
Abstract
PoxtA and OptrA are ATP binding cassette (ABC) proteins of the F subtype (ABCF). They confer resistance to oxazolidinone and phenicol antibiotics, such as linezolid and chloramphenicol, which stall translating ribosomes when certain amino acids are present at a defined position in the nascent polypeptide chain. These proteins are often encoded on mobile genetic elements, facilitating their rapid spread amongst Gram-positive bacteria, and are thought to confer resistance by binding to the ribosome and dislodging the bound antibiotic. However, the mechanistic basis of this resistance remains unclear. Here we refine the PoxtA spectrum of action, demonstrate alleviation of linezolid-induced context-dependent translational stalling, and present cryo-electron microscopy structures of PoxtA in complex with the Enterococcus faecalis 70S ribosome. PoxtA perturbs the CCA-end of the P-site tRNA, causing it to shift by ∼4 Å out of the ribosome, corresponding to a register shift of approximately one amino acid for an attached nascent polypeptide chain. We postulate that the perturbation of the P-site tRNA by PoxtA thereby alters the conformation of the attached nascent chain to disrupt the drug binding site.
Similar content being viewed by others

Introduction
Antibiotic resistance (ARE) is a growing threat to the efficacy of our current arsenal of clinically approved antimicrobial agents. The ATP-binding cassette (ABC) family of proteins are well-known for their role as multidrug resistance transporters, which use the energy of ATP hydrolysis to drive the efflux of antibiotics from the bacterial cytoplasm1,2. In recent years, it has become clear that ARE ABC proteins that belong to subfamily F (ABCF) are not transporters—and thus do not confer resistance via efflux—but rather act via a direct target protection mechanism3,4,5,6,7,8,9,10.
ARE–ABCF proteins confer resistance to a diverse range of antibiotics that inhibit protein synthesis by targeting the large subunit (LSU) of the ribosome. Based on the spectrum of antibiotic resistance that they confer ARE–ABCF proteins fall into three functional groups: (i) those that protect from pleuromutilins, lincosamides and streptogramins A (PLSA), (ii) those that protect from macrolides and streptogramin B (MSB), and, finally, (iii) those that protect from phenicols and oxazolidinones (PhO)3,4,7,8,9,10. These functional groups do not map exactly to the phylogenetic tree of ARE–ABCFs, in which seven subclasses (ARE1-7) were originally distinguished, but are rather scattered amongst non-ARE–ABCFs, implying that these resistance factors have arisen multiple times by convergent evolution6. Despite the divergence in the spectrum of antibiotic resistance, ARE–ABCFs share a common architecture. ARE–ABCFs are comprised of two ABC nucleotide-binding domains (NBD1 and NBD2) that are separated by a helical linker termed an ARD (antibiotic-resistance domain), and, depending on the species, ARE–ABCFs may also have an additional “Arm” subdomain inserted within NBD1 as well as a C-terminal extension (CTE)6. In fact, this architecture is similar to many non-ARE–ABCFs, such as the E. coli housekeeping ABCF ATPase EttA, in which the ARD equivalent is shorter and referred to as a P-site tRNA-interaction motif (PtIM)11,12, thus making it difficult to judge whether many ABCF proteins are actually resistance determinants or endogenous proteins of mostly unknown function6.
Cryo-EM structures of ribosomes in complex with ARE–ABCFs that confer resistance to PLSA (ARE1 VgaALC and VgaL, ARE2 VmlR, ARE3 LsaA) and MSB (ARE1 MsrE) classes of antibiotics have revealed that these proteins bind within the E-site13,14,15, similar to that reported previously for the housekeeping non-ARE–ABCF EttA11. However, in contrast to EttA11,12, the longer ARD of the ARE–ABCF proteins distorts the P-site tRNA, allowing the factor to access the peptidyl transferase centre (PTC) on the LSU of the ribosome and dislodge the relevant antibiotics from their binding sites13,14,15. These structures revealed that there is often no steric overlap between the ARD of the ARE–ABCF and the drugs and even when there is a steric overlap, the mutational analysis indicated that it is not strictly required for resistance13,14,15. Collectively, these results support a model where the ARE–ABCFs dislodge the drugs from the PTC by inducing a cascade of conformational changes within the 23S rRNA nucleotides that comprise the drug-binding site15. For MsrE, drug release was reported to occur in the presence of a so-called EQ2 variant of the protein, which is deficient in ATPase activity14, suggesting that ATP hydrolysis is not essential for drug release, but rather is needed for recycling of the factor from the ribosome.
In contrast to the relatively well-studied PLSA- and MSB-protecting ARE–ABCFs, mechanistic insights into how ARE7 OptrA and ARE8 PoxtA confer resistance to PhO antibiotics are lacking. The first ARE–ABCF from this group to be discovered was OptrA. This factor confers resistance to the phenicols, such as chloramphenicol and florfenicol, as well as the oxazolidinones linezolid and, to a lesser extent, tedizolid16. OptrA was originally found on the conjugative plasmid pE349 from Enterococcus faecalis16, but has subsequently been detected, both plasmid and chromosomal-encoded, across many Gram-positive enterococci, staphylococci and streptococci of human and animal origin16,17,18,19,20,21,22. At least 69 variants of the optrA gene have been reported to date, differing by 1–20 aa substitutions, which corresponds to an amino acid identity of 97.1–99.8% compared to the first reported OptrA sequence22. Moreover, evidence for horizontal transfer of OptrA to Gram-negative bacteria, such as Campylobacter coli, has also recently been described23,24. While oxazolidinones are not clinically effective against Gram-negative pathogens, this raises the concern of possible co-selection of antibiotic co-resistance i.e. selection for simultaneous transfer and spread of several antibiotic-resistance genes encoded by one mobile genetic element. In addition to OptrA, a second ARE–ABCF from this group was detected by bioinformatic analysis of the genome of a methicillin-resistant Staphylococcus aureus (MRSA) strain AOUC-0915 isolated from a cystic fibrosis patient at the Careggi University Hospital in Florence, Italy25. Expression of the resistance determinant in S. aureus, E. faecalis and E. coli was reported to confer resistance to phenicol-oxazolidinone-tetracycline antibiotics, and was therefore termed PoxtA25. To date, PoxtA has been found exclusively in Enterococcus and Staphylococcus species, most frequently in E. faecium isolates of both human and animal origin22.
In the absence of structures of OptrA and PoxtA on the ribosome, it has remained unclear how these ARE–ABCFs confer antibiotic resistance. Both chloramphenicol and linezolid bind at the PTC and inhibit the elongation phase of protein synthesis26. However, their activity is context-specific such that translation arrest is most efficient when the nascent polypeptide chain on the ribosome carries an alanine residue and, to a lesser extent, serine or threonine in its penultimate position27,28,29. Although the ARDs of OptrA and PoxtA are slightly longer (4–5 aa) than the PtIM of non-ARE–ABCFs such as EttA, they are considerably shorter than the ARDs of ARE–ABCFs from other groups, and at least 20 amino acids shorter than other ARE–ABCFs for which structures have been reported (Fig. 1). Thus, assuming OptrA and PoxtA bind similarly to the ribosome as other ARE–ABCFs, the ARDs are unlikely to be able to reach into the PTC to dislodge the drugs from their binding site8. Moreover, it is hard to rationalise how PoxtA also confers resistance to tetracycline antibiotics, which bind near the decoding site on the small subunit (SSU), which is located far from the PTC on the LSU26.

The PoxtA and OptrA ARDs are slightly longer (4–5 amino acids) than the equivalent region in EttA, but significantly shorter than in other ARE–ABCFs. The central region with an orange highlight, which includes the ARD loop and some of the adjacent helices in some proteins, was not aligned but simply ordered by length. Sequences were aligned with MAFFT70 and edited by hand to reduce gap placement. Sequence accession numbers in the title are Uniprot or NCBI protein accessions.
Here, we systematically characterise the PoxtA and OptrA resistance determinants, revealing that both increase the minimum inhibitory concentrations (MICs) to phenicols, such as chloramphenicol, thiamphenicol and florfenicol, as well as to the oxazolidinone linezolid, but not to macrolides, pleuromutilins, lincomycins and streptogramins that bind adjacent to the PTC within the ribosomal exit tunnel. Moreover, we find no evidence for either PoxtA or OptrA conferring resistance to the non-PTC binding antibiotic tetracycline. Cryo-EM structures of PoxtA on the ribosome reveal that it binds in the E-site and, despite the short ARD, still induces a distortion of the P-site tRNA, leading to retraction of its CCA-end from the PTC. Unlike for other ARE–ABCFs, we observe no conformational rearrangements within the 23S rRNA around the drug-binding sites within the A site of the PTC upon binding of PoxtA to the ribosome. This leads us to propose a model whereby the distortion of the P-site tRNA by PoxtA (and OptrA) reduces the affinity of the drugs for their binding site by altering the context and therefore interaction of the nascent polypeptide chain with respect to the drugs.
