
Abstract
Glycosyl phosphosaccharides represent a large and important family of complex glycans. Due to the distinct nature of these complex molecules, efficient approaches to access glycosyl phosphosaccharides are still in great demand. Here, we disclose a highly efficient and stereoselective approach to the synthesis of biologically important and complex α-glycosyl phosphosaccharides, employing direct gold(I)-catalyzed glycosylation of the weakly nucleophilic phosphoric acid acceptors. In this work, the broad substrate scope is demonstrated with more than 45 examples, including glucose, xylose, glucuronate, galactose, mannose, rhamnose, fucose, 2-N3-2-deoxymannose, 2-N3-2-deoxyglucose, 2-N3-2-deoxygalactose and unnatural carbohydrates. Here, we show the glycosyl phosphotriester prepared herein was successfully applied to the one-pot synthesis of a phosphosaccharide from Leishmania donovani, and an effective preparation of a trisaccharide diphosphate of phosphosaccharide fragments from Hansenula capsulate via iterative elongation strategy is realized.
Similar content being viewed by others
Introduction
Glycosyl phosphosaccharides (GPSs) represent a large and important family of complex glycans, which are ubiquitously distributed in bacteria, yeasts, protozoan parasites and animals, and exhibit numerous bio-functions including bacterial infections, cell adhesive, immunoresponse, and antimicrobial (Fig. 1b)1,2,3,4,5. The GPSs consist of anomeric glycosyl phosphates in which the anomeric position of one constituent glycan was linked to another one mainly by α-type phosphodiester linkage (Fig. 1a). In the process of carbohydrate metabolism, the constituent glycosyl phosphates (GPs) are significant intermediates6. Synthetically, protected GPs have been utilized as effective glycosyl donor reagents7, and under the catalysis of bis-thiourea, the armed GPSs proceed an SN-2 glycosylation pathway8. Despite the significance of GPSs, efficient approaches to access GPSs are rather limited: isolation from cell culture hardly affords homogeneous sample and chemical synthesis remains challenging due to the character of complex structure. Accordingly, efficient methods of constructing homogeneous GPSs and GPs are still in great demand.

a Nomenclatures for glycosyl phosphate and phosphosaccharide. b Representative bioactive phosphosaccharides.
In addition to intrinsically labile character, the anomeric stereocontrol of forming α-GPSs, most of which assume 1,2-cis configuration, is regarded as a challenging task9,10,11,12,13,14,15,16,17. The synthetic method of H-phosphonate chemistry has found extensive applications in the synthesis of α-GPSs, while two-step transformations of nucleophilic displacement and oxidation are inevitable (Fig. 2A)2,18,19. Nevertheless, the resulted phosphate anions render product incompatible with follow-up or late-stage chemical modifications to increase molecular complexity and diversity20. The alternative approach employing phosphoramidite displays great success in installation of phosphoester, yet is rarely applied to synthesis of anomeric GPS probably due to issues of diastereo-selectivity and undesired oxidative cleavage reaction (Fig. 2B)2,21,22,23.

In red are the α-configuration of the formed glycosidic bond and the benzyl (Bn) group. In yellow is the gold(I) catalysis. In pink is the indication of the role of gold(I). In blue are the features of the present approach.
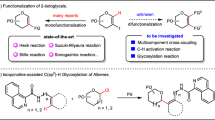
One-step and direct glycosylation of phosphate acceptors with glycosyl donors represents a convergent and concise method for the synthesis of α-GPSs, which does not require oxidation transformation (Fig. 2C). This glycosylation strategy to access α-GPSs was previous realized by utilizing glycosyl trichloroacetimidate as donor and the weak nucleophile of phosphoric acid as acceptor under the action of strong acid24, and later, by a panel of donors with different leaving groups (e.g., SPh, 3-methoxypyridin-2-yl, pentenyloxy)25,26,27,28,29,30,31. The efficiency of these glycosylation reactions with phosphates as acceptors remains unmet: (1) the yield was deteriorated when strong acid was used to realize α-stereoselectivity24, (2) stoichiometric base was applied to preserve the entity of GPs, but leading to poor 1,2-cis stereoselectivity28, (3) only a handful of complex disaccharide GPSs were accessed by using the direct glycosylation strategy with phosphate anion as acceptor25.
Catalytic glycosylation methods have emerged as an appealing approach to the synthesis of carbohydrates, which feature less promoter and waste, and high efficiency32. Among those, alkynylphilic gold(I) catalysis has been extensively applied in the syntheses of numerous complex glycans and glycoconjugates33, along with other natural products34, by exploiting the compatibility with oxygen-containing functionalities35. Especially, glycosyl donor with ortho-alkynylbenzoate as leaving group first introduced by Yu and coworkers can glycosylate a variety of acceptors36,37. Nevertheless, the glycosylation of exceedingly poor nucleophile of phosphoric acid remains elusive, which entails mild conditions free from competitive nucleophilic species.
