
Abstract
Earth-abundant electrocatalysts for the oxygen evolution reaction (OER) able to work in acidic working conditions are elusive. While many first-row transition metal oxides are competitive in alkaline media, most of them just dissolve or become inactive at high proton concentrations where hydrogen evolution is preferred. Only noble-metal catalysts, such as IrO2, are fast and stable enough in acidic media. Herein, we report the excellent activity and long-term stability of Co3O4-based anodes in 1 M H2SO4 (pH 0.1) when processed in a partially hydrophobic carbon-based protecting matrix. These Co3O4@C composites reliably drive O2 evolution a 10 mA cm–2 current density for >40 h without appearance of performance fatigue, successfully passing benchmarking protocols without incorporating noble metals. Our strategy opens an alternative venue towards fast, energy efficient acid-media water oxidation electrodes.
Similar content being viewed by others
Introduction
Hydrogen is considered the most environmentally friendly alternative fuel to replace traditional fossil energy1,2. However, hydrogen production is still dominated worldwide by natural gas steam reforming, a mature technology fixing a very low competitive price in the market. Electrochemical water splitting powered by renewable sources is regarded as the ideal future technology to produce hydrogen, but costs must be lowered to improve its market penetration3,4. The catalysts responsible to improve the efficiency of the process, mainly relying on noble metals, are part of the high cost of the technology, and because of this, many laboratories in the world are working to find viable solutions to develop effective, earth-abundant, robust and scalable catalyst5,6,7,8.
Acidic electrolytes offer many advantages for hydrogen production, given that high H+ concentration improves the hydrogen evolution reaction (HER), and also facilitates fast/low resistance ionic transport9,10. Several electrocatalysts based on low-cost raw materials have shown great promise to substitute the state-of-the-art platinum electrodes responsible for HER6,7,11,12,13,14. On the contrary, no viable candidates are known to substitute noble-metal oxides such as IrO2 at the anode where the oxygen evolution reaction (OER) takes place1,6,9,15,16,17,18,19,20,21. So far, no stable and inexpensive OER catalysts can work under high potential and/or current densities in acidic media, where even the highly active RuO2 presents serious deactivation problems22,23.
Several strategies have been proposed to promote OER at high proton concentration24,25,26,27,28. One strategy deals with the investigation of ternary/complex oxide structures such as nickel-manganese antimonate. This rutile-type oxide was stable for 168 h at 10 mA cm–2 operating in 1 M sulfuric acid, although with the penalty of requiring a large overpotential (η ≥ 700 mV)26. Cobalt-doped hematite thin-film electrocatalysts were also able to sustain a geometric current density of 10 mA cm–2 for up to 50 h at pH 0.1, but again at large overpotentials (η ≥ 650 mV)27. Indeed, cobalt oxides have shown promising OER electrocatalysis in acidic media, although highly limited by its redox potential-dependent instability, since CoOx may rapidly dissolve either at open circuit conditions or under high applied potentials29,30,31,32. Some interesting advances in long-term stability were achieved by doping or processing techniques33,34,35.
In our previous work with polyoxometalate (POM)-supported catalysts, we disclosed how these molecular catalysts showed promising OER catalytic performance in acidic conditions when incorporating active Co centers36. The high activity of these catalysts, even in heterogeneous conditions, was achieved thanks to the synergic stability offered by a partially hydrophobic carbon-based support. However, this strategy was not successful to achieve long-term stability, since these POM-based electrodes could only survive at low current densities and for a very limited time, given their intrinsic instability to the mechanical stress provoked by gas bubbling.
In this work, we present a promising processing protocol, which combines in one single anode two powerful strategies: (i) the incorporation of a nanostructured OER catalyst from earth-abundant metals to maximize active surface area, (ii) supported by a conducting, partially hydrophobic binder made from paraffin oil and graphite powder. Our processing protocol with nitrogen-doped carbon-coated Co3O4 nanoparticles (Co3O4@C) delivers robust and scalable anodes that exhibit excellent acidic OER performances, needing a minimum overpotential (η ≤ 398 mV) to maintain a 10 mA cm–2 current density for >40 h when working in 1 M sulfuric acid solution, without any sign of fatigue or deactivation. The high activity and also great stability demonstrate a performance superior to any other non-noble catalysts reported. Oxygen evolution quantification confirms the Faradaic efficiency (>96%) of these electrodes towards OER, with negligible participation of other oxidation processes. These results open alternative opportunities for stable OER electrocatalysis with earth-abundant raw materials.
Results and discussion
Catalyst and electrode preparation
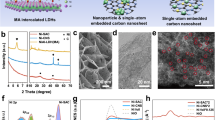
The overall synthetic and processing protocol is summarized in Fig. 1. We synthesized carbon-coated Co (Co@C) nanoparticles, starting from the thermal treatment of Co(bIm)2 (bIm = 2-benzimidazolate), a metal-organic framework (MOF) precursor (ZIF-9)37. Then Co@C was oxidized at low-temperature to achieve its full transformation into cobalt oxide nanoparticles, covered by an amorphous, nitrogen-doped-carbon coating derived from the organic skeleton (Co3O4@C, Fig. 1). Powder X-ray diffraction (PXRD) patterns and Raman spectra confirmed the presence of a Co3O4 phase and the carbon support (Supplementary Figs. 1–4). High-resolution transmission electron microscopy the presence of graphitic-like nanostructures all around the sample, embedding the Co3O4 nanoparticles. Some of these C-nanostructures had a nanosheet-like morphology (Fig. 2), while some others were folded forming onion-like rings around the Co3O4 nanoparticles (Supplementary Fig. 5). Electron energy loss spectroscopy in scanning TEM mode (EELS-STEM) confirmed the chemical composition of the nanoparticles and surrounding nanostructures (Supplementary Fig. 6). The Co3O4@C composition was determined as (Co3O4)(H2O)0.30(OH)0.85C2.00N0.05 by thermogravimetry elemental analysis (Supplementary Fig. 7 and Supplementary Table 1).

Scheme of the synthesis and processing protocol to obtain a Co3O4@C/GPO electrode.

