
Abstract
Disulfides are widely found in natural products and find a wide range of applications in life sciences, materials chemistry and other fields. The preparation of disulfides mainly rely on oxidative couplings of two sulfur containing compounds. This strategy has many side reactions and other shortcomings. Herein, we describe the reductive nickel-catalyzed cross-electrophile coupling of unactivated alkyl bromides with symmetrical alkyl- and aryltetrasulfides to form alkyl-alkyl and aryl-alkyl unsymmetrical disulfides. This approach for disulfide synthesis is practical, relies on easily available, unfunctionalized substrates, and is scalable. We investigated the mechanism of this transformation and found that the tetrasulfide compound does not selectively break the central S–S bond, but regio-selectively generates trisulfide intermediates.
Similar content being viewed by others
Introduction
Disulfides are widely found in natural products and have a wide range of applications in many fields, such as life science1,2,3 and medical chemistry4,5. In the formation of protein tertiary structure, disulfide bonds play a central role2,6,7. The activity of many natural products and drugs depends on the exchange reaction of disulfide bonds in proteins4,8,9. The lipoic acid present in the mitochondria can increase the synthesis of ATP in the cell energy cycle10. Kottamide E has a five-membered ring disulfide structure, which has good anti-tumor and anti-inflammatory effects11. As a consequence, the efficient preparation of various functionalized disulfides is of continued interest (Fig. 1).

The importance of disulfides in life science, natural products, and other fields.
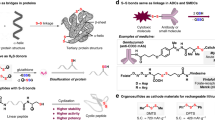
Given the importance and predominance of disulfides in pharmaceuticals and bioactive compounds, chemists are continually seeking for efficient preparation strategies for multifunctional disulfides. While the preparation of symmetrical disulfide has been greatly developed12. Due to the limitation of the substrate structures, the preparation of unsymmetrical disulfide is still challenging. The traditional method for the construction of unsymmetrical disulfide compounds based on oxidative coupling of two different thiols (Fig. 2a)13,14,15,16,17. In view of low yields, poor selectivities, and poor substrate universality, the development of a more efficient method for preparing unsymmetrical disulfide is desirable. In recent years, chemists have designed and synthesized many prefunctionalized/activated disulfurating reagents, and successfully constructed a series of functionalized unsymmetrical disulfides. Xian18, Xu19, and Wang20 have independently disclosed their own disulfide reagents. In this field, Jiang’s group has made excellent achievement, by the introduction of the Mask group into the disulfide reagent, designed and synthesized a series of nucleophilic or electrophilic disulfide reagents, and efficiently constructed a series of highly functionalized polysulfide compounds (Fig. 2b)21,22. On the basis of these studies, they designed and synthesized a series of new bilateral disulfurating reagents, which greatly expanded the construction of unsymmetrical disulfide compounds23,24. In 2020, the Pratt group demonstrated that tetrasulfides undergo efficient homolytic substitution with alkyl radicals, enabling the synthesis of a broad range of unsymmetrical disulfides (Fig. 2c)25,26,27. This protocol showed mild reaction conditions and a large substrate scope.

a Traditional disulfuration by S–S bond formation. b Direct disulfuration via masked strategy. c Radical substitution on tetrasulfides. d Nickel-catalyzed reductive thiolation on tetrasulfides.
Reductive coupling reactions of electrophiles avoid the preparation and use of organometallic reagents, have excellent compatibility with various functional groups, and provide an important method for the construction of C–C bonds28. In recent years, breakthroughs have been made in the research of this strategy, and a series of cross-coupling reactions involving C(sp2)-X and C(sp3)-X electrophiles have been realized29,30,31,32. In recent years, our group has used thiosulfonate as a sulfur source and constructed a series of highly functionalized sulfides and selenides under nickel-catalyzed reductive coupling conditions33,34,35. Inspired by Pratt’s work and combined with our previous work on reductive coupling reactions to synthesize sulfides, we reasoned that we can prepare unsymmetrical disulfides from alkyl halides through Nickel-catalyzed cross-electrophile coupling with tetrasulfide. Herein, we report on the successful development of a cross-coupling reaction of unactivated alkyl bromides with symmetrical tetrasulfides utilizing nickel-catalyzed reductive coupling strategy to construct unsymmetrical alkyl–alkyl disulfides and alkyl–aryl disulfides (Fig. 2d).