In this work, we disclose a stereoselective and general approach to the synthesis of α-GPs and α-GPSs via a gold(I)-catalyzed glycosylation method with glycosyl ortho-alkynylbenzoate as donor and weakly nucleophilic phosphoric acid as acceptor. While the alkynylphilicity of gold(I) catalysis has been widely investigated and applied, the Lewis acid property when oxygen-containing functionalities is activated by gold(I) remains much less explored. Herein, both the alkynylphilicity and weak acidity of gold(I) catalyst are capitalized on to initiate the glycosylation reaction and promote epimerization to α-anomer, respectively (Fig. 2D). Moreover, the anomeric effect, which is conferred by the weak nucleophile of phosphoric acid in the glycosylation reaction, is also exploited to direct the α-selectivity under the present weakly acidic condition38,39. The present gold(I)-catalyzed glycosylation method facilitates the efficient synthesis of more than 45 complex glycosyl phosphates, one-pot synthesis of GPS from Leishmania donovani, and iterative elongation of a trisaccharide diphosphate of GPS fragments from Hansenula capsulate. Mechanistic studies are investigated to indicate the dual role of Ph3PAuNTf2 in the glycosylation-epimerization process, in which Ph3PAuNTf2 not only triggers the glycosylation reaction, but also subsequently promotes the epimerization of the α/β products to enrich the α one.
Results
Reaction optimization
To test the validity of this proposal, tetrabenzyl glucoside 1a was examined with a structurally simple acceptor of phosphoric acid dibenzyl ester 2a (Table 1). Initial experiment gave promising results with a good yield of α/β mixture (88%) and α-selectivity (α/β = 2.8/1, entry 1). Thus, detailed optimizations were subsequently conducted by tuning reaction temperature, solvent and additive. As depicted in Table 1, lowering temperature was not effective (83%, α/β = 2/1, entry 2); replacing the anion of Ph3PAuNTf2 with −OTf diminished α-selectivity (entry 3). The ether solvent and additive of Ph3P = O, which direct α-selectivity when alcohols are used as acceptors, did not lead to satisfactory results in the present reaction (entries 4-6)11. Gratifyingly, the diastereoselective ratio was raised to 10/1 by an added HOTf (0.1 equiv.), which is supposed to thermodynamically equilibrated β-anomer to the α-one, but compromise the overall yield (62%, entry 7). The strong and heterogenous acidic H+ resin did not result in an improvement in diastereoselectivity as that of HOTf (entry 8). Then, the homogeneous and weak Lewis acid of gold(I) catalysis under an elevated temperature was anticipated to trigger the epimerization to enrich α-anomer and maintain a high yield. Indeed, after complete glycosylation of 2a at 0 °C for 0.5 h, keeping the mixture at 60 °C for 2 h in non-coordinating ClCH2CH2Cl (DCE) for anomerization produced the α-anomer in good selectivity (α/β = 9/1) without deteriorating yield (87%, entry 9). Further elevation of temperature (75 °C and 95 °C) resulted in a drop of yield or decomposition of product (entry 10, 11). Fortunately, increasing the equivalence of donor 1a to 1.5 relative to acceptor 2a (1.0 equiv.) reached an exceptional ratio of 16/1 in 62% yield which was calculated based on donor (entry 12). Interestingly, decreasing the equivalence of donor to 1.0 relative to acceptor (1.0 equiv.) significantly lowered the yield and α-selectivity (entry 13). While prolonging the reaction time for epimerization from 2 h to 3 h gave higher α-selectivity of 20/1, the yield dropped to 57% (entry 14). Notably, the yields of α/β mixtures were herein reported owing to that the α/β mixture was not separable by utilizing flash silica gel column chromatography, and the yields were calculated based on the donor which was used in excess (1.5 equiv.).
Reaction scope of donors
Next, we wondered whether this gold (I)-catalyzed glycosylation strategy was amenable to various glycosyl donors outfitted with different protecting groups or configurations (Fig. 3). First, xylosyl donor 1b, of which the BnOCH2 moiety is omitted compared to 1a, delivered the expected xylosyl phosphate 3b in a good diastereoselectivity (11/1) and 55% yield of α/β mixture calculated based on the donor (1.5 equiv.) by utilizing the aforementioned protocol. The electron-withdrawing groups such as AcOCH2 of 6-O-acetyl-glucosyl donor 1c and COOMe of glucuronic acid methyl ester donor 1d may greatly reduce the reactivity of donor, thereby causing difficulty in epimerization. Luckily, the 6-O-Ac product (3c) could be reached in a diastereoselective ratio of 12/1 merely at room temperature, of which α-selectivity is presumably dominated by anomeric effect38,39. For the other one equipped with COOMe, the product (3d) can be epimerized to enrich α-anomer (9.7/1) under a higher temperature of 100 °C with a yield of 58%.

