.P.L. fabricated all samples and devices, and performed and analyzed the SEM, XRD experiments, the device efficiency, and stability experiments. X.W. performed and analyzed the NMR experiments. L.Z.H., Y.R., C.C., and D.C.L. carried out the gas absorbing and desorbing test. Q.Q.Z. and X.H.S. assisted with film fabrication and optical measurement. C.Y.G. and B.Q.Z. helped to perform and analyze NMR measurements. X.Z.W. helped to perform TRPL measurements. Z.P.L., Z.W.W., and S.P.P. took the lead in drafting the manuscript and compiled the figures. All authors discussed the results and provided feedback on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dominik Kubicki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Z., Wang, X., Wang, Z. et al. Ammonia for post-healing of formamidinium-based Perovskite films. Nat Commun 13, 4417 (2022). https://doi.org/10.1038/s41467-022-32047-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32047-z
This article is cited by
-
Strain relaxation and multidentate anchoring in n-type perovskite transistors and logic circuits
Nature Electronics (2024)
-
Molecular cation and low-dimensional perovskite surface passivation in perovskite solar cells
Nature Energy (2024)
-
Unveiling the surface-interface properties of perovskite crystals and pivotal regulation strategies
Nano Research (2024)
-
Dopant-additive synergism enhances perovskite solar modules
Nature (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