Results
PoxtA and OptrA confer resistance to phenicols and oxazolidinones, but not tetracyclines
To systematically characterise the antibiotic-resistance profiles of PoxtA and OptrA, representatives of these ARE–ABCF groups were expressed in an E. faecalis TX5332 strain where the lsaA gene had been disrupted (ΔlsaA), and MICs were determined for phenicol (chloramphenicol, thiamphenicol and florfenicol), oxazolidinone (linezolid), macrolide (erythromycin, azithromycin and leucomycin), lincosamide (lincomycin and clindamycin), pleuromutilin (tiamulin, retapamulin), streptogramin A and B (Virginiamycin M1 and S1, respectively) and tetracycline antibiotics (Table 1). We have characterised OptrA E35048 from E. faecium30, OptrA ST16 from the clinical E. faecalis ST16 isolate21, PoxtA AOUC-0915 from MRSA25 and, finally, PoxtA EF9F6 from a multidrug-resistant ST872 E. faecium clinical isolate 9-F-631. As controls, we determined MICs for the E. faecalis ΔlsaA strain transformed with the empty vector plasmid pCIEspec, as well as expressing LsaA, the native genome-encoded ARE–ABCF from E. faecalis.
In agreement with earlier reports, expression of LsaA confers resistance to PLSA antibiotics, but not PhO, MSB or tetracycline15,32. By contrast, cCF10-inducible expression of either OptrA or PoxtA results in a 2- to 16-fold MIC increase for PhO antibiotics, and does not, as expected, result in resistance against either PLSA or MSB, as observed in earlier reports16,25. While the earlier study25 reported a minor protective effect of PoxtA against tetracycline (a twofold increase in MIC for E. faecalis and S. aureus), in our hands expression of neither of the PoxtA variants resulted in any increase in MIC for this antibiotic. Notably, the lack of effect of PoxtA AOUC-0915 expression on the MIC for the tetracycline antibiotic tigecycline in either E. faecalis or S. aureus25 is consistent with the original E. faecium 9-F-6 strain from which we have isolated PoxtA EF9F6 also being susceptible to tigecycline (Supplementary Table 1). Therefore, we concluded that the antibiotic-resistance spectrum of PoxtA is similar, if not identical, to that of OptrA. This similarity appears to be a case of convergent evolution, as there is no phylogenetic support for PoxtA and OptrA being more closely related to each other than to any other ABCF subfamily member (Supplementary Fig. 1). Indeed, OptrA is more closely related to the vertically inherited and probable housekeeping ABCF YdiF of Firmicutes (84% bootstrap support, Supplementary Fig. 1), while the relationship of PoxtA to other subfamilies is unresolved (bootstrap support below 50%). On these grounds, we conclude that PoxtA-like proteins constitute a separate ARE subfamily which we call ARE8.
PoxtA rescues ribosome stalls associated with linezolid treatment
Extensive in vivo and in vitro studies into the mechanism of action of chloramphenicol and linezolid during E. coli protein synthesis have established that both antibiotics act as context-specific inhibitors of translation elongation27,28,29. Specifically, efficient ribosomal stalling by linezolid is strongly stimulated by the presence of alanine as the penultimate amino acid in the polypeptide nascent chain (i.e. the −1 position), while the codon for this amino acid occupies the ribosomal E-site. Dependence of chloramphenicol-mediated stalling on alanine in the −1 position of the nascent chain was previously shown in Gram-positive Firmicute species Lactobacillus plantarum and Bacillus subtilis using the recently developed bacterial high-throughput 5ʹP sequencing (5Pseq) method33. Analogous to the well-established 5Pseq for eukaryotes34, the bacterial version of the technique detects ribosome occupancy by generating in vivo footprints of the 5ʹ-most ribosome on the mRNA molecule through specific sequencing of the 5ʹ monophosphorylated intermediates of 5ʹ–3ʹ mRNA degradation33.
We used 5Pseq to globally survey the linezolid-induced ribosomal stalls in E. faecalis and directly assess the role of PoxtA in overcoming these stalls. 5Pseq analysis of linezolid-treated E. faecalis robustly detected increased 5Pseq coverage signal −8 nucleotides upstream of the first (5ʹ) nucleotide of the alanine codon (and to lesser extent at serine codons), which is indicative of ribosomal stalling with these codons in the E-site and their respective amino acids in the −1 position of the nascent chain (Fig. 2a and Supplementary Fig. 2). Importantly, all of the alanine iso-codons display near-identical linezolid-induced stalls, which is indicative of the stalling being defined by the nature of the amino acid, not the codon itself (Supplementary Fig 3). There was no particular enrichment of codons in the A- or P-sites (Fig. 2a). The strong stalling at alanine and weaker stalling at serine and threonine is in good agreement with an E. coli ribosome profiling study, indicating that linezolid affects translation similarly in both organisms27. PoxtA expression resulted in the reduction of this context-specific stalling signal in response to linezolid (Fig. 2b, c and Supplementary Fig. 2). The rescue effect is highly reproducible between the three biological replicates (Supplementary Fig. 4a–d), and both the linezolid-induced alanine stalling and its rescue by PoxtA expression are statistically significant (Supplementary Fig. 4e).

a A heatmap of log2-relative change in the 5Pseq coverage of wild-type E. faecalis upon linezolid treatment. The distance (in nucleotides, nt) from the 5ʹ of the sequenced mRNA fragments to synonymous codons encoding a specified amino acid is indicated on the x axis. The 0 nt position corresponds to the first nucleotide of the codon. Dark blue signifies decreased 5PSeq coverage upon linezolid treatment, yellow signifies increased coverage upon linezolid treatment. A specific increase in the 5Pseq coverage associated with synonymous codons located at –8 nucleotides is indicative of a ribosomal stall with the corresponding amino acid in the −1 position of the nascent chain, as illustrated schematically in panels b and c (ref. 33). b, c Metagene analysis of 5Pseq libraries aligned to alanine codons. Linezolid-induced ribosomal stalling results in a specific increase in 5Pseq read counts 8 nt upstream of alanine codons, indicating a ribosomal stall with alanine residues in the −1 position (see schematics). Samples were prepared with E. faecalis harbouring the empty pCIEspec vector (a, b) or upon poxtA expression (c, blue line), with and without linezolid treatment. Exponentially growing cells were treated with 4 μg/mL linezolid (final concentration) 10 min before harvest. All analyses were performed on pooled datasets from three biological replicates.
By contrasting 5PSeq datasets of antibiotic-untreated E. faecalis that do not express PoxtA with that of an untreated PoxtA-overexpressing control, we probed whether sampling of the ribosomes by PoxtA in the absence of antibiotics causes ribosomal stalling. Our analyses revealed slightly increased ribosomal stalling at intrinsically slow initiation and termination steps (Supplementary Fig. 5). Importantly, no stalling signal specific for the −1 nascent chain position (corresponding to the ribosomal E-site) was detectable, consistent with linezolid determining the amino acid specificity of antibiotic-induced stalls.
Collectively, our 5Pseq study confirms the generality of the context-specific mechanism of action of linezolid and indicates that PoxtA alleviates the drug-induced stalling at positions with −1 alanine residues.
Cryo-EM structures of PoxtA–70S complexes
Our attempts to reconstitute OptrA- and PoxtA–70S ribosome complexes in vitro were unsuccessful due to problems obtaining soluble homogenous OptrA and PoxtA proteins. Therefore, we employed an in vivo pull-down approach with strains overexpressing C-terminally FLAG3-tagged OptrA and PoxtA proteins, as used recently to generate other ARE–ABCF–ribosome complexes15. We expressed both the wild-type ATPase-competent OptrA and PoxtA and EQ2 ATPase-deficient variants bearing Glu-to-Gln substitutions in both NBD cassettes. Such EQ2 variants have been successfully employed to trap other ABCF proteins on the ribosome because these substitutions allow binding but prevent hydrolysis of ATP by the ABCF11,13,15,35. The OptrA-EQ2 variant was earlier shown to have a compromised in vitro ATPase activity and unable to confer resistance in vivo36. Consequently, the addition of chloramphenicol or linezolid to growth media would block cCF10-driven overexpression of ATPase-deficient OptrA/PoxtA EQ2 variants. Therefore, in vivo ABCF–ribosome complex formation and its subsequent immuno-purification were performed in the absence of the antibiotic. Affinity purification via the FLAG3 tag was performed in the presence of 0.5 mM ATP from clarified lysates prepared from the E. faecalis ΔlsaA strain expressing the FLAG3-tagged ARE–ABCF, either wild-type or EQ2-variant. Analysis of the elution fractions from the purifications indicated that only the E. faecium PoxtA(EF9F6)-EQ2 factor was bound stably to the ribosome (Supplementary Fig. 6). Furthermore, our attempts with OptrA carrying a single individual EQ substitution (E470Q) were equally unsuccessful. Therefore, we focused on the PotxA(EF9F6)-EQ2–70S sample and subjected it to structural analysis using single-particle cryo-EM.