aTemperature: 0 °C, 30 min then 60 °C, 2 h. bTemperature: 0 °C, 30 min, then 20 °C, 2 h. cTemperature: 0 °C, 30 min, then 100 °C, 2 h. dgalactosyl trichloroacetimidate was used as donor in the presence of TMSOTf (0.1 equiv.) at 0 °C in 30 min. eOn 1.2 g scale with 0.5 mol% Ph3PAuNTf2. f0.2 equiv. of i-Pr2NEt was used as additive. gYields of α/β mixtures (α/β mixtures were not separable, except 3e and 3i), and ratios were determined by crude 31P NMR. Bn: benzyl. Bz: benzoyl. Ph: phenyl. Ac: acetyl. Tf: trifluoromethanesulfonyl. In red are the formed glycosidic bonds. In blue are the acceptors of phosphoric acids and the phosphate moieties in the products.
Consequently, this strategy was extended to a variety of donors including Gal, Man, ManN3, Rha, Fuc, GlcN3, GalN3 and unnatural carbohydrates (Fig. 3). For the production of those with highly anomeric effect and dominated by steric hindrance, such as galactosyl phosphate (3e and 3e’), mannosyl phosphate (3 f and 3 f’), disarmed 2-N3-2-dexoy-mannosyl phosphate (3 g), rhamnosyl phosphate (3 h) and fucosyl phosphate (3i), an operationally simple procedure at room temperature was effective to access desired products in highly diastereoselective manners. However, switching to phenyl tetrabenzyl-1-thio-mannoside which was preactivated with p-TolSCl and AgOTf, followed by addition of 2a, gave only byproduct 3p derived from intramolecular cyclization between 2-O Bn and anomeric position (please see SI)40. Among those, 3 g represents constituent unit derived from capsule polysaccharide of Neisseria meningitidis A41. The straightforward syntheses of extremely unstable diphenyl phosphate 3e’8 and 3 f’ demonstrate the utility of this strategy for directly glycosylating weakly nucleophilic phosphoric acid. In comparison, only 7% yield was obtained when galactosyl trichloroacetimidate was used as donor in the presence of TMSOTf (0.1 equiv.) at 0 °C. Moreover, 3 f can be obtained on a 1.2 g scale with low catalytic loading of Ph3PAuNTf2 (0.5 mol%) albeit in slightly decreased diastereoselectivity (17/1) and yield (58%)42. The deprotected form of 3 f might serve as replacement therapy for the disease of congenital disorder of glycosylation type Ia which is under clinical trial43. Interestingly, 1,2-cis α-fucosyl phosphate 3i was obtained even with neighboring-participating Bz situated at 2-O, demonstrating the strong anomeric effect in the case of phosphoric acid as acceptor. However, the diastereoselectivity can be reversed by adding extra base of iPr2NEt (0.2 equiv.) to form phosphate anion as acceptor.
Because of the distinct nature of N3 substituent in comparison with OBn, formation of 1,2-cis α-D-glycosamine glycosidic bond remains elusive in the case of alcohols as acceptors14,15,16,17. Although the azido substituted glycosyl phosphates are resistant to epimerization at high temperature, donors of tribenzyl GlcN3 and GalN3 underwent smoothly coupling reactions with good diastereoselective ratios of 8.6/1 (3j) and 7.3/1 (3k) which might find utility in the syntheses of Lipid A or other phosphosaccharides2,44
Derivatization and mimicking of natural glycosyl phosphates emerge as attractive tools to elucidate molecular mechanism of glycosyltransferases and discover novel therapeutic reagents45,46. Herein, a panel of fluorine-substituted α-GPs (3l-3o) were readily assembled via gold(I)-catalyzed glycosylation approach. Notably, the highly α-selective outcomes are in stark contrast to the reported results of fluorine-directed glycosylation with alcohol acceptors47.