Results
Reaction conditions survey
Initially, we tried the reaction of 1-bromo-3-phenylpropane (1b) with 1,4-di-tert-butyltetrasulfane (2a), Ni(PPh3)2Cl2 (5 mol%), Mn (1.5 equiv), ligand (10 mol%) in (dry) DMF at 40 °C for 12 h under N2 atmosphere. Gratifyingly, the reaction proceeded smoothly to give a mixture products of 1-(tert-butyl)-2-(3-phenylpropyl)disulfane (3b) and 1-(tert-butyl)-3-(3-phenylpropyl)trisulfane (3b′) (2.8:1) (However, the disulfide could not be separated from the trisulfide). Surprisingly, when we extended the reaction time to 24 h, the ratio of 3b and 3b′ increased to 31:1. With this promising results in hand, we tried to further optimize the reaction conditions (see Supplementary Tables 1–3 for more details). As briefly illustrated in Table 1, after systematic exploring the reductive coupling with L1 as the ligand, Ni(acac)2 was chosen as catalyst as its use lead to the highest yield and selectivity (Table 1, entries 1–5). Next, we conducted the ligand optimization with a series of bipyridine ligands and phenanthroline ligands. As a result, L1 provided superior result (Table 1, entries 6–12). Then, we tried the reactions in several different solvents, such as DMF, DMA, DMSO, and MeCN. It was found that DMF was crucial for the successful transformation of 1b (Table 1, entries 13–15). When Zn was used instead of Mn, the target product 3b could not be obtained (Table 1, entry 16).
Generality of protocol
With the optimized reaction conditions in hand, we set out to assess the generality of this protocol (Fig. 3). Hence, primary alkyl bromides bearing various functional groups were employed in the reactions with 1,4-di-tert-butyltetrasulfane 2a. To our delight, the length of the carbon chain had no effect on the reaction, the desired disulfide products 3a–3f could be obtained in good to excellent yields. The reaction of 3-(2-bromoethyl)-1H-indole 1g with 1,4-di-tert-butyltetrasulfane (2a) gave the target product 3g in excellent yield. Satisfactorily, a wide range of substituents, including alcohol, siloxane, phosphonate were well-tolerated under this mild reaction condition. The target products 3h–3j were isolated in medium to good yields. However, the reaction of ethyl 6-bromohexanoate 1k with 1,4-di-tert-butyltetrasulfane (2a) gave the target product 3k in only 34% yield. It should be noted that the reaction of methyl(R)-3-bromo-2-((tert-butoxycarbonyl)amino)propanoate (1l) with 1,4-di-tert-butyltetrasulfane (2a) furnished the desired product 3l in 82% yield. This greatly expanded the application of this strategy in the construction of unsymmetrical disulfide compounds. Surprisingly, the reaction of 1-bromo-6-chlorohexane (1m) with tetrasulfane 2a not only could delivered the expected product 3m in 67% yield, but also led to 3m′ in 20% yield. This demonstrated that this strategy is also applicable to alkyl chlorides. When tert-butyl (3-bromopropyl)carbamate (1n), 2-(3-bromopropyl)isoindoline-1,3-dione (1o) and 4-bromobutanenitrile (1p) were applied to the reaction respectively, a mixture of trisulfide compounds and disulfides were obtained (3n–3p, 3n′–3p′). In contrast, the reaction of secondary bromide, tertiary bromide and dibromide failed to give the desired products. According to the related works of Prof. Gong36,37,38, we speculate that the possible reason is that the oxidative addition of nickel(0) and tetrasulfide furnishes nickel(II) intermediate, this intermediate is very sterically hindered due to the coordination of the ligand. Therefore, when secondary bromine and tertiary bromine participate in the reaction, the target products could not be obtained due to the steric hindrance effect.

aReaction conditions: primary alkyl bromides 1 (0.20 mmol, 1.0 equiv.), 2a (0.24 mmol, 1.2 equiv.), Ni(acac)2 (5.0 mol%); L1 (10 mol%), Mn (0.30 mmol, 1.5 equiv.), DMF (1 mL), N2 atmosphere, 40 °C, 24 h. bIsolated yield. c80 °C.