Using a Titan Krios transmission electron microscope with a K2 direct electron detector, we collected 3640 micrographs which, after 2D classification, yielded 140,310 ribosomal particles (Supplementary Fig. 7). In silico sorting revealed that 80% of these particles contained an additional density for PoxtA and/or tRNAs, which after 3D refinement resulted in a cryo-EM map of E. faecalis 70S ribosome with an average resolution of 2.4 Å (Supplementary Fig. 8a). Subsequent multibody refinement yielded average resolutions of 2.2 Å and 2.5 Å for the LSU and SSU, respectively (Supplementary Fig. 8b–e). The increase in resolution compared to the previous E. faecalis 70S ribosome models at 2.8–2.9 Å15,37 is evident from improved quality and features of the cryo-EM density, including visualisation of some rRNA modifications (e.g. N2-methylguanosine), water molecules and hydrated magnesium ions (Supplementary Fig. 9a–f and Supplementary Table 2).
Further subsorting of ribosomal particles using a mask focused on the intersubunit space yielded four defined classes, which we refer to as states I–IV (Supplementary Fig. 7). States I and II contained density for PoxtA bound in the E-site and tRNA in the P site, and had average resolutions of 2.9 and 3.0 Å, respectively (Fig. 3a, Supplementary Fig. 10a, b and Supplementary Table 2). State II (not shown in Fig. 3) differed from the state I by only a slight rotation of the SSU relative to the LSU, and in particular the conformation of PoxtA was nearly identical to that of state I (Supplementary Fig. 11). State III was similar to state II, but additionally contained an A-site tRNA and PoxtA differed only slightly in NBD2, close to the SSU (Fig. 3b and Supplementary Fig. 11), whereas state IV contained P-site tRNA only (Fig. 3c), presumably because PoxtA dissociated during sample preparation. States III and IV were also refined, resulting in final reconstructions with average resolutions of 2.9 Å and 3.1 Å, respectively (Supplementary Fig. 10a, b and Supplementary Table 2). The cryo-EM density for PoxtA in states I–III was generally well-resolved (Supplementary Fig. 10c), enabling a reliable model to be built for NBD1, NBD2 and the ARD (Fig. 3d). By contrast, the Arm domain, which interacts with uL1, appeared flexible (Supplementary Fig. 10c) and could only be modelled as a rigid body fit of the two α-helices (Fig. 3d). The best-resolved region of PoxtA was the ARD, consisting of two α-helices (α1 and α2) and the ARD loop (Fig. 3d and Supplementary Fig. 10c), where the majority of sidechains could be modelled unambiguously (Fig. 3e). Additional density located between NBD1 and NBD2 of PoxtA was attributed to two ATP molecules (ATP-1 and ATP-2) and a likely magnesium ion (Fig. 3f), as expected from the use of the ATPase-deficient PoxtA-EQ2 variant. As observed for other ribosome-bound ARE–ABCF structures13,14,15, the NBDs of PoxtA adopt a closed conformation, which is also consistent with the inability to hydrolyse ATP. In all states, the anticodon stem loop (ASL) and acceptor arm, including the CCA-end, are well-resolved, whereas the elbow region of the tRNAs exhibits some flexibility (Supplementary Fig. 10d).

a–c Cryo-EM maps with isolated densities for (a, b) E. faecium PoxtA (red) in complex with the E. faecalis 70S ribosome and a P-tRNA (cyan) or b P-tRNA (cyan) and A-tRNA (tan), c P-tRNA (cyan) only, with small subunit (SSU, yellow) and large subunit (LSU, grey). d Density (grey isosurface) with molecular model of PoxtA from a coloured according to domain as represented in the associated schematics: nucleotide-binding domain 1 (NBD1, tan), antibiotic-resistance domain (ARD, pink), nucleotide-binding domain 2 (NBD2, green) and C-terminal extension (CTE, grey, not modelled). α1 and α2 indicate the two α-helices of the ARD interdomain linker. In d, the ATP nucleotides are coloured blue. e Close view of the ARD tip from state I with sharpened map. f Close view of ATPs bound by PoxtA (state I).
In all states, the density for the P-site tRNA is consistent with initiator tRNAfMet, indicating that in the absence of chloramphenicol- or linezolid-stalled ribosomes, the PoxtA-EQ2 variants bind to the vacant E-site of initiation complexes, as observed previously for other ARE–ABCF-EQ2–ribosome complexes prepared by an in vivo pull-down approach15. Specific recruitment of PoxtA-EQ2 to initiation complexes is likely due to the availability of the E-site rather than due to the specific affinity to initiating ribosomes as such. In state IV, clear density for the fMet moiety attached to the P-site tRNA is evident, whereas state III appears to be a post-peptide bond formation state with a deacylated P-site tRNA and A-site tRNA bearing a dipeptide. In states I and II, which contain PoxtA, but lack A-site tRNA, some density for the fMet moiety on the distorted P-site tRNA is evident but is poorly resolved.
Interaction of PoxtA with the ribosome and P-site tRNA
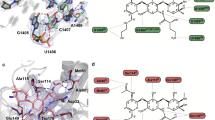
In states I–III, PoxtA is located within the ribosomal E-site (Fig. 4a) and generally binds similarly to that observed for other ARE–ABCF proteins, such as VmlR, MsrE, LsaA, VgaL and VgaALC13,14,15, as well as the non-ARE–ABCF protein EttA11. Unlike these ARE–ABCFs that lack an Arm subdomain (or have a short Arm in the case of LsaA)6, the Arm subdomain of PoxtA (and OptrA6) is prominent, like that of EttA11,12, and stabilises an open conformation of the L1 stalk via direct interaction with domain II of uL1 (Fig. 4a). Additional contacts to the 23S rRNA helices H77/H78 of the L1 stalk are evident from the NBD1 of PoxtA, as are interactions for NBD1 with H68 and bL33 on the LSU (Fig. 4a). By contrast, NBD2 of PoxtA spans across the intersubunit interface, establishing interactions with uL5 on the LSU as well as uS7 and h41 on the SSU (Fig. 4A). NBD2 also interacts directly with the elbow region of the P-site tRNA, namely, with the G19-C56 basepair that links the D- and T-loops (Fig. 4a, b). Here, Ser430 of PoxtA is within hydrogen bonding distance of the N7 of G19 and the sidechain of Arg426 of PoxtA stacks upon the nucleobase of C56 of the T-loop (Fig. 4b). However, it is the ARD that makes the most extensive interactions with the P-site tRNA, establishing a complex network of hydrogen bonding interactions with the acceptor arm and CCA-end (Fig. 4a). In particular, two glutamine residues, Gln275 and Gln279, located at the distal end of the α-helix α1 of the ARD insert into the minor groove of the acceptor’s arm where hydrogen bond interactions can form with the C3-G70 basepair (Fig. 4c). Hydrogen bonding is also possible from the ε-amino group of Lys278 and the backbone carbonyl of Gln275 of PoxtA with the phosphate-oxygen of G4 and the ribose-oxygen of C71 of the P-tRNA, respectively. The loop region of the ARD of PoxtA interacts predominantly with the single-stranded CCA-3ʹ end of the P-site tRNA (Fig. 4d). Ser281 is within H-bonding distance to the phosphate-oxygen of A73, whereas the sidechain of Gln282 stacks upon the base of C74 and can interact with the ribose-hydroxyl of A72 (Fig. 4d). C75 of the P-site tRNA is also stabilised by indirect contacts with the backbone carbonyl of Thr283 via a water molecule, as well as a direct H-bond with the sidechain of His285, the first residue of α-helix α2 of the ARD of PoxtA (Fig. 4e). The ARD is stabilised by multiple contacts between residues within α-helix α2 and rRNA nucleotides located in H74 and H93. For example, the sidechains of Arg294 and Arg297 contact nucleotides A2595–G2598 (E. coli numbering used throughout) located within the loop of H93 of the 23S rRNA, and Glu293 interacts with G2597 directly as well as G2598 via a putative water molecule (Fig. 4f).

a Overview of PoxtA interactions with the 23S rRNA (grey), 16S rRNA h41 (yellow), uL1 (gold), uS7 (green), uL5 (pink), bL33 (tan) and the P-tRNA (light blue). The P-tRNA CCA 3ʹ end, acceptor stem (Acc.), and elbow are indicated. b–e Interactions between the P-tRNA elbow (light blue) and the PoxtA NBD2 (b), the P-tRNA acceptor stem (light blue) and the PoxtA ARD (c), the ARD and the P-tRNA CCA-end, including a modelled water molecule (labelled W) (d, e). f An interaction between the PoxtA ARD α2 and the 23S rRNA. The high-resolution model from the combined 70S volume was used.