HAADF STEM (a) and HRTEM (b) micrographs from a fresh Co3O4@C/GPO composite. The power spectrum analysis confirms the presence of Co3O4 nanoparticles with a cubic FD3-MZ (s.g. 227) spinel structure, oriented along its [011] axis (yellow circles) superimposed to the reflections corresponding to graphite layers with a ≈ 0.34 nm spacing and here oriented along the [–1–12–3] (turquoise circles). To highlight the different systems, we show a frequency filtered map (on the bottom right) where the Co3O4 corresponding lattice fringes are in yellow and the graphite layer ones in turquoise . Source data are provided as a Source Data file.
For the preparation of the electrode composites, Co3O4@C was mixed with graphite (G) and paraffin oil (PO) in the desired ratio (see Methods section) to prepare a homogeneous composite (Co3O4@C/GPO) with the desired Co3O4@C content up to 40% (40-Co3O4@C). Composites above 40% were mechanically too fragile for further processing into the working electrode pocket. HRTEM images and EELS-STEM maps showed similar nanostructures within Co3O4@C/GPO and close contact between Co3O4@C and GPO (Fig. 2 and Supplementary Fig. 8). X-ray photoemission spectroscopy (XPS) analysis was employed to further identify the surface chemical composition and the mixed oxidation state Co2+/3+ (Supplementary Fig. 9) consistent with the presence of the Co3O4, as confirmed by PXRD and HR-TEM data38. XPS spectra from the Co3O4@C/GPO composite show no differences respect to the Co3O4@C precursor, demonstrating the absence of chemical modification during composite preparation.
OER electrocatalytic activity in 1 M H2SO4
The x-Co3O4@C/GPO composites (x corresponds to the % in weight for the metal oxide) were inserted into the pocket of a working electrode and used as anode during electrochemical water oxidation in 1 M H2SO4 (pH ≈ 0.1). The cyclic voltammetry (CV) showed the appearance of a catalytic current density on the Co3O4@C/GPO electrode at relatively low overpotentials, which was sustained after successive cycling curves (Supplementary Figs. 10 and 11). Comparative linear sweep voltammetry (LSV) showed an enhanced electrochemical activity upon increasing Co3O4@C content, reaching a very low onset overpotential (ηonset = 194 ± 4 mV) for the 40-Co3O4@C/GPO electrode (Fig. 3 and Supplementary Fig. 12). These electrodes reach 10 mA cm–2 currents at just 360 ± 4 mV overpotential. Interestingly, no sign of a transport-limited regime appeared in the studied potential range, reaching over 20 mA cm–2 at η = 397 ± 4 mV. A current density limit of 100 mA cm−2 was found in these x-Co3O4@C/GPO electrodes.

a LSV curves for x-Co3O4@C/GPO electrodes in 1 M H2SO4 (pH 0.1), at x = 0 (GPO blank, light blue), 5 (blue), 10 (yellow), 20 (grey), 30 (pink), 40 (red) and with 1 mV s–1 scan rate; b LSV curves of x-IrO2/GPO electrodes in 1 M H2SO4 (pH 0.1), at x = 5 (pink), 10 (gray), 20 (orange), 30 (blue), 40 (black) compared to 40-Co3O4@C/GPO (red); c Tafel plots of IrO2/GPO (black) and Co3O4@C/GPO (red) extracted from LSV data. Source data are provided as a Source Data file.
We prepared analogous IrO2/GPO working electrodes to benchmark our results in the same conditions with the state-of-the-art IrO2. The IrO2/GPO anodes delivered higher overpotentials, \({{{{{{\rm{\eta }}}}}}}_{{{{{{\rm{j}}}}}}=10{{{{{\rm{mA}}}}}}{{{{{{\rm{cm}}}}}}}^{-2}}=368\,{{{{{\rm{mV}}}}}}\) at 10 mA cm–2 and \({{{{{{\rm{\eta }}}}}}}_{{{{{{\rm{j}}}}}}=20{{{{{\rm{mA}}}}}}{{{{{{\rm{cm}}}}}}}^{-2}}=396\,{{{{{\rm{mV}}}}}}\) at 20 mA cm–2, slightly above those obtained for the Co3O4@C-based electrode (Supplementary Table 2).
Tafel analyses of the LSV data yielded slopes of 143 mV dec–1 for Co3O4@C and 82 mV dec–1 for IrO2 (Fig. 3c), suggesting a different reaction mechanism (rate-limiting step) for these two catalysts, and indicating a faster increment of current density with the applied potential for IrO239,40. Interestingly, this is compensated by the lower onset potential of Co3O4@C/GPO. The electrochemical double-layer capacitance (EDLC) of Co3O4@C/GPO and IrO2/GPO were calculated as 25 and 2 mF cm–2, respectively, with 0.03 mF cm–2 for the blank GPO (Supplementary Fig. 13). This indicates a greater electrochemical active surface area for Co3O4@C/GPO, due to its higher density of active sites in Co3O4@C/GPO, thanks to its nanostructuration, favouring the higher current densities observed in the potential range studied41,42. It is important to point out that nanostructured IrO2 can perform significantly better than our IrO2/GPO electrodes20,43,44. This must be related to the relatively low electrochemical surface area (ECSA) of our IrO2/GPO composite. When we normalize current densities vs. ECSA (Supplementary Fig. 14), IrO2/GPO electrodes reach higher values at lower potentials. Still, the Co3O4@C catalyst remains competitive as a non-noble-metal catalysts, and especially when current density is normalized per gram (Supplementary Fig. 14), as a more practical parameter for applications.
Finally, we measured anodic oxygen evolution during chronopotentiometry experiments with Co3O4@C/GPO electrodes (Supplementary Fig. 15). We found over >96% Faradaic efficiency, confirming that OER is the dominant process at these electrodes’ surface, and confirming no significant oxidation of the carbon-based matrix is taking place in these conditions.