Encouraged by the above achievements, we turned our attention to exploring other symmetrical tetrasulfide compounds (Fig. 4). However, under the previously optimized conditions, only 1,4-dibenzyltetrasulfane and 1,4-dicyclohexyltetrasulfane gave the target products 4a and 4b with a high selectivity. When other tetrasulfides were applied to the reaction, a mixture products of trisulfide and tetrasulfide was obtained. Therefore, we adjusted the reaction conditions (see Supplementary Tables 4–6 for more details). Next, we reexamined the activity of various symmetric tetrasulfide substrates. It is exciting that regardless of whether it is branched liner tetrasulfides, the desired products could be obtained with high chemoselectivity (4c–4e). The reaction of 1-(2-bromoethyl)-4-methoxybenzene with 1,2-bis(furan-2-ylmethyl)disulfane gave the desired product 4f in moderate yield. However, when 1,4-diisopropyltetrasulfane and 1,4-di-p-tolyltetrasulfane were applied to the reaction, respectively, a mixture product of trisulfide and disulfide were formed (4g–4h, 4g′–4h′). To demonstrate the synthetic value of our method, we explored the scale-up with (3-bromopropyl)benzene (1b) and 1,4-di-tert-butyltetrasulfane (2a) under the otherwise identical reaction conditions. To our delight, the desired product 3b could be obtained in 90% yield.

Reaction conditions: Primary alkyl bromides 1 (0.20 mmol, 1.0 equiv.), symmetrical tetrasulfides 2 (0.24 mmol, 1.2 equiv.), Mn (0.30 mmol, 1.5 equiv.), DMF (1 mL), N2 atmosphere. aNi(acac)2 (5.0 mol%), L1 (10 mol%); 40 °C, 24 h. bNiF2 (5.0 mol%), L2 (10 mol%), 80 °C; 24 h. cIsolated yield.
Mechanistic studies
Subsequently, mechanistic studies were conducted to gain insights into the reaction pathway (see Supplementary Figs. 1–3 for more details). When Ni(PPh3)2Cl2 was applied as the catalyst, the reaction proceeded to give a mixture products of 3b and 3b′ (2.8:1) (Fig. 5a). Surprisingly, when we extended the reaction time to 24 h, the ratio of 3b and 3b′ changed 31:1 (Fig. 5b). This indicated that product 3b′ might be a reaction intermediate. Therefore, we prepared the trisulfide product 2i, regrettably, the reaction of (3-bromopropyl)benzene (1b) with 1,3-dibenzyltrisulfane (2i) did not give the target product 7, but the trisulfide product 3i itself was converted into disulfide compound 2j (2i:2j = 1:13) (Fig. 5c). This finding indicated that trisulfide is more likely to transform into disulfide.

a The reaction of 1b and 2a under standard conditions for 12 h. b The reaction of 1b and 2a under standard conditions for 24 h. c The reaction of 1b and 2j under standard conditions for 24 h. d The catalytic system was analyzed by high-resolution mass spectrometry. e Investigation on the reaction mode of trisulfide 6a under different conditions. f Investigation on the reaction mode of unsymmetrical trisulfide 8 under different conditions. g The reaction of 1,2-dibenzyldisulfane 7a and 1,2-di-tert-butyldisulfane 9 under standard conditions for 24 h.
Next, we performed high-resolution mass spectrometry to analyse the catalytic system. Under the standard conditions, only trace amount of the disulfide product 9 and the trisulfide product 10 were detected by careful HRMS analysis (Fig. 5d). We likewise investigated the viability for a conversion of trisulfides to disulfides (Fig. 5e) (see Supplementary Figs. 4, 5 and Table 7 for more details). Through detailed experimentation, it was demonstrated that both the nickel catalyst and the manganese play a crucial role in the conversion of trisulfides to disulfides. According to a previous study19, we furthermore prepared unsymmetric trisulfide 8. Under standard conditions, 1-benzyl-2-(tert-butyl)disulfane 8b and 1,2-dibenzyldisulfane 7a could be obtained in 23% and 15% yields, respectively (with a conversion of 8 being 77%). Only trace amounts of the disulfide product 9 was detected by HRMS analysis. We investigated the conditions for the conversion of trisulfide 8 to disulfide 8b. It was found that when the reaction conditions were modified, only trace amounts of unsymmetric disulfide 8b and symmetric disulfide 9 were detected and 7a was formed as the main product (Fig. 5f). We further investigated the cross reaction of 1,2-dibenzyldisulfane 7a and 1,2-di-tert-butyldisulfane 9 under otherwise identical reaction conditions, which notably failed to give unsymmetrical disulfide 8b. This result provides strong support against an initial formation of two symmetrical disulfides from the trisulfide. Therefore, a subsequent cross reaction to form the unsymmetric disulfide is unlikely to be operative (Fig. 5g).