PoxtA perturbs the position of the CCA-end of the P-site tRNA at the PTC
Despite the short ARD, binding of PoxtA to the E-site nevertheless causes a distortion of the P-site tRNA when compared to the canonical P-site tRNA binding position, such as that observed in state IV (Fig. 5a, b). While the ASL remains fixed in position on the SSU where it decodes the P-site codon of the mRNA, the elbow region shifts towards the E-site by ~6–7 Å, thus bringing it into contact with NBD2 of PoxtA (Fig. 5b). The shift of the elbow region is very similar to that observed for the distorted P-site tRNAs observed on the ribosome in the presence of the other ARE–ABCFs13,14,15 such as LsaA (Fig. 5c). However, in the PoxtA–70S complex, the distortion is accompanied by a smaller ~4 Å shift of the acceptor arm of the P-site tRNA away from the PTC (Fig. 5d), whereas for other ARE–ABCF complexes, the CCA-end of the P-site tRNA completely vacates the PTC due to the presence of the longer ARD (as illustrated here for LsaA in Fig. 5c). Although PoxtA contains a shorter ARD than other ARE–ABCFs, the loop of the ARD still contacts the acceptor stem of the P-tRNA, precluding canonical interactions between the tRNA and the ribosome (Fig. 5d). As a consequence, the single-stranded CCA-3ʹ end of the P-site tRNA becomes contorted in the presence of PoxtA and the canonical interactions of the C75 and C74 of a P-site tRNA with 23S nucleotides G2251 and G2252, respectively, of the P-loop (H80) are disrupted (Fig. 5e, f). This results in a shift in register such that C75 basepairs with G2252 and C74 stacks upon Gln282 of PoxtA and interacts with G2253 and C2254 (Fig. 5f). The register shift is reminiscent of, but distinct to, that observed for the Pint-tRNA in E. coli 70S ribosome complexes formed in the presence of a derivative of the antimicrobial peptide apidaecin and the termination release factor RF338.

a–c Comparison of of the P-site tRNA from (a) E. faecalis 70S–P-tRNA only complex (grey), (b) PoxtA–70S complex (state I, PoxtA in red and P-tRNA in light blue) and (c) LsaA–70S complex (PDB ID 7NHK15, LsaA in green and P-tRNA in light blue). Models of chloramphenicol for PoxtA (PDB ID 6ND577) and lincomycin for LsaA (PDB ID 5HKV)78 are superimposed for reference. d close-up of (b) showing P-tRNA acceptor stem distortion induced by PoxtA binding. e, f Interaction between the 23S rRNA P-loop (grey) and P-tRNA (light blue) for (e) the P-tRNA-only complex (state IV) and f with bound PoxtA (state I, state IV P-tRNA is overlayed in transparent grey for comparison). g, h Indirect modulation of the chloramphenicol binding site by PoxtA. g The structure of chloramphenicol (cam) stabilised by a nascent peptide chain with alanine in the −1 position39. h Modelled shift of the nascent chain shown in g induced by PoxtA displacement of the P-tRNA CCA-end. The P-tRNA from g is overlaid in transparent grey for comparison.
A comparison of the binding position of the ARD of PoxtA with that of the phenicol (chloramphenicol) and oxazolidinone (linezolid) antibiotics reveals that there is no steric overlap between PoxtA and either drug-binding site (Fig. 5b). Indeed, His285 of PoxtA, which comes closest to the drugs, is still located >16 Å away, and furthermore the ARD of PoxtA is partitioned from the drugs by the CCA-3ʹ end of the P-site tRNA (Fig. 5b). This raises the question as to whether PoxtA dislodges these drugs from the ribosome using an indirect mechanism, such as by inducing conformational changes within the 23S rRNA nucleotides comprising the drug-binding site(s), as proposed for other ARE–ABCFs13,14,15. To examine this, we compared the conformation of the PTC nucleotides comprising the drug-binding sites in our structures with those in the presence of chloramphenicol and linezolid. We could find no evidence that the presence of PoxtA induces any conformational changes within the drug-binding sites that would lead to dissociation of the drugs (Supplementary Figs. 12 and 13). In fact, we note that the conformation of the A-site pocket where the drugs bind is very similar, if not identical, for states I–IV, regardless of whether PoxtA is present (state I–III) or absent (state IV), or whether the A-site is vacant (state I–II, IV) or occupied (state III) (Supplementary Figs. 12 and 13). Although this suggests that PoxtA does not dislodge the drugs from the ribosome by altering the 23S rRNA in the binding site, we cannot exclude that such conformational changes occur upon ATP hydrolysis or upon dissociation of PoxtA from the ribosome.
An alternative scenario is that the distortion of the P-site tRNA induced by PoxtA binding indirectly reduces the affinity of the drugs for their A-site binding pocket. Both chloramphenicol and linezolid are context-specific inhibitors of translation elongation, such that the highest translation arrest activity occurs when the penultimate amino acid (−1 position) attached to the CCA-end of the P-site tRNA is alanine, and to a lesser extent, serine or threonine27,28,29. Structures of chloramphenicol on the ribosome with peptidyl-tRNA mimics reveal an intimate interaction between the drug and the nascent polypeptide chain, illustrating how alanine in the −1 position stabilises drug binding via a CH-π interaction (Fig. 5g)39. Since the distortion of the P-site tRNA by PoxtA involves a shift out of the PTC by 4 Å, effectively altering the nascent chain register by one amino acid, this would also result in a shift of the alanine away from chloramphenicol (Fig. 5h) and thereby perturb the interactions and reduce the affinity of the drug for the ribosome.
Discussion
Based on the structures of the PoxtA–ribosome complexes determined here, as well as the available literature on the mechanism of action of oxazolidinones and phenicols, we propose a model for how PoxtA can confer antibiotic resistance to these antibiotic classes (Fig. 6). As mentioned, linezolid and chloramphenicol are context-specific inhibitors that stall the ribosome during translation elongation with the peptidyl-tRNA in the P site and the drug bound within the A-site pocket of the PTC (Fig. 6a)27,28,29. The amino acid in the −1 position of the nascent polypeptide chain influences the strength of the arrest. Specifically, alanine elicited the strongest arrest27,28,29, apparently due to direct interaction with the ribosome-bound drug, as was recently shown for both chloramphenicol (Fig. 6a)39 and the oxazolidinones linezolid and radezolid40.

a Elongating ribosomes are stalled by PhO antibiotics with a peptidyl-tRNA in the P-site. The sidechain of the amino acid at position −1 contributes to the PhO-binding site. b Stalled ribosomes are recognised by PoxtA, which induces a shifted P-site tRNA and nascent chain, thereby disrupting the PhO-binding site. c After dissociation of the PhO drug and PoxtA, the P-tRNA and nascent chain return to the regular conformation. Accommodation of an A-tRNA occludes the drug-binding site. d After peptidyl transfer and translocation, amino acid at position −1 would change, thereby resetting the PhO-binding site.
We propose that PoxtA recognises these drug-arrested ribosomes and binds to the vacant E-site (Fig. 6b). In contrast to other ARE–ABCFs where the CCA-end is completely removed from the PTC13,14,15, the binding of PoxtA causes the CCA-end to shift by only ~4 Å. Although modest, such a shift would be sufficient to change the register of the nascent polypeptide chain with respect to the drug, such that the drug can no longer interact with the amino acid in the −1 position, thereby decreasing its affinity for the ribosome and leading to its dissociation (Fig. 6b). It is possible that the shift of the nascent chain upon distortion of the P-site tRNA by PoxtA also contributes to “brushing” the drug from the ribosome7, analogous to the mechanism proposed for leader peptides that confer resistance to macrolides41. We also cannot exclude the possibility that conformational changes in PoxtA upon ATP hydrolysis play a role in drug release, in addition to facilitating dissociation of PoxtA from the ribosome (Fig. 6c).
What prevents PhO antibiotics from rebinding following their dissociation by PoxtA? As suggested previously for other ARE–ABCFs13,15, we favour a model where, following PoxtA dissociation, the peptidyl-tRNA can reaccommodate in the P site, thus allowing accommodation of a tRNA in the A site, which, in turn, would occlude the PhO-binding site (Fig. 6c). In this respect, we note that in contrast to previous ARE–ABCF structures13,15, we observe here a subpopulation of PoxtA–70S complexes where the A-site tRNA is fully accommodated at the PTC despite the presence of PoxtA and the distorted P-site tRNA. Thus, once the drug has been released, the A-site tRNA could accommodate at the PTC and following peptide bond formation and translocation, the context of the nascent chain would be shifted by one amino acid i.e. alanine in the −1 position would now be located at the −2 position and therefore disfavour drug rebinding (Fig. 6d). PoxtA and OptrA are often found on the same mobile genetic element as drug efflux pumps22, which may also contribute to preventing drug rebinding by transporting the dissociated drug directly out of the cell.