OER electrocatalytic stability in 1 M H2SO4
As mentioned before, stability is a critical issue for earth-abundant OER catalysts in acidic media45,46,47,48,49. To determine the stability of our Co3O4@C/GPO electrodes, we take advantage of the benchmarking protocol designed by Jaramillo’s group that uses as figure of merit the overpotential required to achieve and maintain a 10 mA cm–2 current density, as this is approximately the current density expected at the anode in a 10% efficient solar water-splitting device under 1 sun illumination6,50. The corresponding chronopotentiometry data (Fig. 4a–c) show very good stability for all electrodes, independently of their Co3O4@C content. In all cases, \({{{{{{\rm{\eta }}}}}}}_{{{{{{\rm{j}}}}}}=10{{{{{\rm{mA}}}}}}{{{{{{\rm{cm}}}}}}}^{-2}}\) after 2 h shows just a small increment. In the case of our best electrodes, the 40-Co3O4@C/GPO, this increment is of just 3 mV, and the stability is maintained for long times. After 43 h of continuous electrolysis, the overpotential is essentially identical to the starting value (Fig. 4a). Meanwhile, even to keep 100 mA cm–2 current density, only 27 mV increment was needed after 2 h catalysis (Supplementary Fig. 11b). We also applied more aggressive stability tests under cycling applied potentials. In this accelerated degradation testing (ADT), complete CV cycles were collected, thus exposing the electrodes sequentially to catalytic and non-catalytic potentials. These conditions simulate better the different degradation mechanisms during electrode and electrolyzer operation. The 40-Co3O4@C/GPO electrodes showed also good stability after ~600 CV cycles with only a 14 mV increment in the overpotential to reach 10 mA cm–2 current density (Supplementary Fig. 11c).

Chronopotentiometry measurements at j = 10 mA cm–2 in 1 M H2SO4 electrolyte. a 40-Co3O4@C/GPO (>40 h). b 20-Co3O4@C/GPO (2 h). c 30-Co3O4@C/GPO (2 h). d Benchmarking of their activity/stability features (red stars) in comparison with other OER electrocatalysts (black squares) in the same electrolysis conditions according to the data from a previous benchmarking study6. Source data are provided as a Source Data file.
The benchmarking of these electrodes with previous literature is highlighted in the \({{{{{{\rm{\eta }}}}}}}_{j=10{{{{{\rm{mA}}}}}}{{{{{{\rm{cm}}}}}}}^{-2},t=2{{{{{\rm{h}}}}}}}\) vs. \({{{{{{\rm{\eta }}}}}}}_{j=10{{{{{\rm{mA}}}}}}{{{{{{\rm{cm}}}}}}}^{-2},t=0{{{{{\rm{h}}}}}}}\) plot (Fig. 4d). This comparative plot illustrates the high activity and stability of our electrodes. The three of them appear at the diagonal of the graph, as expected for sustainable performance, and very close and competitive to the results obtained with noble-metal counterparts, indicating that earth-abundant anodes may successfully pass this benchmarking protocol for OER performance in acidic media.
Stability number (S-number) and activity-stability factor (ASF) were also proposed as key metrics for estimating lifetime and long-term stability for electrocatalysts51,52,53. Thus, we analyzed the electrolyte after stability tests to check for Co leaching (Supplementary Table 3). We found the presence of Co but at the ppb level, corresponding to just ≈0.4 % of the total. Based on this number, we can estimate a 25 S-number, an ASF of 101 and a lifetime of 462 h. These estimations are comparable even to Ir-based catalysts such as SrIrO3 in analogous conditions, and confirm the promising performance/stability of these electrodes. It is worthy to mention that this small Co loss does not significantly affect performance. This may be due to Co leaching only from non-catalytically areas, or to the 3D nature of our electrodes. Thus, after leaching, the new surface exposed brings additional active sites, keeping the performance. Both may explain why activity is so stable, but a relatively modest S-number is obtained.
Post-electrolysis Co3O4@C/GPO characterization
To further confirm the stability of Co3O4@C as a genuine OER catalyst, we characterized the structural and chemical evolution of the electrodes after these 2 h electrolysis at 10 mA cm–2 in 1 M H2SO4. The powder XRD patterns did not show any significant change nor shift in the observed peaks, still typical of Co3O4@C and graphite (Supplementary Fig. 16). This suggests no major structural changes are occurring to the bulk of the material Co3O4@C.
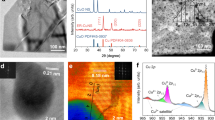
We explored potential changes on the chemical composition of the catalyst due to OER process by XPS characterization of the fresh electrode and after water electrolysis under different conditions. The Co 2p XPS spectrum of the fresh Co3O4@C/GPO electrode (Fig. 5a) shows two peaks located at 794.9 eV (Co 2p 1/2) and 779.7 eV (Co 2p3/2), corresponding to the spin–orbit splitting of the 2p orbital. Both components contain equivalent chemical information. The deconvoluted analysis of the peaks reveals the presence of two different chemical components, which we attribute to the Co3+ (blue) and Co2+ (green) states, in agreement with the presence of Co3O438. In addition, we observe two doubled satellite peaks arising from charge transfer and final states effects from Co2+ (satellite A, yellow) and Co3+ (satellite B, pink)54, again characteristic of Co3O4. We also analyzed the O 1 s peak (Fig. 5a). In addition to the Co-O component (brown) related to the Co3O4, we observe a higher binding energy component attributed to residual OH/H2O (purple). Quantitative analysis of the Co 2p and O 1 s core levels of Co3O4@C/GPO after chronopotentiometry at 10 mA cm–2 for 2 h (Fig. 5b) and 5 mA cm–2 for 24 h (Fig. 5c) revealed no shifts in the binding energies of the components respect to the fresh sample. Crucially, the main spectral features attributed to a Co3O4@C catalyst remain unaltered after water electrolysis, which demonstrates the preservation of the oxidation state of the catalyst. Changes in the intensity of the OH/H2O component on the O 1s can be fairly attributed to the different environmental conditions of the emersed electrode (see Methods section). XPS analysis of the C component does not evidence significant changes in the oxidation state of the C 1s peak, supporting the preservation of the carbon-based matrix (Supplementary Fig. 17) in agreement to the obtained Faradaic efficiencies. Nitrogen detection in the system is below our resolution limit; therefore, no discussion is referred to this element. Based on the current analysis, the most important finding is that the oxide film is stable and no cobalt oxide is lost nor further oxidized during the electrolysis process. In summary, XRD and XPS strongly support the bulk and surface stability of Co3O4@C before and after acidic OER electrocatalysis, and its genuine catalytic activity.