Reaction scope of acceptors
After determining the generality of various glycosyl donors which glycosylated with phosphoric acid 2a, we explored the possibility of extension to more complex phosphate nucleophiles. Thus, a set of structurally diverse acceptors of phosphoric acid glycosyl esters were readily prepared through a straightforward route of phosphorylation of alcohol and subsequent debenzylation (see SI), including 6-O-benzyloxyphosphoryl glucoside 2b and 2c, sterically hindered 4-O-benzyloxyphosphoryl glucoside 2d and galactoside 2e, and 3-O-benzyloxyphosphoryl glucoside 2 f outfitted with labile groups of TBS and benzylidene, and serinyl phosphate 2 g (Fig. 4). The carbohydrates widely distributed in natural GPSs were selected as glycosyl donors (glucose (Glc, 1a), galactose (Gal, 1e), mannose (Man, 1 f), rhamnose (Rha, 1 h), 2-N3-2-deoxyglucose (GlcN3, 1j), 2-N3-2-deoxygalactose (GalN3, 1k)), which led to twenty-seven bis-glycosyl benzylphosphotriesters. For convenience of characterization, the phosphorus chirality was eliminated by hydrogenolysis of benzyl phosphates (4a-4za), which simultaneously resulted in reduction of N3 (4t-4z, 4za, 4e, 4j, 4o). In detail, by using the protocol of glycosylation and subsequent anomerization, condensation of Glc donor (1a) and all the five acceptors (2b-2f) delivered the corresponding GPSs (4a-4e) in highly diastereoselective manners. The donors of Gal (1e), Man (1 f) and Rha (1 h) with highly anomeric effect produced GPSs (4f-4s) in invariably high α-selectivities. Although azido substituted substrates of GlcN3 and GalN3 are resistant to epimerization and display weaker α-configured bias, good results were attained with consistent stereoselectivities (4t-4v, 4x-4z, 4za), except 4w which was formed in low stereoselectivity (3/1). Furthermore, one of phosphoglycoserines (4zb), which are found in parasites, was concisely assembled via glycosylation of serinyl phosphate 2 g in a stereospecific manner, albeit in a low yield of 40% because of limited solubility of 2 g in DCE48 (Fig. 4).

aCondition: 0 °C, 30 min then 60 °C, 2 h. bCondition: 0 °C, 30 min, then 20 °C, 2 h. cCondition: 0 °C, 30 min then 60 °C, 3 h. dYields of α/β mixtures (α/β mixtures not separable), and ratios were determined by 31P NMR. TBS: tert-butyldimethylsilyl. Bn: benzyl. Bz: benzoyl. Tf: trifluoromethanesulfonyl. Fmoc: 9-fluorenylmethyloxycarbonyl. In red are the formed glycosidic bonds. In blue are the acceptors of phosphoric acid and the phosphate moieties in the products.
Reaction scope of large donors
The successful glycosylation-epimerization process with mono glucosyl donor encouraged us to evaluate large glucosyl donors (one disaccharide donor and one trisaccharide donor) with a non-participating group at 2-O position (Fig. 5). As shown in Fig. 5, while the glycosyl phosphoesters (4zc, 4zd) with less hindered environment around the motif of glucosyl phosphate possessed good selectivity, higher steric hindrance had a detrimental effect on the α/β selectivity (4ze (2.8/1), 4zf (4.6/1), 4zg (1.1/1)).

aYields of α/β mixtures (α/β mixtures were not separable), and α/β ratios were determined by 31P NMR. Bn: benzyl. Bz: benzoyl. In red are the formed glycosidic bonds. In blue are the acceptors of phosphoric acids and the phosphate moieties in the products.
Global deprotection reactions
Although the prepared glycosyl phosphotriesters are rather labile toward acid, the corresponding anions readily prepared through hydrogenolysis exhibit remarkable inertness toward acid. Thus, the deprotection sequence for these glycosyl phosphotriesters could be carried out through a two-step protocol: removal of benzyl group of benzyl phosphate and global deprotection of other protecting groups (Fig. 6). The phosphotriester 3a and 3e were hydrogenated in the presence of Et3N to produce quantitatively intermediate compounds of glycosyl phosphomonoester, which were then subjected to global deprotection of all other benzyl groups even in the presence of acidic HCOOH to afford cleanly the product 5 and 6. Similarly, the glycosyl phosphodiester 4c were directly deprotected under the acidic hydrogenation condition to furnish 7 in excellent yield (90%). For the benzoyl protected 4d, one additional step of deacetylation by using hydrazine after hydrogenation reaction was required to release the bis-glycosyl phosphoester 8 in high yield (89%).

aYields of isolated products. Bn: benzyl. Bz: benzoyl.