Our insights into the mechanism of action of PoxtA also provide a possible explanation for why these ARE–ABCFs do not confer resistance to MSB or PLSA antibiotics. Firstly, the ARD of PoxtA is too short to sterically overlap with these drugs and directly displace them from the ribosome, and secondly, binding of PoxtA does not perturb the rRNA portion of the drug-binding sites and therefore could not induce their dissociation by conformational relays in the 23S rRNA. For PLSA antibiotics, which stall translation after initiation, there is no nascent chain that forms part of the drug-binding site. In the case of MSB antibiotics, which stall translation elongation but bind deeper in the nascent peptide exit channel, perhaps the “pulling” effect of PoxtA on the nascent chain is mitigated by conformation elasticity of the several amino acids between the P-tRNA and the drug-binding site. Interestingly, ARE–ABCF antibiotics, such as MsrE or LsaA that confer resistance to MSB and PLSA antibiotics, respectively, do not confer resistance to PhO antibiotics, despite direct overlap between the ARDs and the drug-binding sites. We speculate that in these cases, the peptidyl-tRNA of the PhO-stalled ribosomes are refractory to the action of these ARE–ABCFs to distort the P-site tRNA; however, further investigations will be needed to validate this.
Given the similarity in ARD length and antibiotic spectrum, we believe that the findings and model presented here for PoxtA are likely to also be applicable for OptrA. Indeed, like OptrA, our MIC analysis provides no evidence for PoxtA conferring resistance to tetracycline antibiotics, consistent with the binding site of PoxtA in the E-site being far from the tetracycline-binding site located in the decoding A-site of the small subunit. We also see no evidence for conformational differences in the 16S rRNA nucleotides that comprise the tetracycline-binding site in the absence or presence of PoxtA. For this reason, we suggest reassigning letters from the PoxtA acronym from phenicol-oxazolidinone tetracycline A to phenicol-oxazolidinone transmissible A, analogous to OptrA.
Methods
Identification of poxtA EF9F6 and characterisation of E. faecium 9-F-6 antibiotic susceptibility
The poxtA EF9F6 gene was identified in the multidrug-resistant ST872 E. faecium 9-F-6 isolated in 2012 from faeces of a patient in a Norwegian hospital who had recently also been hospitalised in India31. ST872 is a single locus variant of ST80 which is a pandemic hospital-adapted genetic lineage. Transferable linezolid resistance (optrA and cfr(D) has recently been described in blood culture E. faecium ST872 strain from Australia42. Species identification was performed by MALDI-TOF (Bruker, Billerica, USA) according to the manufacturer’s instructions and later confirmed by whole-genome sequencing31. Antimicrobial susceptibility testing was performed by broth microdilution using the EUENCF Sensititre plate (Thermo Fisher Scientific, Waltham, Massachusetts, USA), and further by gradient tests for vancomycin, teicoplanin, ampicillin, clindamycin, chloramphenicol and gentamicin (MIC Test strip, Liofilchem, Roseto Degli Abruzzi, Italy). The results (MICs) were interpreted according to EUCAST clinical breakpoints v. 10.0 2020 and MICs were within the accepted range for quality control strain E. faecalis ATCC 29212. The E. faecium 9-F-6 is resistant to linezolid (MIC = 8 mg/L), ampicillin (>256 mg/L), ciprofloxacin (>16 mg/L), high-level gentamicin (HLGR > 256 mg/L), high-level streptomycin (HLSR > 1024 mg/L) and high-level glycopeptides (vancomycin >256 mg/L and teicoplanin 128 mg/L), but susceptible to quinupristin/dalfopristin (MIC 4 mg/L) and tigecycline (MIC 0.12 mg/L) (Supplementary Table 1). Whole-genome sequences confirmed vanA and the aac(6’)-Ie-aph(2”)-Ia genes conferring high-level glycopeptide and gentamicin resistance, respectively, as well as a defunct, functionally inactive cfr pseudogene.
Strains and plasmids
All bacterial strains and plasmids used in the study are listed in Supplementary Table 3. E. faecalis TX5332 (Rifr Fusr Kanr; lsa gene disruption mutant (OG1RF lsa::pTEX4577)) is a lsa knockout strain that was received from Barbara Murray32. pCIEspec plasmid was constructed on the basis of the pCIEcam plasmid, where the chloramphenicol resistance gene was substituted with spectinomycin, due to intrinsic resistance of PoxtA to phenicol antibiotics. All cloning was performed by the PEP facility (Umeå University). Plasmids encoding OptrA-ST16 (ref. 21) and OptrA-E35048 (ref. 30) were kindly provided by Anette M. Hammerum and Alberto Antonelli, respectively. In each case, the optrA gene was amplified with ribosome-binding sequence (RBS) (TAAGAGGAGGAGATAAAC) and inserted into pCIEspec plasmid, resulting in pCIEspec:optrA-ST16 and pCIEspec:optrA-E35048, respectively. The poxtA AOUC-0915 gene25 was PCR amplified from genomic DNA of S. aureus AOUC-0915 (kindly provided by Alberto Antonelli) and inserted in pCIEspec plasmid using the BamHI and HindIII restriction sites. The RBS (TAAGAGGAGGAGATAAAC) was inserted directly in front of the ATG to improve expression levels. Genetic material encoding PoxtA-EF9F6 (OZN12776.1) was obtained from an E. faecium (9-F-6) isolated from a Norwegian patient in 2012, the genome of which has been sequenced (GCA_002263195.1)31. The poxtA-EF9F6 gene with added RBS was inserted in the pCIEspec plasmid resulting in pCIEspec:poxtA-EF9F6. These plasmids were used for MIC determinations. For use in pull-down experiments we introduced His6-TEV-FLAG3 tags at the C-terminus of either OptrA or PoxtA protein, with and without EQ mutations (E184Q and E471Q for PoxtA EQ2, E190Q and E470Q for OptrA EQ2, E470Q for OptrA EQ1) expressed from either pCIEcam (OptrA variants) or pCIEspec (PoxtA variants) plasmid.
Bacterial transformation
E. faecalis electrocompetent cells were prepared as described previously43. Briefly, an overnight culture E. faecalis TX5332 grown in the presence of 2 mg/mL of kanamycin was diluted to OD600 of 0.05 in 50 mL of Brain Heart Infusion (BHI) media and grown further to an OD600 of 0.6–0.7 at 37 °C with moderate shaking (160 rpm/10 × g). Cells were collected by centrifugation at 4000 rpm (1200 × g) at 4 °C for 10 min. Cells were resuspended in 0.5 mL of sterile lysozyme buffer (10 mM Tris-HCl pH 8; 50 mM NaCl, 10 mM EDTA, 35 µg/mL lysozyme), transferred to 0.5 mL Eppendorf tube and incubated at 37 °C for 30 min. Cells were pelleted at 10,000 rpm (8000 × g) at 4 °C for 10 min and washed three times with 1.5 mL of ice-cold electroporation buffer (0.5 M sucrose, 10% (w/v) glycerol). After the last wash, the cells were resuspended in 500 µL of ice-cold electroporation buffer and aliquoted and stored at −80 °C. For electroporation, 35 µL of electrocompetent cells were supplemented with 0.5–1 µg of plasmid DNA, transferred to ice-cold 1 mm electroporation cuvette and electroporated at 1.8 keV. Immediately after electroporation 1 mL of ice-cold BHI media was added to the cells, the content of the cuvette was transferred to 1.5-mL Eppendorf tubes and the cells were recovered at 37 °C for 2.5 h and plated to BHI plates containing appropriate antibiotics.
Antibiotic susceptibility testing
E. faecalis cells were grown in BHI media supplemented with 2 mg/mL kanamycin (to prevent lsa revertants), 0.1 mg/mL spectinomycin (to maintain the pCIEspec plasmid), 100 ng/mL of cCF10 peptide (to induce expression of proteins of interest) as well as increasing concentrations of antibiotics. The media was inoculated with 5 × 105 CFU/mL (OD600 of approximately 0.0005) of E. faecalis ΔlsaA (lsa::Kan) strain TX5332 transformed either with empty pCIEspec plasmid or with pCIEspec encoding indicated protein of interest. After 16–20 h at 37 °C without shaking, the presence or absence of bacterial growth was scored by eye.
Preparation of bacterial biomass
For cryo-EM: E. faecalis TX5332 transformed with pCIEspec/pCIEcam-based expression constructs (either empty vector or expressing wild-type/EQ2 variants of both PoxtA-HTF and OptrA-HTF as well as EQ1 variant of OptrA-HTF) were grown overnight from a single colony in BHI media supplemented with appropriate antibiotics (100 µg/mL of spectinomycin for pCIEspec-based constructs, 10 µg/mL chloramphenicol for pCIEcam-based constructs). Overnight cultures were then diluted to a starting OD600 of 0.05 in 200 mL of the same media. Cells were grown with intensive shaking at 37 °C till OD600 of 0.6 and were induced with 100 ng/mL of cCF10 peptide for 30 min prior harvesting by centrifugation at 10,000 × g for 15 min at 4 °C.