a X-ray photoelectron spectra of the Co 2p (left) and O 1s (right) core levels for a 20-Co3O4@C/GPO electrode. b Same spectra for a 20-Co3O4@C/GPO electrode after OER chronopotentiometry at 10 mA cm–2 for 2 h. c Same spectra for a 20-Co3O4@C/GPO electrode after OER chronopotentiometry at 5 mA cm–2 for 24 h. Color assignment for the different areas: Co3+ (blue), Co3+ satellite (pink), Co2+ (green), Co2+ satellite (yellow), OH/H2O (purple), O–Co (brown). Source data are provided as a Source Data file.
We also investigated the Co3O4@C/GPO composite after 2 h electrolysis at 10 mA cm–2 by means of HR-TEM (Fig. 6). The images and power spectra (FFT) analyses also confirm a high structural and chemical stability. Neither crystallinity nor particle size are affected by the electrochemical process.

HRTEM micrograph (a) from a Co3O4@C/GPO composite, recovered after 2 h water electrolysis at 10 mA cm–2. To highlight the different systems, we show a frequency filtered map (b) where the Co3O4 nanoparticle lattice fringes are in yellow and the ones corresponding to the surrounding graphitic layers in pink and turquoise. The power spectra (FFT) confirm the high stability of these electrodes, showing again Co3O4 nanoparticles with a cubic FD3-MZ (s.g. 227) spinel structure (yellow cirlces) here oriented along the [2–11] zone axis. No changes in crystallinity or particle size are observed for the Co3O4@C composite when compared to the fresh samples (Fig. 2). Notice that the graphitic layers may show multiple rotated domains as shown by the highlighted dotted green circles in the power spectrum, but this effect also happened in the as-prepared sample. Source data are provided as a Source Data file.
Critical role of GPO
To investigate the actual role of the carbon paste in the stability of the electrodes, we carried out additional alternative experiments. First, we directly deposited Co3O4@C on a glassy carbon (GC) electrode as a Nafion-based ink. This electrode showed a significantly lower electrocatalytic activity when compared with the Co3O4@C/GPO (Figs. 3–4 vs. Supplementary Fig. 18). More importantly, after 30-min of the benchmarking test in 1 M H2SO4, the Co3O4@C/GC electrode is apparently deactivated. This suggests that the GPO binder is fundamental to confer the acidic stability and activity of the Co3O4@C component.
This effect of the GPO binder could be due to a modified local pH at the electrode/electrolyte interface55,56,57. To check this hypothesis, we decided to investigate the effect of the GPO binder on the local pH through the reversible H+/H2 pair as catalyzed with commercial Pt/C58. The reversible potential for this model reaction differs when the Pt/C is directly deposited on a graphite electrode, or when incorporated into a GPO electrode as observed in their CV plots in a hydrogen saturated 1 M H2SO4 electrolyte (Supplementary Fig. 19). An average value of –0.002 V vs. ERHE was estimated for the Pt/C catalyst, in good agreement with the theoretical +0.0 V value. A + 0.031 V vs. ERHE was found for the (Pt/C)/GPO electrode. If we associate this potential difference only to a theoretical local pH difference, ∆E = 0.059 ∆pH, we can estimate a pH difference of just 0.52 unit between both electrodes. Of course, this number cannot be assigned to the actual pH at the surface, since this difference may be caused by pH and other parameters, including Pt surface states. However, such small difference indicates that the higher stability and activity found in GPO electrodes cannot arise exclusively from the modification of the local pH. Therefore, we associate the protective function to the hydrophobic environment, which avoids proper solvation of the oxides, precluding its dissolution. The high hydrophobicity of GPO is corroborated by contact angle tests (Supplementary Fig. 20). A contact angle of 125° was found for a 1 M H2SO4 solution on GPO, significantly higher than on GC (86°).
To prove the general importance of this protective strategy, typical oxides based on Ni, Co, and Fe, which were active in alkaline solution but unstable in acid for oxygen evolution, were synthesized and then their electrochemical performances were measured in 1 M H2SO4 (Supplementary Fig. 21). It was showed that all of these oxides were active for oxygen evolution and kept their performance at 10 mA cm–2 during 2 h catalysis with the small change of ≤13 mV (Supplementary Fig. 22).
We also compared the activity/stability of Co3O4@C vs. Co3O4 (Supplementary Fig. 10). The corresponding x-Co3O4/GPO electrodes showed good stability during preliminary CV cycles and chronopotentiometry measurement, but at higher overpotentials. A 5 mF cm–2 EDLC was determined, just 1/5 that of Co3O4@C/GPO (Supplementary Fig. 13). Specific surface area from N2 sorption isotherm curves for Co3O4@C was also about five times greater than that of Co3O4 (Supplementary Fig. 23). These results suggest that the role of the carbon coating is to improve the nanostructuration of the active Co3O4 material.
In addition, the Co3O4@C/GPO, Co3O4/GPO, and IrO2/GPO electrodes were studied by Electrochemical Impedance Spectroscopy (EIS) at different applied potentials. Supplementary Fig. 24 shows the obtained Nyquist plots, which systematically feature two arcs (or distorted arc for IrO2), consistent with two simultaneous/consecutive charge-transfer channels59. Fitting the experimental data to a suitable equivalent circuit model (Supplementary Fig. 25) revealed that the best ohmic contact (reflected by the series resistance, RS) is obtained for the Co3O4@C/GPO (Supplementary Fig. 25a). On the other hand, the charge transfer resistance (Rct), scales inversely with the electrocatalytic activity of the different electrodes, being the lowest one for the Co3O4@C/GPO electrode (Supplementary Fig. 25b). This is consistent with the estimated surface capacitance, which scales with electrode performance, as a result of higher surface area and hence, higher density of catalytic sites (Supplementary Fig. 25c).
In summary, we are reporting the activity and promising stability of carbon-decorated Co3O4@C nanoparticles (Co3O4@C) for electrocatalytic OER under acidic conditions when protected by a hydrophobic binder support. Electrodes built from Co3O4@C, graphite and paraffin oil are able to evolve oxygen from water during the electrolysis of a concentrated (1 M) sulfuric acid solution (pH < 0.1).