One-pot glycosylation
While one-pot glycosylation protocol emerges as versatile strategy to synthesize complex oligo/polysaccharides of which the units are tethered by acetal linkages49,50,51, this strategy is not applicable to the assembly of conventionally synthesized bis-glycosyl phosphodiester which incorporate reactive functionality of phosphate anion20. Gratifyingly, bis-glycosyl benzyl phosphotriesters readily prepared in our system could serve as attractive substrates for one-pot glycosylation, and described in Fig. 7 is an example, in which linker-tethered 9 was assembled in one pot via gold(I)-catalyzed glycosylation reaction and a follow-up orthogonal coupling reaction promoted by NIS and TMSOTf. The chirality of phosphorus atom can be eliminated by converting OBn to O−, generating a single stereomer (10, 97%) derived from phosphosaccharide of Leishmania donovani1.

NIS: N-iodosuccinimide. Tol: tolyl. Lev: levulinoyl. Bn: benzyl. Bz: benzoyl. TMSOTf: trimethylsilyl trifluoromethanesulfonate. In red are the formed glycosidic bonds. In blue is the phosphate moiety in product.
Iterative elongation
Finally, the utility of this gold(I)-catalyzed one-step approach to synthesis of GPSs was further illustrated by iterative elongation of phosphomannosyl fragments from Hansenula capsulate (Fig. 8)52. First, condensation of donor 1q and acceptor 2i furnished the desired phosphotriester 4zh. Because 4zh was resistant to preactivation by using p-TolSCl/AgOTf or BSP/Tf2O which led to only 30% yield and armed donor with benzyl groups was prone to give cyclized byproduct (e.g., 3p)40,53, SPh was converted to ortho-alkynylbenzoate as leaving group via two steps. Next, condensation of 11 and 2i generated a trisaccharide 12 consisting of two phosphotriester funcitonalities in 47% yield based on donor 11 (1.5 equiv.), which was subsequently converted to trisaccharide donor 13 in a procedure similar to that for 11. As a late-stage chemical modification on this trisaccharide, a third glycosylation reaction between donor 13 and 3-azidopropanol was performed to install a linker with the two present phosphotriesters intact. Finally, the resulting trisaccharide was globally deprotected under mild conditions to afford an amino-linker tethered trisaccharide diphosphate 14.

a Ph3PAuNTf2 (10 mol%), 5 Å MS, DCE. b 1) NBS, acetone, H2O; 2) HX, EDCl, DMAP, CH2Cl2. c 2i, Ph3PAuNTf2, DCE. d 1) NBS, acetone, H2O; 2) HX, EDCl, DMAP, CH2Cl2. e 1) 3-azidopropanol, Ph3PAuNTf2 (10 mol%); 2) H2, Pd/C, HCOOH, HCOONH4, THF, t-BuOH; 3) NH2NH2•H2O, MeOH. NBS: N-bromosuccinimide. EDCl: 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. DMAP: 4-(dimethylamino)pyridine. Bz: benzoyl. In red are the formed glycosidic bonds. In blue are the phosphate moieties in the products.
Mechanistic studies
To probe the role of Ph3PAuNTf2 in the epimerization reaction, the epimerization process to enrich α-anomer was monitored at elevated temperature (60 °C) by HPLC. As depicted in Fig. 9, the ratio of α/β increased along with the time under the catalysis of Ph3PAuNTf2 to give the α/β ratio of 12/1 in approximately 3 h (Fig. 9a). Further prolonging the epimerization time can improve the α/β ratio to 18/1 (entry 2, Fig. 9b). Interestingly, in the presence of sterically hindered base (2,6-di-tert-butyl-4-methylpyridine) which is supposed to not coordinate the gold(I) catalyst, the epimerization process was substantially impeded to give a ratio of 5.8/1 (entry 3, Fig. 9b). Other Lewis acid promoted epimerization afforded the same α/β selectivity as that when Ph3PAuNTf2 was utilized (Fig. 9b and Supplementary Fig. 1). On the basis of these results, we speculated the acid was the real specie to trigger the epimerization process, and the gold(I) catalyst probably acted as an acid reservoir which can slowly release acid through possible disproportionation reaction to generate strong acidic Au3+ at elevated temperature (Supplementary Fig. 2)54. The strong Lewis acid Au3+ itself, or the H+ which could be generated by reaction of Au3+ with trace of H2O in the reaction system could promote the epimerization process.

a The plot of α/β ratio versus reaction time. b The epimerization reaction with various acids. c The proposed mechanism. In red are the formed glycosidic bonds. In blue are the phosphoric acid and the phosphate moiety in product. In pink are the reaction temperatures. In yellow is the gold(I) catalysis. In orange is the curve of α/β ratio versus reaction time.