For 5Pseq: E. faecalis TX5332 Δlsa::kan was transformed with either the empty vector pCIEspec (VHp426) or a vector expressing poxtA-EF9F6 (VHp506). 50 mL cultures were inoculated from an overnight culture to 0.005 OD600, in filtered BHI media containing 100 ng/mL cCF10 peptide (to induce poxtA expression) and the cells were grown at 37 °C with shaking at 160 rpm (10 × g). Once cells reached 0.3 OD600 the cells were treated with either linezolid at 4 µg/mL (final concentration) or corresponding volume of ethanol (for as the untreated control) for 10 min, harvested via centrifugation (4000 rpm/1200 × g, 2 min), and flash-frozen in liquid nitrogen for storage at −80 °C and subsequent processing.
RNA extraction
RNA was extracted via phenol-chloroform extraction44. Briefly, 300 μL LET-phenol (LET: 25 mM Tris-HCl pH 8.0, 100 mM LiCl, 20 mM EDTA) was added to cell pellets and vortexed with an equal volume of 0.1-mm glass beads to lyse cells. In total, 250 μL of water and 250 μL of acidic phenol/chloroform was added before vortexing for a further 2 min. RNA was then extracted from the aqueous phase with an acidic phenol/chloroform step followed by a final chloroform extraction step. RNA was then ethanol precipitated in the presence of NaOAc (pH 5.2). RNA was assessed by running a 2% agarose gel and quantified by fluorescence (Qubit, Thermo Fisher Scientific).
5Pseq library preparation and data analysis
HT-5Pseq libraries were prepared as described earlier45. Briefly, 4 µg total RNA from E. faecalis was DNase treated (TURBO Dnase kit, Thermo Fisher Scientific), ligated overnight at 16 °C to r5P_RNA_ oligo (CrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrNrNrNrNrN) and subsequently purified using 1.8 volumes of RNAClean XP beads (Beckman Coulter). Ligated RNA was then reverse transcribed using random hexamer (5Pseq-RT, GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNN, 20 µM) to prime. Leftover RNA was degraded using NaOH. Ribosomal RNA was removed using previously described rRNA DNA oligo depletion mixes33, following duplex-specific nuclease (DSN, Evrogen) degradation. rRNA-depleted cDNA was amplified by PCR (17 cycles) using Illumina dual index (i5, i7) PCR primers (Supplementary Table 4) and enriched for fragments with the range of 300–500 nt using Ampure XP. Size-selected HT-5P Libraries were quantified by fluorescence (Qubit, Thermo Fisher Scientific), size estimated using an Agilent Bioanalyzer and sequenced using a NextSeq2000 Illumina sequencer (100 cycles P3 kit). BCL sequencing files were demultiplexed and converted to fastq using bcl2fastq software (bcl2fastq v2.20.0.422) with default settings (allowing for 1 barcode-mismatch, minimum trimmed read length: 35, mask short adapter reads: 22). FASTQC and MultiQC (v1.9) tools were used for quality control. In addition to standard Illumina dual index (i5, i7), UMI barcode was extracted using UMI-tools (version 1) with default options (using –bc-pattern NNNNNNNN)46, and reads were mapped to E. faecalis genome index (build GCF003319925.1_ ASM331992v) using STAR (version 2.7.2b, options –alignEndsType Extend5pOfRead1; --outFilterMatchNminOverLread 0.9; --outFilterMultimapNmax 3; --limitBAMsortRAM 100000000000; alignIntronMax 2500)47. Aligned reads were deduplicated (i.e. potential artefactual PCR duplicates are removed of the analysis) with UMI-tools (version 1, options --soft-clip-threshold 1 --edit-distance 2 --method unique). Alignment BAM files were supplied as input to the fivepseq application for 5P-end distribution analysis and figure generation48. To generate the heat maps, cumulative counts at relative distance from each amino acid were taken and normalised to library size to derive RPM values, then the log2-fold change of the RPM values between the experimental condition (linezolid treatment or PoxtA expression) and the untreated control was used to compare the pairs of samples in each case.
Preparation of clarified lysates
Cell pellets were resuspended in 1.5 mL of cell opening buffer (95 mM KCl, 5 mM NH4Cl, 20 mM HEPES pH 7.5, 1 mM DTT, 5 mM Mg(OAc)2, 0.5 mM CaCl2, 8 mM putrescine, 0.5–0.75 mM ATP, 1 mM spermidine, 1 tablet of cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche) per 10 mL of buffer). Resuspended cells were opened by FastPrep homogeniser (MP Biomedicals) with 0.1-mm Zirconium beads (Techtum) in four cycles by 20 s with a 1 min chill on ice. Cell debris was removed after centrifugation at 14,800 × g for 15 min at 4 °C. Total protein concentration in the supernatant was measured by Bradford assay (Bio-Rad), the supernatant was aliquoted and frozen in liquid nitrogen.
Affinity purification on anti-FLAG M2 affinity gel
In all, 100 µL of well-mixed anti-FLAG M2 Affinity Gel aliquots (Sigma) were loaded on columns (Micro Bio-Spin Columns, Bio-Rad) and washed twice with 1 mL of cell opening buffer by gravity flow. All incubations, washings and elutions were done at 4 °C. The total protein concentration of each individual lysate was separately adjusted to 2 mg/mL with cell opening buffer pH-adjusted with either HEPES pH 7.5 (OptrA samples) or glycine pH 9.0 (PoxtA samples). The pH of the opening buffer was selected through a series of small-scale pull-down experiments aimed at optimisation of the pull-down efficiency. 1 mL of each lysate was loaded on columns and incubated for 2 h with end-over-end mixing for binding. The columns were washed five times with 1 mL of cell opening buffer (pH 7.5 in the case of OptrA samples, pH 9.0 in the case of PoxtA samples) by gravity flow. For elution of FLAG-tagged proteins and their complexes 100 µL of 0.2 mg/mL FLAG3 peptide (Sigma) was added to samples, the solutions were incubated at 4 °C for 20 min with end-over-end mixing. Elutions were collected by centrifugation at 2000 × g for 2 min at 4 °C. 20 µL aliquots of collected samples (flow-through, washes and elutions) were mixed with 5 µL of 5× SDS loading buffer and heated up at 95 °C for 15 min. The beads remaining in the column were resuspended in 100 µL of 1× SDS loading buffer. Denatured samples were loaded on 15% SDS-PAGE. SDS-gels were stained by “Blue-Silver” Coomassie Staining49 and destained in water for at least one hour (or overnight) before imaging with LAS4000 (GE Healthcare).
Preparation of cryo-EM grids
Elutions from pull-downs were kept on ice until being applied within 2 h to glow discharged cryo-grids (Quantifoil 2/2 Cu300 coated with 2 nm continuous carbon). In total, 3.5 µL of sample was loaded on grids three times manually at room temperature conditions and a fourth time in Vitrobot (FEI) under conditions of 100% humidity at 4 °C, blotted for 5 s and vitrified by plunge-freezing in liquid ethane. Samples were imaged on a Titan Krios (FEI) operated at 300 kV at a nominal magnification of ×165,000 (0.86 Å/pixel) with a Gatan K2 Summit camera at an exposure rate of 5.85 electrons/pixel/s with a 4 s of exposure and 20 frames using the EPU software.
Single-particle reconstruction
Processing was performed in RELION 3.1 unless otherwise specified50. MotionCor2 with 5 × 5 patches and CTFFIND4 (using power spectra) were used for motion correction and initial CTF estimation51,52. Micrographs with estimated CTF fits beyond 4.5 Å and CTF figure of merits >0.04 were selected for further processing. Particles were picked with crYOLO using the general model53. After 2D classification, all ribosome-like classes were selected, particles extracted with a 3× reduced pixel size (2.46 Å), and an initial model created ab initio. After 3D refinement using the ab initio model as a reference54, 3D classification with eight classes and without angular searching was performed. The majority of particles (~80%) clustered into two classes that contained protein-like density in the E-site, and which were selected for further processing. Particles were re-extracted at the original pixel size, and serial 3D refinements with CTF refinement and Bayesian polishing55 were performed until the resolution did not improve further, resulting in the “combined 70 S volume”. A mask around the A, P and E sites was created and used for partial signal subtraction with re-extraction at a pixel size of 2.46 Å. These particles were used for 3D classification with six classes, T = 40 and the resolution of the expectation step limited to 12 Å. Four of the six resulting classes, labelled states I–IV, were chosen for refinement with the original pixel size. For the multibody refinement of the combined 70S class56, volumes corresponding to the LSU core, CP, SSU body, SSU head and E-site were isolated using the volume eraser tool in UCSF ChimeraX57, and masks created from the densities low-pass-filtered to 30 Å. Bsoft was used to estimate local resolution58.