Although previous reports on acidic water splitting with earth-abundant raw materials had achieved either high activity or high stability, our working anodes fulfill both requirements (Supplementary Table 4). These electrodes operate during more than 40 h, at a relatively high current density (10 mA cm–2) and at a low overpotential of η < 398 mV, very close to the benchmarking performance of state-of-the-art IrO2.
The synergy between active catalytic phase, Co3O4 nanoparticles, and the carbon support (doped-carbon cover, graphite and paraffin oil) is crucial to reach this robust performance. We assign this protective effect to the hydrophobicity of the electrode surface, which could avoid proper solvation of the metal oxides precluding their dissolutions. Indeed, we found preliminary data that supports this approach to be general, also effective when applied to other non-noble-metal-based OER catalysts for working in acid.
Despite the promising performance of our electrodes, a few challenges will need to be addressed before their implementation into commercial electrolyzers. To start with, the carbon content may become an issue at high current densities, as those expected from commercial devices (>500 mA cm–2). However, our successful corrosion-protection opens an interesting strategy that can be translated into full cell devices, looking for alternative approaches to incorporate hydrophobic species at the electrode surfaces.
In addition, it is worthy to mention that the current densities reached and sustained by the Co3O4@C/GPO electrodes are high enough to satisfy the needs of photoelectrochemical (PEC) devices, well in line with the maximum currents provided by photoanodes. Investigations to combine these acid-stable electrodes as co-catalyst for photoactive semi-conductors are under way.
Methods
Materials and chemicals
All the chemical reagents and solvents were of commercial grade and used directly without any further purification Experimental Details.
Synthesis
ZIF-9 was prepared via a solvothermal method according to the previous literature with some modifications60,61. Co(NO3)2·6H2O (0.175 g) and benzimidazole (0.142 g) were dissolved into 15 mL DMF and then the homogeneous solution was transferred into a Teflon-lined stainless autoclave. The sealed autoclave was put into an oven and kept at 140 °C for 24 h. When it was cooled down to room temperature, the purple product was filtered out and washed with acetone, and then dried at 60 °C.
To obtain the target material, Co@C was firstly synthesized using ZIF-9 as precursor by heating at 500 and then 750 °C for 2 h, respectively, under Ar flow while Co3O4 was obtained under air. Afterwards, the pyrolysis product was oxidized in air at 230 °C for 48 h to generate Co3O4@C.
Mixed metal oxides (NiOx, FeOx, NiFeOx, CoFeOx, and NiCoOx) were prepared by modified method available in the literature62. Metal nitrates with ratio 1:1 were dissolved in 50 mL of distilled water with constant stirring until a clear solution was obtained. The whole metal concentration was fixed to 0.0125 M. Glycine was added into the aqueous solution (glycine/metal molar ratio is 1.2) and stirred until total dissolution. Afterwards, the solution was heated up to 210 °C until total solvent evaporation and glycine combustion. The resulting porous dark solid was recovered and calcined at 1100 °C in a tubular oven for 1 h.
The composite electrodes were prepared by 2-h ball-milling at 20 s−1 of a mixture of paraffin oil (20 mg), graphite powder (80 mg) and the desired weight of metal oxide (5, 10, 20, 30 or 40 mg), namely, x-Co3O4@C/GPO. x-IrO2/GPO (commercial IrO2 from AlfaAesar), x-Co3O4/GPO and other oxide electrodes were also prepared using the same process for comparison purposes.
Structural characterization
Powder X-ray diffraction (PXRD) data were recorded with a Bruker D8 Advance Series equipped with a VANTEC-1 PSD3 detector. Elemental analyses were carried out with an Agilent 725-ES inductively coupled plasma optical emission spectrometer (ICP-OES) at University of Valladolid (Co) and LECO CHNS-932 elementary microanalyzers (C, H, N) at Complutense University of Madrid. Thermogravimetric analysis was conducted with a thermogravimetric balance of Mettler Toledo. Nitrogen adsorption-desorption isotherms at 77 K were measured on a Quantachrome Autosorb iQ gas adsorption analyzer. Prior to analysis, the sample was degassed in vacuum. The BET method was applied to calculate the total surface area.
Electrochemistry
All electrochemical experiments were performed under ambient conditions (≈293 K) with a Bio-Logic VMP3 multichannel potentiostat and implemented with a three-electrode configuration using 1 M H2SO4 (pH 0.1) as filling solution, Pt mesh as counter electrode, Ag/AgCl (3 M KCl) as reference electrode and a pocket working electrode (0.07 cm2 surface area and 4 mm depth) filled with the GPO composites. The actual mass amounts of the x-Co3O4@C/GPO composites in the electrode pocket were measured with a weight balance and are indicated in Supplementary Table 5. Although it is difficult to estimate the actual active layer, an estimation is suggested that 1/8 of the total electrode pocket volume is used as the maximum limit in contact with the solution in order to determine the mass loading for comparison (Supplementary Table 5)36. All potentials were measured vs. Ag/AgCl electrode and converted to the RHE reference scale using ERHE = EAg/AgCl + 0.21 + 0.059 pH (V) while overpotentials η = ERHE – 1.229 V. All current densities were calculated based on the geometrical surface area of the electrodes. Ohmic drop (R) was determined by using the automatic current interrupt (CI) software and the corresponding ohmic drops were included in Supplementary Table 6. iR-compensations were applied to all electrochemical data. CV experiments were carried out with 10 or 100 mV s–1 scan rates. Single LSV curve was recorded with a 1 mV s–1 scan rate for activity comparison after 10-CV cycle activation in the 0–1.4 V vs. Ag/AgCl potential range. Tafel slopes were estimated from the LSV curves by plotting overpotential η vs. log j (j = current density). The potential vs. RHE to drive 1 mA cm–2 was used to define onset potential and corresponding ηonset. Chronopotentiometry tests were carried out at fixed current densities of 10 or 100 mA cm–2. Accelerated degradation testing (ADT) was measured between 0–1.4 V vs. Ag/AgCl using 100 mV s−1 scan rate63. For the electrochemical double-layer capacitance (EDLC) measurements, open circuit potentials (OCPs) vs. the Ag/AgCl were firstly recorded for 30 min to reach rather stable values. Combined with above CV measurements, the 100 mV potential windows centered at OCPs could be determined and cyclic voltammetries were then carried out under scanning rates of 20, 40, 60, 80, and 100 mV s–1. The current density differences between the minimum and maximum values at OCPs vs. the Ag/AgCl and the corresponding scanning rates were plotted to calculate the EDLC value (1/2 of the slope of current density-scan rate plots)64. Co3O4@C and commercial Pt/C from AlfaAesar were also deposited on the glassy carbon (GC, 0.07 cm−2) disk electrodes. The inks were prepared by sonicating 10 mg of catalyst, 25 μL Nafion 117 containing solution and 975 μL ethanol aqueous solution (3:1 in volume) for 30 min.