In conclusion, we have developed a highly efficient and stereoselective approach to the synthesis of GPSs by employing gold(I)-catalyzed glycosylation of phosphoric acid acceptors. The efficiency of this protocol was demonstrated by its universal application in preparing more than 45 complex GPSs, one-pot synthesis of linker-tethered GPS from Leishmania donovani, and an effective preparation of trisaccharide diphosphate from Hansenula capsulate via iterative elongation. Because of its exceptionally broad substrate scope, high α-selectivity and inertness of phosphotriester toward chemical manipulations in comparison to phosphodiester, this strategy will offer new opportunities to create complex phosphosaccharides and install diverse phosphosaccharides on bioactive molecules.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information file, including experimental details, characterization data, and NMR spectra of new compounds.
References
Oka, N., Sato, K. & Wada, T. Recent progress in the synthesis of glycosyl phosphate derivatives. Trends Glycosci. Glyc. 24, 152–168 (2012).
Nikolaev, A. V., Botvinko, I. V. & Ross, A. J. Natural phosphoglycans containing glycosyl phosphate units: structural diversity and chemical synthesis. Carbohydr. Res. 342, 297–344 (2007).
Simpson, B. W. & Trent, M. S. Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 17, 403–416 (2019).
Ostash, B. & Walker, S. Moenomycin family antibiotics: chemical synthesis, biosynthesis, and biological activity. Nat. Prod. Rep. 27, 1594–1617 (2010).
Jennings, J. H., Rosell, K. G. & Carlo, D. J. Structual determination of the capsular polysaccharide of Streptococcus pneumoniae type 19 (19F). Can. J. Chem. 58, 1069–1074 (1980).
Thibodeaux, C. J., Melançon, C. E. III & Liu, H.-W. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. 47, 9814–9859 (2008).
Plante, O. J., Palmacci, E. R., Andrade, R. B. & Seeberger, P. H. Oligosaccharide synthesis with glycosyl phosphate and dithiophosphate triesters as glycosylating agents. J. Am. Chem. Soc. 123, 9545–9554 (2001).
Levi, S. M., Li, Q., Rötheli, A. R. & Jacobsen, E. N. Catalytic activation of glycosyl phosphates for stereoselective coupling reactions. Proc. Natl. Acad. Sci. USA 116, 35–39 (2019).
Lu, S.-R., Lai, Y.-H., Chen, J.-H., Liu, C.-Y. & Mong, K.-K. T. Dimethylformamide: an unusual glycosylation modulator. Angew. Chem. Int. Ed. 50, 7315–7320 (2011).
Fang, T., Mo, K.-F. & Boons, G.-J. Stereoselective assembly of complex oligosaccharides using anomeric sulfonium ions as glycosyl donors. J. Am. Chem. Soc. 134, 7545–7552 (2012).
Wang, L., Overkleeft, H. S., van der Marel, G. A. & Codée, J. D. C. Reagent controlled stereoselective synthesis of α-Glucans. J. Am. Chem. Soc. 140, 4632–4638 (2018).
Yasomanee, J. P. & Demchenko, A. V. Effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc. 134, 20097–20102 (2012).
Le Mai Hoang, K. & Liu, X.-W. The intriguing dual-directing effect of 2-cyanobenzyl ether for a highly stereospecific glycosylation reaction. Nat. Commun. 5, 5051 (2014).
Benakli, K., Zha, C. & Kerns, R. J. Oxazolidinone protected 2-amino-2-deoxy-D-glucose derivatives as versatile intermediates in stereoselective oligosaccharide synthesis and the formation of α-linked glycosides. J. Am. Chem. Soc. 123, 9461–9462 (2001).
Mensah, E. A. & Nguyen, H. M. Nickel-catalyzed stereoselective formation of α-2-deoxy-2-amino glycosides. J. Am. Chem. Soc. 131, 8778–8780 (2009).
Zulueta, M. M. et al. α-Glycosylation by D-glucosamine-derived donors: synthesis of heparosan and heparin analogues that interact with mycobacterial heparin-binding hemagglutinin. J. Am. Chem. Soc. 134, 8988–8995 (2012).
Park, J., Kawatkar, S., Kim, J.-H. & Boons, G.-J. Stereoselective glycosylations of 2-azido-2-deoxy-glucosides using intermediate sulfonium Ions. Org. Lett. 9, 1959–1962 (2007).
Garegg, P. J., Regberg, T., Stawinski, J. & Strömberg, R. Nucleoside hydrogenphophonates in oligonucleotide synthesis. Chem. Scr. 25, 59–62 (1986).
Westerduin, P., Veeneman, G. H., van der Marel, G. A. & van Boom, J. H. Synthesis of the fragment GlcNAc-α(1→P→6)-GlcNAc of the cell wall polymer of Staphylococcus lactis having repeating N-acetyl-D-glucosamine phosphate units. Tetrahedron Lett. 27, 6271–6274 (1986).