Subsequently, we investigated the standard reaction at different reaction times (Fig. 6). Through detailed GC-MS analysis, we found that when the reaction proceeded for 1 h, 3b could not be detected, while instead only the trisulfide product 3b′ was detected. When the reaction time was prolonged to 3 h, the ratio of products 3b and 3b′ was close to 1:1. Thereafter, the yield of 3b′ gradually decreased. These experimental findings render the possibility of nickel inserting into the central S–S bond of the tetrasulfide unlikely. Instead our observations provide further strong support for trisulfide serving as the key intermediate.

The investigation of the template reaction under standard conditions at different time by GC-Ms.
There is increasing literature precedence for S–S bonds in polysulfides serving as strong Raman scatterers39. Thus, we exploited the enormous enhancement in the Raman intensity of polysulfides in order to understand the working mode of our reductive coupling. To aid the assignment, Raman frequency calculations for 2a (Fig. 7, A), 3a (Fig. 7, I), and 3a′ (Fig. 7, C) were carried out at the UB3LYP/aug-cc-pV(Q + d)Z-DK level. Thus, Raman frequency calculations for 2a featured peaks at 476 cm−1, 518 cm−1, and 581 cm−1 corresponding to the S(2)–S(3), S(1)–S(2), and (tBu)C-S stretching of tetrasulfide, respectively. As Raman frequency calculations for 3a′, peaks at 500 cm−1, 524 cm−1, and 588 cm−1 correspond to the S(1)–S(2), S(2)–S(3), and (tBu)C-S stretching of trisulfide 3a′, respectively. As Raman frequency for 3a′, peaks at 476 cm−1, 508 cm−1, and 569 cm−1 correspond to the S(1)–S(2), S(2)–S(3), and (tBu)C-S stretching of trisulfide 3a′, respectively. This result is most likely match with the calculated data with −24 cm−1 relative error. As Raman frequency for 3a, peaks at 528 cm−1, and 569 cm−1 correspond to the S–S, and C-S stretching of disulfide 3a, respectively. We further monitored the reaction of 1a and 2a under the standard reaction conditions with the Raman spectroscopy (Fig. 7, E–H). It was found that 476 cm−1 correspond to the S(2)–S(3) of tetrasulfide 2a decreased and a significant increase in 528 cm−1 peak (ν1), which is the characteristic S–S stretching of 3a. We also investigated the Raman spectroscopy of 3a′ under the standard reaction conditions (Fig. 8, C–F). It was found that peak at 476 cm−1 and 508 cm−1 corresponds to the S(1)–S(2) and S(2)–S(3) of trisulfide 3a′ decreased dramatically. This result means that S–S–S bond of trisulfide 3a′ could be quickly activated under the reaction conditions.

Raman caculation spectra of (A) 2a calculated data. (B) 2a. (C) 3a′ calculated data. (D) 3a′. (E) 1a with 2a under optimized reaction conditions, t = 1 h, (F) 1a with 2a under optimized reaction conditions, t = 4 h, (G) 1a with 2a under optimized reaction conditions, t = 8 h, (H) 1b with 2a under optimized reaction conditions, t = 1 h. (I) 3a.

Raman spectra of (A) 3a′ calculated data, (B) 3a′. (C) 3a′ under optimized reaction conditions, t = 1 h, (D) 3a′ under optimized reaction conditions, t = 4 h, (E) 3a′ under optimized reaction conditions, t = 8 h, (F) 3a′ under optimized reaction conditions, t = 12 h, (G) 3a.