Electrochemical impedance spectroscopy (EIS) was performed by means of a typical three-electrode cell in the frequency range from 100 kHz to 0.1 Hz with 8 points per decade. The AC perturbation was 5 mV. Experimental data were fitted to the selected equivalent circuit model using Zview software (Scriber Associates) for extracting both capacitances and resistances.
Faradaic efficiency
In order to evaluate the faradaic efficiency towards oxygen production, the chronopotentiometric experiment (Supplementary Fig. 15b) was carried out applying a fixed current (1.4 mA, 2 h) while oxygen concentration in the headspace was in situ measured by using an Unisense sensing system equipped with an oxygen microsensor based on voltage polarization. The experiment was performed under continuous flow conditions by bubbling Ar as carrier gas in both anodic and cathodic compartments. For this purpose, an H-type cell was used containing a frit glass separating both compartments, a connection for the sensor to be inserted in the anodic gas headspace, and connections for the inlet and outlet Ar streams in both compartments (Supplementary Fig. 26). The oxygen microsensor was in situ two-point calibrated by feeding to the H-cell with certified standard of Ar (≥99.999%) and compressed air, being the gas flow rate controlled by a set of mass flowmeters (Bronkhorst EL-FLOW).
After purging the cell headspace with argon, the chronopotentiometry test was started and the oxygen concentration was monitored until reaching stabilization. The expected faradaic oxygen production rate (\({F}_{{{{{{{\mathrm{O}}}}}}}_{2},{{{{{{{\mathrm{far}}}}}}}}}\) in mol s−1) is calculated with the following equation:
where I is the applied current (in A), ne is the number of mols of electrons involved in the water oxidation reaction to generate one mol of oxygen (4) and F is the Faraday constant (96,485 C mol–1).
The experimental O2 flow rate (\({F}_{{O}_{2},{\exp }}\) in mol s–1) was calculated considering ideal gas behavior with the following equation:
where P is the total gas pressure (1 atm), C (O2) is the steady-state oxygen concentration provided by the sensor (in %), FAr is the Ar carrier flow (in L s–1), R is the ideal gas constant (0.082 atm L K–1 mol–1) and T is the cell temperature (293 K).
Then Faradaic efficiency (in %), FE, is calculated as follows:
X-ray photoemission spectroscopy
X-Ray photoemission (XPS) experiments were performed inside an ultra-high vacuum chamber (base pressure of 10–10 mbar) using a Phoibos 100 photoelectron spectrometer equipped with an Al Kα X-ray source (16 mA, 1486.6 V) as the incident photon radiation. XPS spectra of Co 2p, O 1s, N 1s, and C 1s core levels were measured for as received samples deposited on top of indium tape. The spectra are well described by the superposition of several Doniach-Sunjic curve-components. The intensities of the XPS core levels were evaluated by the peak areas, after a standard background subtraction according to Shirley procedure65. The spin–orbit splitting for every component into the Co-2p core level has been set to D = 15.2 eV with a branching ratio of 0.5. The metallic cobalt peak, Co 2p3/2 = 779.7 eV, was used for a final calibration of the spectra38. Co3O4@C/GPO after OER chronopotentiometry at 10 mA cm–2 for 2 h at was washed with acetone in order to remove the paraffin oil prior to XPS measurements. As expected, the treatment do not affect the oxidation state of the composite, as evidenced by the measured spectra.
Transmission electron microscopy
High-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy (STEM) investigations were performed on a field emission gun FEI Tecnai F20 microscope. High-angle annular dark-field (HAADF) STEM was combined with electron energy loss spectroscopy (EELS) in the Tecnai microscope by using a GATAN QUANTUM energy filter in order to obtain compositional maps. STEM-EELS maps were performed using the O K-edge at 532 eV (green), the Co L-edge at 779 eV (red) and C K-edge at 284 eV (blue).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data is available in the main text or in the supplementary materials. Source data are provided with this paper.
Change history
10 August 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41467-022-32399-6
References
Wei, C. et al. Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 31, 1–24 (2019).
Staffell, I. et al. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 12, 463–491 (2019).
Singh, S. et al. Hydrogen: a sustainable fuel for future of the transport sector. Renew. Sustain. Energy Rev. 51, 623–633 (2015).
Sharma, S. & Ghoshal, S. K. Hydrogen the future transportation fuel: from production to applications. Renew. Sustain. Energy Rev. 43, 1151–1158 (2015).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, 1–12 (2017).
McCrory, C. C. L. et al. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 137, 4347–4357 (2015).
Zhu, J., Hu, L., Zhao, P., Lee, L. Y. S. & Wong, K. Y. Recent advances in electrocatalytic hydrogen evolution using nanoparticles. Chem. Rev. 120, 851–918 (2020).
Lyu, F., Wang, Q., Choi, S. M. & Yin, Y. Noble-metal-free electrocatalysts for oxygen evolution. Small 15, 1–17 (2019).
Spöri, C., Kwan, J. T. H., Bonakdarpour, A., Wilkinson, D. P. & Strasser, P. The stability challenges of oxygen evolving catalysts: towards a common fundamental understanding and mitigation of catalyst degradation. Angew. Chem. Int. Ed. 56, 5994–6021 (2017).
Han, B. et al. Screening oxide support materials for OER catalysts in acid. J. Electrochem. Soc. 165, 813–820 (2018).