Molecular modelling
Molecular models were created/adjusted with Coot59 and ISOLDE60, and refined with Phenix61 against unsharpened maps. A previous structure of the E. faecalis 70S ribosome in complex with the ABCF-ARE LsaA (PDB 7NHK)15 was used as the starting model for the ribosome, initially into multibody-refined maps from the combined 70S volume. PDB IDs 7K00 (ref. 62) and 6O90 (ref. 37) were also used as templates in parts of the 70S, and PDB ID 3U4M63 was used as a template for the L1 stalk region. Likely metal ions were assigned by the presence of typical coordination shapes, as well as strength of density and estimated coordination distances. However, we caution that these assignments could not always be made unambiguously. Phenix.douse was used to place water molecules. A recent structure of the S. aureus ribosome with modified nucleotides (PDB ID 6YEF)64 was compared to the density to check for RNA modifications, and where the density matched the modification with high confidence, that modification was inserted into the E. faecalis 70S model. We note that, despite the use of an inactivating resistance marker, density for the selection agent spectinomycin was observed in each map. Spectinomycin was modelled based on PDB ID 4V56 (ref. 65). For PoxtA, homology models were generated by SWISS-MODEL66 using the crystal structure of EttA (PDB ID 4FIN)12 and the previous cryo-EM structure of LsaA (PDB ID 7NHK)15. The ARD was sufficiently well-resolved that it could be built manually. The Arm insertion in NBD1 was built manually with low confidence (especially in the loop connecting the two α-helices). PSI-PRED secondary structure predictions were used to help define secondary structure boundaries67. The models for states I–IV were assembled using the model from the combined 70S as a template. For the A-tRNA in state III, tRNA-Lys from PDB ID 5E7K (ref. 68) was used as a template for E. faecalis tRNA-Lys-UUU-1-1 (gtRNAdb nomenclature69) based on density.
Sequence analysis methods
Representative ABCF sequences were aligned with Mafft L-INS-i 7.453 (ref. 70) and visualised with Jalview 2.11.1.4 (ref. 71) and Aliview 1.26 (ref. 72). Phylogenetic analysis was carried out with IQTree version 2.1.2 on the CIPRES server with 1000 rapid bootstrap replicates and automatic model detection73,74. Positions with more than 50% gaps were removed with TrimAL v1.4.rev6 before phylogenetic analysis75.
Figures
Figures were created using UCSF ChimeraX, PyMol v2.4, and assembled with Adobe Illustrator (Adobe Inc.) and Inkscape (latest development release, regularly updated). For difference vectors, the PyMol modevectors.py script (modified by Jamie Cate, University of California, Berkeley, USA) was used. For measuring SSU rotation compared to the LsaA–70S volume, the SSU head and body from PoxtA combined 70S volume were separately aligned to the LsaA–70S model, then used to draw the vectors.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Micrographs have been deposited as uncorrected frames in the Electron Microscopy Public Image Archive (EMPIAR) with the accession codes EMPIAR-10764. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) with accession codes EMD-13241 (E. faecalis combined 70S volume), EMD-13242 (PoxtA–70S state I), EMD-13243 (PoxtA–70S state II), EMD-13244 (PoxtA–70S state III with A-site tRNA) and EMD-13245 (E. faecalis 70S with P-tRNA state IV). Molecular models have been deposited in the Protein Data Bank with accession codes 7P7Q (E. faecalis combined 70S volume), 7P7R (PoxtA–70S state I) 7P7S (PoxtA–70S state II), 7P7T (PoxtA–70S state III with A-site tRNA) and 7P7U (E. faecalis 70S with P-tRNA, state IV). Source data are provided with this paper. Sequencing data are deposited at GEO with accession code GSE179348. Source data are provided with this paper.
References
Lubelski, J., Konings, W. N. & Driessen, A. J. Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiol Mol. Biol. Rev. 71, 463–476 (2007).
Orelle, C., Mathieu, K. & Jault, J. M. Multidrug ABC transporters in bacteria. Res Microbiol. 170, 381–391 (2019).
Sharkey, L. K., Edwards, T. A. & O’Neill, A. J. ABC-F proteins mediate antibiotic resistance through ribosomal protection. MBio 7, e01975 (2016).
Sharkey, L. K. R. & O’Neill, A. J. Antibiotic resistance ABC-F proteins: bringing target protection into the limelight. ACS Infect. Dis. 4, 239–246 (2018).
Murina, V., Kasari, M., Hauryliuk, V. & Atkinson, G. C. Antibiotic resistance ABCF proteins reset the peptidyl transferase centre of the ribosome to counter translational arrest. Nucleic Acids Res. 46, 3753–3763 (2018).
Murina, V. et al. ABCF ATPases involved in protein synthesis, ribosome assembly and antibiotic resistance: structural and functional diversification across the tree of life. J. Mol. Biol. 431, 3568–3590 (2019).
Ousalem, F., Singh, S., Chesneau, O., Hunt, J. F. & Boel, G. ABC-F proteins in mRNA translation and antibiotic resistance. Res. Microbiol. 170, 435–447 (2019).
Wilson, D. N., Hauryliuk, V., Atkinson, G. C. & O’Neill, A. J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 18, 637–648 (2020).
Ero, R., Yan, X. F. & Gao, Y. G. Ribosome protection proteins - “new” players in the global arms race with antibiotic-resistant pathogens. Int. J. Mol. Sci. 22, 5356 (2021).
Fostier, C. R. et al. ABC-F translation factors: from antibiotic resistance to immune response. FEBS Lett. 595, 675–706 (2021).
Chen, B. et al. EttA regulates translation by binding the ribosomal E site and restricting ribosome-tRNA dynamics. Nat. Struct. Mol. Biol. 21, 152–159 (2014).
Böel, G. et al. The ABC-F protein EttA gates ribosome entry into the translation elongation cycle. Nat. Struct. Mol. Biol. 21, 143–151 (2014).
Crowe-McAuliffe, C. et al. Structural basis for antibiotic resistance mediated by the Bacillus subtilis ABCF ATPase VmlR. Proc. Natl Acad. Sci. USA 115, 8978–8983 (2018).
Su, W. et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl Acad. Sci. USA 115, 5157–5162 (2018).
Crowe-McAuliffe, C. et al. Structural basis of ABCF-mediated resistance to pleuromutilin, lincosamide, and streptogramin A antibiotics in Gram-positive pathogens. Nat. Commun. 12, 3577 (2021).
Wang, Y. et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicrob. Chemother. 70, 2182–2190 (2015).
Cai, J. et al. Enterococcal isolates carrying the novel oxazolidinone resistance gene optrA from hospitals in Zhejiang, Guangdong, and Henan, China, 2010-2014. Clin. Microbiol Infect. 21, e1091–e1094 (2015). 1095.
Li, D. et al. Co-location of the oxazolidinone resistance genes optrA and cfr on a multiresistance plasmid from Staphylococcus sciuri. J. Antimicrob. Chemother. 71, 1474–1478 (2016).
Fan, R. et al. Distribution of optrA and cfr in florfenicol-resistant Staphylococcus sciuri of pig origin. Vet. Microbiol. 210, 43–48 (2017).
Lazaris, A. et al. Novel multiresistance cfr plasmids in linezolid-resistant methicillin-resistant Staphylococcus epidermidis and vancomycin-resistant Enterococcus faecium (VRE) from a hospital outbreak: co-location of cfr and optrA in VRE. J. Antimicrob. Chemother. 72, 3252–3257 (2017).
Vorobieva, V. et al. Detection of the optrA gene in a clinical ST16 Enterococcus faecalis isolate in Denmark. J. Glob. Antimicrob. Resist. 10, 12–13 (2017).
Schwarz, S. et al. Mobile oxazolidinone resistance genes in Gram-positive and Gram-negative bacteria. Clin. Microbiol. Rev. 34, e0018820 (2021).
Liu, D. et al. Detection of the enterococcal oxazolidinone/phenicol resistance gene optrA in Campylobacter coli. Vet. Microbiol. 246, 108731 (2020).
Tang, Y. et al. Novel insertion sequence ISChh1-like mediating acquisition of optrA gene in foodborne pathogen Campylobacter coli of swine origin. Vet. Microbiol. 252, 108934 (2020).
Antonelli, A. et al. Characterization of poxtA, a novel phenicol-oxazolidinone-tetracycline resistance gene from an MRSA of clinical origin. J. Antimicrob. Chemother. 73, 1763–1769 (2018).
Wilson, D. N. Ribosome-targeting antibiotics and bacterial resistance mechanisms. Nat. Rev. Microbiol. 12, 35–48 (2014).
Marks, J. et al. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl Acad. Sci. USA 113, 12150–12155 (2016).
Vazquez-Laslop, N. & Mankin, A. S. Context-specific action of ribosomal antibiotics. Annu. Rev. Microbiol. 72, 185–207 (2018).
Choi, J. et al. Dynamics of the context-specific translation arrest by chloramphenicol and linezolid. Nat. Chem. Biol. 16, 310–317 (2020).