Based on our detailed experimental and computational findings and the literature reports33,34,35,40,41,42, we propose a plausible reaction mechanism in Fig. 9. The in situ reduction of nickel(II) by manganese affords Ni(0)(L)2 I. The tetrasulfide 2 could be activated by Ni(0) or Mn. The oxidative addition of I and tetrasulfide 2 in the presence of Ni(0) or Mn furnishes intermediate II, which reacts with an alkyl radical to form nickel(III)(L) intermediate III. The reductive elimination of III leads to trisulfide intermediate IV and the nickel(I)(L)2 species V. Next, the nickel(I) V reacts with alkyl bromide 1 to give the alkyl radical and nickel(II)(L) VI. Finally, the reduction of intermediate VI regenerates complex I and disulfide by-product. On the other hand, the oxidative addition of I and trisulfide IV in the presence of Ni(0) affords intermediate VII (both intermediates VII-1 and intermediate VII-2 are possible). VII is converted into intermediate VIII with releasing polysulfur anion and S8 (polysulfur anion and S8 were confirmed by XRD. For details, please see Supplementary Fig. 5). The reductive elimination of VIII furnish disulfide and complex I.

Possible reaction mechanism for nickel-catalyzed reduction of disulfide.
Discussion
In conclusion, we have reported on a nickel-catalyzed reductive disulfideization of unactivated alkyl bromides with organic tetrasulfides. The reaction proceeded with excellent chemoselectivity under mild conditions, and enabled the efficient assembly of diversely decorated disulfides. Detailed mechanistic studies revealed that, in contrast to literature precedence, the tetrasulfide compound did not undergo scission of the central S–S bond, but selectively formed substituted trisulfides as key intermediates.
Methods
General procedures
Synthesis of 3b
In a glovebox, an oven-dried screw-capped 8 mL vial equipped with a magnetic stir bar was charged with (3-bromopropyl)benzene (1b, 39.6 mg, 0.2 mmol) and 1,4-di-tert-butyltetrasulfane (2a, 58.1 mg 0.24 mmol), Ni(acac)2 (2.6 mg, 5.0 mol%), L1 (10 mol%), Mn (1.5 equiv), DMF (1.0 mL) was added via syringe. The reaction mixture was stirred for 24 h at 40 °C. After 24 h, the crude reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (20 mL × 3). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to afford pure product 3b (99% yield).
Synthesis of 4h
In a glovebox, an oven-dried screw-capped 8 mL vial equipped with a magnetic stir bar was charged with 1-(2-bromoethyl)-4-methoxybenzene (1f, 42.8 mg, 0.2 mmol) and 1,4-di-p-tolyltetrasulfane (2h, 74.4 mg 0.24 mmol), NiF2 (1.0 mg, 5.0 mol%), L1 (10 mol%), Mn (1.5 equiv), DMF (1.0 mL) was added via syringe. The reaction mixture was stirred at 80 °C for 24 h. After 24 h, the crude reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (20 mL × 3). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography to afford products of 4h and 4h′.
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author on request.
References
Narayan, M., Welker, E., Wedemeyer, W. J. & Scheraga, H. D. Oxidative folding of proteins. Acc. Chem. Res. 33, 805–812 (2000).
Wommack, A. J. et al. Discovery and characterization of a disulfide-locked C2-symmetric defensin peptide. J. Am. Chem. Soc. 136, 13494–13497 (2014).
Lu, S. et al. Mapping native disulfide bonds at a proteome scale. Nat. Methods 12, 329–331 (2015).
Jiang, C.-S., Müller, W. E. G., Schröder, H. C. & Guo., Y.-W. Disulfide- and multisulfide-containing metabolites from marine organisms. Chem. Rev. 112, 2179–2207 (2012).
Chankhamjon, P. et al. Biosynthesis of the halogenated mycotoxin aspirochlorine in koji mold involves a cryptic amino acid conversion. Angew. Chem. Int. Ed. 53, 13409–13413 (2014).
A.-Cebollada, J., Kosuri, P., R.-Pardo, J. A. & Fernández., J. M. Direct observation of disulfide isomerization in a single protein. Nat. Chem. 3, 882–887 (2011).
Góngora-Benítez, M., Tulla-Puche, J. & Albericio, F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem. Rev. 114, 901–926 (2014).
Moura, F. A., de Andrade, K. Q., dos Santos, J. C. & Goulart., M. O. Lipoic acid: its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 15, 458–483 (2015).
Silva, F. et al. Short total synthesis of ajoene. Angew. Chem., Int. Ed. 57, 12290–12293 (2018).