Ohira, S., Yamaguchi, Y., Takahashi, T. & Tanaka, H. The chemoselective O-glycosylation of alcohols in the presence of a phosphate diester and its application to the synthesis of oligomannosylated phosphatidyl inositols. Tetrahedron 71, 6602–6611 (2015).
Sim, M. M., Kondo, H. & Wong, C. H. Synthesis and use glycosyl phosphites: an effective route to glycosyl phosphates, nucleotides, and glycosides. J. Am. Chem. Soc. 115, 2260–2267 (1993).
Fujita, S., Oka, N., Matsumura, F. & Wada, T. Synthesis of oligo (α-D-glycosyl phosphate) derivatives by a phosphoramidite method via boranophosphate intermediates. J. Org. Chem. 76, 2648–2659 (2011).
Westerduin, P., Veeneman, G. H., Marugg, J. E., van der Marel, G. A. & van Boom, J. H. An approach to the synthesis of α-L-fucopyranosyl phosphoric mono- and diesters via phosphite intermediates. Tetrahedron Lett. 27, 1211–1214 (1986).
Schmidt, R. R., Stumpp, M. & Michel, J. α- and β-D-glucopyranosyl phosphates from α-D-glucoypyranosyl trichloroacetimidates. Tetrahedron Lett. 23, 405–408 (1982).
Crich, D. & Picard, S. Highly stereoselective synthesis of α-D-mannopyranosyl phosphosugars. J. Org. Chem. 74, 9576–9579 (2009).
Hanessian, S., Lu, P.-P. & Ishida, H. One-step, stereocontrolled synthesis of glycosyl 1-phosphates, uridine-5’-diphosphogalactose, and uridine-5’-diphosphoglucose from unprotected glycosyl donors. J. Am. Chem. Soc. 120, 13296–13300 (1998).
Pale, P. & Whitesides, G. M. Synthesis of glycosyl phosphates ssing the Fraser-Reid activation. J. Org. Chem. 56, 4547–4549 (1991).
Garcia, B. A. & Gin, D. Y. Synthesis of glycosyl-1-phosphates via dehydrative glycosylation. Org. Lett. 2, 2135–2138 (2000).
Edgar, L. J. G., Dasgupta, S. & Nitz, M. Protecting-group-free synthesis of glycosyl 1-phosphates. Org. Lett. 14, 4226–4229 (2012).
Plante, O. J., Andrade, R. B. & Seeberger, P. H. Synthesis and use of glycosyl phosphates as glycosyl donors. Org. Lett. 1, 211–214 (1999).
Ravidà, A., Liu, X., Kovacs, L. & Seeberger, P. H. Synthesis of glycosyl phosphates from 1,2-orthoesters and application to in situ glycosylation reactions. Org. Lett. 8, 1815–1818 (2006).
Nielsen, M. M. & Pedersen, C. M. Catalytic glycosylations in oligosaccharide Synthesis. Chem. Rev. 118, 8285–8358 (2018).
Li, W. & Yu, B. Gold-catalyzed glycosylation in the synthesis of complex carbohydrate-containing natural products. Chem. Soc. Rev. 47, 7954–7984 (2018).
Dorel, R. & Echavarren, A. M. Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 115, 9028–9072 (2015).
Fürstner, A. & Davies, P. W. Catalytic carbophilic activation: catalysis by platinum and gold π acids. Angew. Chem. Int. Ed. 46, 3410–3449 (2007).
Li, Y., Yang, Y. & Yu, B. An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf. Tetrahedron Lett. 49, 3604–3608 (2008).
Yu, B. Gold(I)-catalyzed glycosylation with glycosyl o-alkynylbenzoates as donors. Acc. Chem. Res. 51, 507–516 (2018).
Tvaroŝka, I. & Bleha, T. In Adv. Carbohydr. Chem. Biochem. Vol. 47 (eds R. Stuart Tipson & Derek Horton) 45-123 (Academic Press, 1989).
Schumann, B. et al. Nucleophile-directed stereocontrol over glycosylations using geminal-difluorinated nucleophiles. Angew. Chem. Int. Ed. 55, 14431–14434 (2016).
Huang, X., Huang, L., Wang, H. & Ye, X.-S. Iterative one-pot synthesis of oligosaccharides. Angew. Chem. Int. Ed. 43, 5221–5224 (2004).
Slättegård, R. et al. Synthesis of structures corresponding to the capsular polysaccharide of Neisseria meningitidis group A. Org. Biomol. Chem. 3, 3782–3787 (2005).