Morroni, G. et al. Characterization of a multiresistance plasmid carrying the optrA and cfr resistance genes from an Enterococcus faecium clinical isolate. Front. Microbiol. 9, 2189 (2018).
Sivertsen, A. et al. The enterococcus cassette chromosome, a genomic variation enabler in enterococci. mSphere 3, e00402-18 (2018).
Singh, K. V., Weinstock, G. M. & Murray, B. E. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin-dalfopristin. Antimicrob. Agents Chemother. 46, 1845–1850 (2002).
Huch, S. et al. RNA degradation analysis reveals ribosome dynamics in complex microbiome samples. Preprint at https://www.biorxiv.org/content/10.1101/2021.04.08.439066v1 (2021).
Pelechano, V., Wei, W. & Steinmetz, L. M. Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161, 1400–1412 (2015).
Kasari, V. et al. A role for the Saccharomyces cerevisiae ABCF protein New1 in translation termination/recycling. Nucleic Acids Res. 47, 8807–8820 (2019).
Zhong, X. et al. A novel inhibitor of the new antibiotic resistance protein OptrA. Chem. Biol. Drug Des. 92, 1458–1467 (2018).
Murphy, E. L. et al. Cryo-electron microscopy structure of the 70S ribosome from Enterococcus faecalis. Sci. Rep. 10, 16301 (2020).
Graf, M. et al. Visualization of translation termination intermediates trapped by the Apidaecin 137 peptide during RF3-mediated recycling of RF1. Nat. Commun. 9, 3053 (2018).
Syroegin, E. A. et al. Structural basis for the context-specific action of a classic peptidyl transferase inhibitor. Nat. Struct. Mol. Biol. 29, 152–161 (2022).
Tsai, K. et al. Structural basis for context-specific inhibition of translation by oxazolidinone antibiotics. Nat. Struct. Mol. Biol. 29, 162–171 (2022).
Tenson, T. & Mankin, A. Short peptides conferring resistance to macrolide antibiotics. Peptides 22, 1661–1668 (2001).
Pang, S. et al. Linezolid-resistant ST872 Enteroccocus faecium harbouring optrA and cfr(D) oxazolidinone resistance genes. Int J. Antimicrob. Agents 55, 105831 (2020).
Bhardwaj, P., Ziegler, E. & Palmer, K. L. Chlorhexidine induces vanA-type vancomycin resistance genes in enterococci. Antimicrobial Agents Chemother. 60, 2209–2221 (2016).
Caponigro, G., Muhlrad, D. & Parker, R. A small segment of the MAT alpha 1 transcript promotes mRNA decay in Saccharomyces cerevisiae: a stimulatory role for rare codons. Mol. Cell Biol. 13, 5141–5148 (1993).
Zhang, Y. & Pelechano, V. High-throughput 5′P sequencing enables the study of degradation-associated ribosome stalls. Cell Rep. Methods 1, 100001 (2021).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Nersisyan, L., Ropat, M. & Pelechano, V. Improved computational analysis of ribosome dynamics from 5′P degradome data using fivepseq. NAR Genom. Bioinform 2, lqaa099 (2020).
Candiano, G. et al. Blue silver: a very sensitive colloidal Coomassie G‐250 staining for proteome analysis. Electrophoresis 25, 1327–1333 (2004).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Wagner, T. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218 (2019).
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Nakane, T., Kimanius, D., Lindahl, E. & Scheres, S. H. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Heymann, J. B. Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171 (2018).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol. 74, 519–530 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Watson, Z. L. et al. Structure of the bacterial ribosome at 2 Å resolution. eLife 9, e60482 (2020).
Tishchenko, S. et al. High-resolution crystal structure of the isolated ribosomal L1 stalk. Acta Crystallogr. D. Biol. Crystallogr. 68, 1051–1057 (2012).
Golubev, A. et al. Cryo-EM structure of the ribosome functional complex of the human pathogen Staphylococcus aureus at 3.2 Å resolution. FEBS Lett. 594, 3551–3567 (2020).
Borovinskaya, M. A., Shoji, S., Holton, J. M., Fredrick, K. & Cate, J. H. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem. Biol. 2, 545–552 (2007).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Buchan, D. W. A. & Jones, D. T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 47, W402–W407 (2019).
Rozov, A. et al. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 7, 10457 (2016).
Chan, P. P. & Lowe, T. M. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. in Gateway Computing Environments Workshop (GCE, https://doi.org/10.1109/GCE.2010.5676129. 2010).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Weaver, K. E. et al. Examination of Enterococcus faecalis toxin-antitoxin system toxin fst function utilizing a pheromone-inducible expression vector with tight repression and broad dynamic range. J. Bacteriol. 199, e00065-17 (2017).
Svetlov, M. S. et al. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 25, 600–606 (2019).
Matzov, D. et al. Structural insights of lincosamides targeting the ribosome of Staphylococcus aureus. Nucleic Acids Res. 45, 10284–10292 (2017).
Acknowledgements
We are grateful to Barbara E. Murray for sharing E. faecalis ΔlsaA (lsa::Kan) strain TX5332 (ref. 32), Jose A. Lemos for sharing pCIE plasmid76, Alberto Antonelli for sharing genetic material containing poxtA AOUC-0915 and optrA-E35048 and Anette M. Hammerum for sharing the plasmid containing optrA ST16 (ref. 21) and to Yury S. Polikanov for providing structural insights into the mechanism of chloramphenicol action. We thank Michael Hall for help with cryo-EM data collection. The electron microscopy data was collected at the Umeå Core Facility for Electron Microscopy, a node of the Cryo-EM Swedish National Facility, funded by the Knut and Alice Wallenberg, Family Erling Persson and Kempe Foundations, SciLifeLab, Stockholm University and Umeå University. Genomic analysis was performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (grant WI3285/8-1 to D.N.W.), Swedish Research Council (Vetenskapsrådet) grants (2017-03783 and 2021-01146 to V.H., 2019-01085 to G.C.A.), Ragnar Söderbergs Stiftelse (to V.H.), postdoctoral grant from the Umeå Centre for Microbial Research, UCMR (to H.T.), the European Union from the European Regional Development Fund through the Centre of Excellence in Molecular Cell Engineering (2014-2020.4.01.15-0013 to V.H.); and the Estonian Research Council (PRG335 to V.H.). D.N.W. and V.H. groups are also supported by the Deutsche Zentrum für Luft- und Raumfahrt (DLR01Kl1820 to D.N.W.) and the Swedish Research Council (2018-00956 to V.H.) within the RIBOTARGET consortium under the framework of JPIAMR. GCA and VH were also supported by a project grant from the Knut and Alice Wallenberg Foundation (2020-0037 to G.C.A.). V.P. group is supported by Swedish Foundation’s Starting Grant (Ragnar Söderberg Foundation), the Swedish Research Council [VR 2016-01842, 2020-01480 and 2021-06112], a Wallenberg Academy Fellowship [KAW2016.0123], Vinnova [2020-03620] and Karolinska Institutet (SciLifeLab Fellowship, SFO and KI funds). LN was supported by the EU H2020-MSCA-IF-2018 program under grant agreement [845495 – TERMINATOR].
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
C.C.M. processed the microscopy data, generated and refined the molecular models and made the structure figures. V.M., H.T. and M.K. cloned the ARE constructs, performed genetic manipulations of E. faecalis, performed polysome fractionations and immunoblotting as well as performed MICs. V.M., K.J.T. and M.K. performed immunoprecipitations. V.M. prepared cryo-EM grids and collected cryo-EM datasets. K.J.T. and H.T. prepared the RNA samples for HT-5Pseq. S.H., L.N. and V.P. performed and analysed HT-5Pseq data. A.S. and K.H. provided research materials and performed MICs. G.C.A. performed sequence conservation analyses. C.C.M. and D.N.W. wrote the manuscript with input from all authors. D.N.W. and V.H. conceived and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
S.H., L.N. and V.P. are co-founders of 3N Bio AB. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crowe-McAuliffe, C., Murina, V., Turnbull, K.J. et al. Structural basis for PoxtA-mediated resistance to phenicol and oxazolidinone antibiotics. Nat Commun 13, 1860 (2022). https://doi.org/10.1038/s41467-022-29274-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29274-9
This article is cited by
-
Global regulation via modulation of ribosome pausing by the ABC-F protein EttA
Nature Communications (2024)
-
A Comprehensive Review of Molecular Mechanisms Leading to the Emergence of Multidrug Resistance in Bacteria
Indian Journal of Microbiology (2024)
-
Structural conservation of antibiotic interaction with ribosomes
Nature Structural & Molecular Biology (2023)
-
Regulation of the macrolide resistance ABC-F translation factor MsrD
Nature Communications (2023)
-
Gene rppA co-regulated by LRR, SigA, and CcpA mediates antibiotic resistance in Bacillus thuringiensis
Applied Microbiology and Biotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