Paulina, K. & Rafal., P. The influence of apocynin, lipoic acid and probiotics on antioxidant enzyme levels in the pulmonary tissues of obese asthmatic mice. Life Sci. 234, 116780 (2019).
Fukaya, M. et al. Cyclic sulfur-containing compounds from Allium fistulosum ‘Kujou’. J. Nat. Med. 73, 397–403 (2019).
Witt, D. Recent developments in disulfide bond formation. Synthesis 16, 2491–2509 (2008).
Mays, J. R., Restituyo, J. A., Katzenberger, R. J., Wassarman, D. A. & Rajski, S. R. Tetrahedron Lett. 48, 4579–4583 (2007).
Sun, Q., Cai, S.-T. & Peterson, B. R. Selective disruption of early/recycling endosomes: release of disulfide-linked cargo mediated by a N-alkyl-3β-cholesterylamine-capped peptide. J. Am. Chem. Soc. 130, 10064–10065 (2008).
Görmer, K., Waldmann, H. & Triola, G. Efficient microwave-assisted synthesis of unsymmetrical disulfides. J. Org. Chem. 75, 1811–1813 (2010).
Smith, R., Zeng, X.-J., Müller-Bunz, H. & Zhu, X.-M. Synthesis of glycosyl disulfides containing an α-glycosidic linkage. Tetrahedron Lett. 54, 5348–5350 (2013).
Harusawa, S., Yoshida, K., Kojima, C., Araki, L. & Kurihara., T. Design and synthesis of an aminobenzo-15-crown-5-labeled estradiol tethered with disulfide linkage. Tetrahedron 60, 11911–11922 (2004).
Park, C.-M. et al. 9-Fluorenylmethyl (Fm) disulfides: biomimetic precursors for persulfides. Org. Lett. 18, 904–907 (2016).
Wang, W.-G., Lin, Y.-Z., Ma, Y.-D., Tung, C.-H. & Xu., Z.-H. Cu-catalyzed electrophilic disulfur transfer: synthesis of unsymmetrical disulfides. Org. Lett. 20, 3829–3832 (2018).
Zou, J.-X. et al. Phthalimide-carried disulfur transfer to synthesize unsymmetrical disulfanes via copper catalysis. ACS Catal. 9, 11426–11430 (2019).
Xiao, X., Feng, M.-H. & Jiang., X.-F. New design of a disulfurating reagent: facile and straightforward pathway to unsymmetrical disulfanes by copper-catalyzed oxidative cross-coupling. Angew. Chem., Int. Ed. 55, 14121–14125 (2016).
Xiao, X., Xue, J.-H. & Jiang., X.-F. Polysulfurating reagent design for unsymmetrical polysulfide construction. Nat. Commun. 9, 2191–2200 (2018).
Xue, J.-H. & Jiang, X.-F. Unsymmetrical polysulfidation via designed bilateral disulfurating reagents. Nat. Commun. 11, 4170–4177 (2020).
Xue, J.-H. & Jiang, X.-F. Polysulfuration via a bilateral thiamine disulfurating reagent. Org. Lett. 22, 8044–8048 (2020).
Wu, Z.-J. & Pratt, D. A. Radical substitution provides a unique route to disulfides. J. Am. Chem. Soc. 142, 10284–10290 (2020).
Chauvin, J. P. R., Haidasz, E. A., Griesser., M. & Pratt., D. A. Polysulfide-1-oxides react with peroxyl radicals as quickly as hindered phenolic antioxidants and do so by a surprising concerted homolytic substitution. Chem. Sci. 7, 6347–6356 (2016).
Chauvin, J. P. R., Griesser., M. & Pratt., D. A. The antioxidant activity of polysulfides: it’s radical! Chem. Sci. 10, 4999–5010 (2019).
Wang, X., Dai, Y.-J. & Gong, H.-G. Nickel-catalyzed reductive couplings. Top. Curr. Chem. 374, 43 (2016).
Diccianni, J. B. & Diao, T.-N. Mechanisms of nickel-catalyzed cross-coupling reactions. Trends Chem. 1, 830–844 (2019).