Zhu, Y. & Yu, B. Characterization of the isochromen-4-yl-gold(I) intermediate in the gold(I)-catalyzed glycosidation of glycosyl ortho-alkynylbenzoates and enhancement of the catalytic efficiency thereof. Angew. Chem. Int. Ed. 50, 8329–8332 (2011).
Matthijs, G. et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13 in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat. Genet. 16, 88–92 (1997).
Hollaus, R. et al. Chemical synthesis of Burkholderia Lipid A modified with glycosyl phosphodiester-linked 4-amino-4-deoxy-β-L-arabinose and its immunomodulatory potential. Chem. Eur. J. 21, 4102–4114 (2015).
Schengrund, C.-L. & Kováč, P. UDP-6-deoxy-6-fluoro-α-D-galactose binds to two different galactosyltransferases, but neither can effectively catalyze transfer of the modified galactose to the appropriate acceptor. Carbohydr. Res. 319, 24–28 (1999).
Jambal, I., Kefurt, K., Hlaváčková, M. & Moravcová, J. Synthesis of 2-fluoro and 4-fluoro galactopyranosyl phosphonate analogues of UDP-Gal. Carbohydr. Res. 360, 31–39 (2012).
Bucher, C. & Gilmour, R. Fluorine-directed glycosylation. Angew. Chem. Int. Ed. 49, 8724–8728 (2010).
Majumdar, D., Elsayed, G. A., Buskas, T. & Boons, G.-J. Synthesis of proteophosphoglycans of Leishmania major and Leishmania mexicana. J. Org. Chem. 70, 1691–1697 (2005).
Zhang, Z. et al. Programmable one-pot oligosaccharide synthesis. J. Am. Chem. Soc. 121, 734–753 (1999).
Wu, Y., Xiong, D.-C., Chen, S.-C., Wang, Y.-S. & Ye, X.-S. Total synthesis of Mycobacterial Arabinogalactan containing 92 monosaccharide units. Nat. Commun. 8, 14851 (2017).
Zhang, Y. et al. Modular synthesis of nona-decasaccharide motif from Psidium guajava polysaccharides: orthogonal one-pot glycosylation strategy. Angew. Chem. Int. Ed. 59, 7576–7584 (2020).
Nikolaev, A. V., Ivanova, I. A. & Shibaev, V. N. The stepwise synthesis of oligo (glycosyl phosphates) via glycosyl hydrogenphosphonates. The chemical synthesis of oligomeric fragments from Hansenula capsulata Y-1842 exophosphomannan and from Escherichia coli K51 capsular antigen. Carbohydr. Res. 242, 91–107 (1993).
Crich, D. & Smith, M. 1-Benzenesulfinyl piperidine/trifluoromethanesulfonic anhydride: a potent combination of shelf-stable reagents for the low-temperature conversion of thioglycosides to glycosyl triflates and for the formation of diverse glycosidic linkages. J. Am. Chem. Soc. 123, 9015–9020 (2001).
Kumar, M., Jasinski, J., Hammond, G. B. & Xu, B. Alkyne/alkene/allene-induced disproportionation of cationic gold(I) catalyst. Chem. Eur. J. 20, 3113–3119 (2014).
Acknowledgements
We thank the National Natural Science Foundation of China (22061042, Y. Zhu, 22007080, Y. Zhu), Yunnan Provincial Science and Technology Department and Yunnan University for financial support (2019FY003014, Y. Zhu, C176220100065, Y. Zhu). Authors thank Advanced Analysis and Measurement Center of Yunnan University for the sample testing service. We are grateful to Prof. Tiehai Li (SIMM, CAS), Prof. Ming Li (OUC) and Jiansong Sun (Jiangxi Normal Univ.) for helpful discussions. We appreciate Prof. Biao Yu (SIOC) for revising the manuscript.
Author information
Authors and Affiliations
Contributions
Y. Zhu conceived the project and wrote the manuscript. X.Z., Y.Y., J.D., Y. Zhao and Y. Zhu performed the experiments. X.Z., Y.Y., J.D., Y. Zhao and Y. Zhu conducted the data analysis. Y. Zhu, X.Z. and H.Z. revised the manuscript. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jeroen Codée, Daniel Silva and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Yang, Y., Ding, J. et al. Stereoselective gold(I)-catalyzed approach to the synthesis of complex α-glycosyl phosphosaccharides. Nat Commun 13, 421 (2022). https://doi.org/10.1038/s41467-022-28025-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28025-0
This article is cited by
-
Photosensitizer-free visible-light-promoted glycosylation enabled by 2-glycosyloxy tropone donors
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