Chung, D. Y. et al. Large-scale synthesis of carbon-shell-coated FeP nanoparticles for robust hydrogen evolution reaction electrocatalyst. J. Am. Chem. Soc. 139, 6669–6674 (2017).
Schipper, D. E. et al. Effects of catalyst phase on the hydrogen evolution reaction of water splitting: preparation of phase-pure films of FeP, Fe2P, and Fe3P and their relative catalytic activities. Chem. Mater. 30, 3588–3598 (2018).
Sun, J. et al. Robust hydrogen-evolving electrocatalyst from heterogeneous molybdenum disulfide-based catalyst. ACS Catal. 10, 1511–1519 (2020).
Yu, J., Cheng, G. & Luo, W. Ternary nickel-iron sulfide microflowers as a robust electrocatalyst for bifunctional water splitting. J. Mater. Chem. A 5, 15838–15844 (2017).
Kuo, D. Y. et al. Influence of surface adsorption on the oxygen evolution reaction on IrO2 (110). J. Am. Chem. Soc. 139, 3473–3479 (2017).
Cherevko, S. et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: a comparative study on activity and stability. Catal. Today 262, 170–180 (2016).
Chen, S. et al. Mn-doped RuO2 nanocrystals as highly active electrocatalysts for enhanced oxygen evolution in acidic media. ACS Catal. 10, 1152–1160 (2020).
Shan, J., Zheng, Y., Shi, B., Davey, K. & Qiao, S. Z. Regulating electrocatalysts via surface and interface engineering for acidic water electrooxidation. ACS Energy Lett. 4, 2719–2730 (2019).
Escudero-Escribano, M. et al. Importance of surface IrOx in stabilizing RuO2 for oxygen evolution. J. Phys. Chem. B 122, 947–955 (2018).
Seitz, L. C. et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353, 1011–1014 (2016).
Ouattara, L., Fierro, S., Frey, O., Koudelka, M. & Comninellis, C. Electrochemical comparison of IrO2 prepared by anodic oxidation of pure iridium and IrO2 prepared by thermal decomposition of H2IrCl6 precursor solution. J. Appl. Electrochem. 39, 1361–1367 (2009).
Lyons, M. E. G. & Floquet, S. Mechanism of oxygen reactions at porous oxide electrodes. Part 2—Oxygen evolution at RuO2, IrO2 and IrxRu1-xO2 electrodes in aqueous acid and alkaline solution. Phys. Chem. Chem. Phys. 13, 5314–5335 (2011).
Stoerzinger, K. A. et al. Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2, 876–881 (2017).
Rodríguez-García, B. et al. Cobalt hexacyanoferrate supported on Sb-doped SnO2 as a non-noble catalyst for oxygen evolution in acidic medium. Sustain. Energy Fuels 2, 589–597 (2018).
Li, A. et al. Stable potential windows for long-term electrocatalysis by manganese oxides under acidic conditions. Angew. Chem. Int. Ed. 58, 5054–5058 (2019).
Moreno-Hernandez, I. A. et al. Crystalline nickel manganese antimonate as a stable water-oxidation catalyst in aqueous 1.0 M H2SO4. Energy Environ. Sci. 10, 2103–2108 (2017).
Kwong, W. L., Lee, C. C., Shchukarev, A. & Messinger, J. Cobalt-doped hematite thin films for electrocatalytic water oxidation in highly acidic media. Chem. Commun. 55, 5017–5020 (2019).
Zhou, L. et al. Rutile alloys in the Mn–Sb–O system stabilize Mn3+ to enable oxygen evolution in strong acid. ACS Catal. 8, 10938–10948 (2018).
Chatti, M. et al. Intrinsically stable in situ generated electrocatalyst for long-term oxidation of acidic water at up to 80 °C. Nat. Catal. 2, 457–465 (2019).
Bloor, L. G., Molina, P. I., Symes, M. D. & Cronin, L. Low pH electrolytic water splitting using earth-abundant metastable catalysts that self-assemble in situ. J. Am. Chem. Soc. 136, 3304–3311 (2014).
Mondschein, J. S. et al. Crystalline cobalt oxide films for sustained electrocatalytic oxygen evolution under strongly acidic conditions. Chem. Mater. 29, 950–957 (2017).
Etzi Coller Pascuzzi, M., van Velzen, M., Hofmann, J. P. & Hensen, E. J. M. On the stability of Co3O4 oxygen evolution electrocatalysts in acid. ChemCatChem 13, 459–467 (2021).
Yang, X. et al. Highly acid-durable carbon coated Co3O4 nanoarrays as efficient oxygen evolution electrocatalysts. Nano Energy 25, 42–50 (2016).
Yan, K. L. et al. Probing the active sites of Co3O4 for the acidic oxygen evolution reaction by modulating the Co2+/Co3+ ratio. J. Mater. Chem. A 6, 5678–5686 (2018).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109–118 (2022).
Arens, J. T. et al. Water oxidation electrocatalysis in acidic media with Co-containing polyoxometalates. J. Catal. 389, 345–351 (2020).
Park, K. S. et al. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl Acad. Sci. USA 103, 10186–10191 (2006).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 257, 2717–2730 (2011).
Anantharaj, S. et al. Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe, Co, and Ni: a review. ACS Catal. 6, 8069–8097 (2016).
Reier, T., Oezaslan, M. & Strasser, P. Electrocatalytic oxygen evolution reaction (OER) on Ru, Ir, and pt catalysts: a comparative study of nanoparticles and bulk materials. ACS Catal. 2, 1765–1772 (2012).
Li, D., Shi, J. & Li, C. Transition-metal-based electrocatalysts as cocatalysts for photoelectrochemical water splitting: a mini review. Small 14, 1–22 (2018).
Tamirat, A. G., Rick, J., Dubale, A. A., Su, W. N. & Hwang, B. J. Using hematite for photoelectrochemical water splitting: a review of current progress and challenges. Nanoscale Horiz. 1, 243–267 (2016).
Oh, H. S., Nong, H. N., Reier, T., Gliech, M. & Strasser, P. Oxide-supported Ir nanodendrites with high activity and durability for the oxygen evolution reaction in acid PEM water electrolyzers. Chem. Sci. 6, 3321–3328 (2015).