Li, Y.-Q., Fan, Y.-H. & Jia, Q.-F. Recent advance in Ni-catalyzed reductive cross-coupling to construct C(sp2)–C(sp2) and C(sp2)–C(sp3) bonds. Chin. J. Org. Chem. 39, 350–362 (2019).
Wu, Y., Luo, F., Pan, S.-M., Pan, Y.-H. & He., S.-H. Nickel-catalyzed coupling of 1,2-diarylthio-1,2-diarylalkenes with grignard reagents for synthesis of multi-substituted alkenes. Chin. J. Org. Chem. 39, 2946–2951 (2019).
Gao, X., He, X. & Zhang., X.-G. Nickel-catalyzed difluoromethylation of (hetero)aryl bromides with BrCF2H. Chin. J. Org. Chem. 39, 215–222 (2019).
Fang, Y., Rogge, T., Ackermann, L., Wang, S.-Y. & Ji., S.-J. Nickel-catalyzed reductive thiolation and selenylation of unactivated alkyl bromides. Nat. Commun. 9, 2240 (2018).
Li, J., Rao, W.-D., Wang, S.-Y. & Ji., S.-J. Nickel-catalyzed defluorinative reductive cross-coupling reaction of gem-difluoroalkenes with thiosulfonate or selenium sulfonate. J. Org. Chem. 84, 11542–11552 (2019).
Li, J., Wang, S.-Y. & Ji., S.-J. Nickel-catalyzed thiolation and selenylation of cycloketone oxime esters with thiosulfonate or seleniumsulfonate. J. Org. Chem. 84, 16147–16156 (2019).
Zhao, C.-L., Jia, X., Wang, X. & Gong., H.-G. Ni-catalyzed reductive coupling of alkyl acids with unactivated tertiary alkyl and glycosyl halides. J. Am. Chem. Soc. 136, 17645–17651 (2014).
Wang, X. et al. Ni-catalyzed reductive coupling of electron-rich aryl iodides with tertiary alkyl halides. J. Am. Chem. Soc. 140, 14490–14497 (2018).
Ye, Y., Chen, H.-F., Sessler, J. L. & Gong., H.-G. Zn-mediated fragmentation of tertiary alkyl oxalates enabling formation of alkylated and arylated quaternary carbon centers. J. Am. Chem. Soc. 141, 820–824 (2019).
Steudel, R. & Chivers, T. The role of polysulfide dianions and radical anions in the chemical, physical and biological sciences, including sulfur-based batteries. Chem. Soc. Rev. 48, 3279–3319 (2019).
Biswas, S. & Weix, D. J. Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 135, 16192–16197 (2013).
Venkanna, G. T., Arman, H. D. & Tonzetich, Z. J. Catalytic C–S cross-coupling reactions employing Ni complexes of pyrrole-based pincer ligands. ACS Catal. 4, 2941–2950 (2014).
Oderinde, M. S., Frenette, M., Robbins, D. W., Aquila, B. & Johannes., J. W. Photoredox mediated nickel catalyzed cross-coupling of thiols with aryl and heteroaryl iodides via thiyl radicals. J. Am. Chem. Soc. 138, 1760–1763 (2016).
Acknowledgements
We gratefully acknowledge Prof. J.-L. Yao and Prof. X.-G. Bao from Soochow University for helpful discussion. We also gratefully acknowledge the National Natural Science Foundation of China (Nos. 21971174, 21772137), PAPD, Cyrus Tang (Zhongying) scholar (award to S.-Y. Wang) and Soochow University for financial support. And generous support by the DFG (Gottfried-Wilhelm-Leibniz award to L.A.).
Author information
Authors and Affiliations
Contributions
F.W., Y.C., W.R., L.A., and S.-Y.W. planned, conducted, and analyzed the experiments. L.A. and S.-Y.W. directed the project. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhen Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, F., Chen, Y., Rao, W. et al. Efficient preparation of unsymmetrical disulfides by nickel-catalyzed reductive coupling strategy. Nat Commun 13, 2588 (2022). https://doi.org/10.1038/s41467-022-30256-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-30256-0
This article is cited by
-
Photo-induced decarboxylative C-S bond formation to access sterically hindered unsymmetric S-alkyl thiosulfonates and SS-alkyl thiosulfonates
Nature Communications (2024)
-
Synthesis of N-acyl sulfenamides via copper catalysis and their use as S-sulfenylating reagents of thiols
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