Oh, H. S. et al. Electrochemical catalyst-support effects and their stabilizing role for IrOx nanoparticle catalysts during the oxygen evolution reaction. J. Am. Chem. Soc. 138, 12552–12563 (2016).
Reier, T., Nong, H. N., Teschner, D., Schlögl, R. & Strasser, P. Electrocatalytic oxygen evolution reaction in acidic environments—reaction mechanisms and catalysts. Adv. Energy Mater. 7, 1601275 (2017).
Ashton, S. J. & Arenz, M. A DEMS study on the electrochemical oxidation of a high surface area carbon black. Electrochem. Commun. 13, 1473–1475 (2011).
Hu, Q. et al. Coupling pentlandite nanoparticles and dual-doped carbon networks to yield efficient and stable electrocatalysts for acid water oxidation. J. Mater. Chem. A 7, 461–468 (2019).
Cheng, W. et al. A metal-vacancy-solid-solution NiAlP nanowall array bifunctional electrocatalyst for exceptional all-pH overall water splitting. J. Mater. Chem. A 6, 9420–9427 (2018).
Mondschein, J. S. et al. Intermetallic Ni2Ta electrocatalyst for the oxygen evolution reaction in highly acidic electrolytes. Inorg. Chem. 57, 6010–6015 (2018).
McCrory, C. C. L., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508–515 (2018).
Kim, Y. T. et al. Balancing activity, stability and conductivity of nanoporous core-shell iridium/iridium oxide oxygen evolution catalysts. Nat. Commun. 8, 1–8 (2017).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 1–31 (2021).
Díaz-Fernández, D. et al. The growth of cobalt oxides on HOPG and SiO2 surfaces: A comparative study. Surf. Sci. 624, 145–153 (2014).
Wang, J., Musameh, M. & Mo, J. W. Acid stability of carbon paste enzyme electrodes. Anal. Chem. 78, 7044–7047 (2006).
Fang, Z. et al. Hybrid organic-inorganic gel electrocatalyst for stable acidic water oxidation. ACS Nano 13, 14368–14376 (2019).
Sempionatto, J. R. et al. Enzymatic biofuel cells based on protective hydrophobic carbon paste electrodes: towards epidermal bioenergy harvesting in the acidic sweat environment. Chem. Commun. 56, 2004–2007 (2020).
Liang, Y. et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786 (2011).
Han, L. et al. Cobalt hexacyanoferrate as a selective and high current density formate oxidation electrocatalyst. ACS Appl. Energy Mater. 3, 9198–9207 (2020).
Yang, F., Chen, Y., Cheng, G., Chen, S. & Luo, W. Ultrathin nitrogen-doped carbon coated with CoP for efficient hydrogen evolution. ACS Catal. 7, 3824–3831 (2017).
Yang, F. et al. Design of active and stable oxygen reduction reaction catalysts by embedding CoxOy nanoparticles into nitrogen-doped carbon. Nano Res. 10, 97–107 (2017).
Chen, J. Y. C., Miller, J. T., Gerken, J. B. & Stahl, S. S. Inverse spinel NiFeAlO4 as a highly active oxygen evolution electrocatalyst: Promotion of activity by a redox-inert metal ion. Energy Environ. Sci. 7, 1382–1386 (2014).
Petzoldt, P. J., Kwan, J. T. H., Bonakdarpour, A. & Wilkinson, D. P. Deconvoluting reversible and irreversible degradation phenomena in OER catalyst coated membranes using a modified RDE technique. J. Electrochem. Soc. 168, 026507 (2021).
Morales, D. M. & Risch, M. Seven steps to reliable cyclic voltammetry measurements for the determination of double layer capacitance. J. Phys. Energy 3, 34013 (2021).
Shirley, D. A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 5, 4709–4714 (1972).
Acknowledgements
The authors thank the support from projects RED2018-102459-T, RTI2018-095618-B-I00, PID2020-116093RB-C41,C43&C44, PID2019-107338RB-C63 and PID2020-114252GB-I00 funded by MCIN/AEI/10.13039/501100011033/ and by “ERDF A way of making Europe”; the Generalitat de Catalunya (2017-SGR-1406 and 2017-SGR-327), and the Basque Government (grant IT1591-22). J.Y. thanks the China Scholarship Council (CSC) for predoctoral fellowships (File No. 201806270234). S.B. acknowledges Grant RYC-2017-21931 funded by MCIN/AEI/ 10.13039/501100011033 and by ESF Investing in your future, and UPV/EHU project EHUrOPE19/01 and IKERBASQUE, Basque Foundation for Science. ICIQ and ICN2 are supported by the Ministerio de Ciencia e Innovación through the Severo Ochoa Excellence Accreditations CEX2019-000925-S (MIC/AEI) and SEV-2017-0706; and by the CERCA Programme/Generalitat de Catalunya. M.C.S. has received funding from the European Union’s Horizon 2020 research and innovation programme under Marie Skodowska-Curie grant 754510 (PROBIST) and the Severo Ochoa programme. M.C.S. has received funding from the postdoctoral fellowship Juan de la Cierva Incorporation from MICINN (JCI-2019) and the Severo Ochoa programme. M.P.D. acknowledges funding for a Ph.D. fellowship from the MPC Foundation.
Author information
Authors and Affiliations
Contributions
J.R.G.M. conceived the original concept and with S.B. designed the project and coordinated the efforts of the research team. J.Y. synthesized, processed and structurally characterized the materials and electrodes. J.Y. and F.G.P. characterized the compounds electrochemically and analyzed the data. J.Y. and J.G.C. measured the oxygen evolution. M.P.D., C.R., and S.B. collected and analyzed the XPS data. M.C.S. and J. A. collected and analyzed the TEM data. S.G. collected and analyzed the EIS data. J.Y., S.B., and J.R.G.M. co-wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, J., Garcés-Pineda, F.A., González-Cobos, J. et al. Sustainable oxygen evolution electrocatalysis in aqueous 1 M H2SO4 with earth abundant nanostructured Co3O4. Nat Commun 13, 4341 (2022). https://doi.org/10.1038/s41467-022-32024-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32024-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
