
Abstract
Catalytic asymmetric dearomatization (CADA) is a powerful tool for the rapid construction of diverse chiral cyclic molecules from cheap and easily available arenes. This work reports an organocatalytic enantioselective dearomatization of substituted thiophenes in the context of a rare remote asymmetric 1,10-conjugate addition. By suitable stabilization of the thiophenyl carbocation with an indole motif in the form of indole imine methide, excellent remote chemo-, regio-, and stereocontrol in the nucleophilic addition can be achieved with chiral phosphoric acid catalysis under mild conditions. This protocol can be successfully extended to the asymmetric dearomatization of other heteroarenes including selenophenes and furans. Control experiments and DFT calculations demonstrate a possible pathway in which hydrogen bonding plays an important role in selectivity control.
Similar content being viewed by others

Introduction
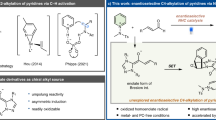
Catalytic asymmetric dearomatization (CADA) reactions have gained attention in the past few years since they offer direct and rapid access to enantioenriched functionalized ring systems and complex heterocyclic skeletons from simple and readily available arenes1,2,3,4,5,6,7. For example, CADA has been a pivotal step in the syntheses of many natural products and drugs8,9,10,11,12,13. Generally, thiophene is less prone to dearomative transformations than other common heteroarenes (e.g., furan, pyrrole, pyridine), owing to its relatively high resonance stabilization energy (Fig. 1a)6,14. Consequently, a high energy barrier is typically encountered, resulting in often harsh conditions for this type of transformations and imposing formidable challenges in controlling regioselectivity and stereoselectivity15,16,17,18. Beyond that, the strong coordination ability of the generated sulfur-containing product may deactivate the metal catalyst or interfere with stereocontrol by competing for binding, thus representing another important issue to address. These challenges have hampered the development of CADA of thiophenes. To the best of our knowledge, the only general example of this type was achieved by Glorius and coworkers via metal-catalyzed hydrogenation, leading to enantioenriched tetrahydrothiophenes (Fig. 1b)19. In contrast, the application of organocatalysis for such processes would be natural owning to the metal-free nature. However, challenges still remain in order to overcome the high barrier and achieve good stereocontrol20. Herein we report an organocatalytic approach (Fig. 1c).

a Resonance stabilization of simple aromatic compounds. b Metal-catalyzed asymmetric dearomatization of thiophenes (by Glorius). c Organocatalytic asymmetric dearomatization of thiophenes (our strategy). d Our reaction design and the challenges (this work).
As depicted in Fig. 1d, we hypothesized that, upon chiral acid activation, a 2-thiophenyl tertiary alcohol might generate a benzylic carbocation IP-a, with a chiral counter anion. The adjacent cation should activate the thiophene ring, for example, with charge delocalization represented in the resonance form IP-b. Subsequently, nucleophilic attack might take place in the fifth position of the thiophene ring. Furthermore, the chiral counter anion might induce asymmetric control in this step, leading to enantioenriched dearomatization product 3. Notably, in addition to the inevitable remote enantiocontrol, this process will encounter other challenges, including regiocontrol on the nucleophilic sites and control over the double bond Z/E-configuration18. Product side reactions, such as rearomatization21, could provide chemoselectivity challenges. To address these potential problems, we envisioned that further stabilization of the positive charge by extended conjugation (e.g., quinone methide (QM) or indole imine methide) in a rigid framework might help selectivity control.
Indole-based imine methides have been demonstrated to be versatile intermediates in a range of organocatalytic asymmemtric transformations, particularly in 1,4- or 1,6-conjugate addition22,23,24,25,26,27,28,29 and cycloaddition30,31,32,33,34, leading to diverse enantioenriched indole derivatives35,36,37,38. Among them, we have reported an asymmetric 1,6-conjugate addition of such intermediates for the synthesis of chiral tetraarylmethanes containing an indole unit29. Recently, Antilla’s group and our group have developed remote asymmetric 1,8-addition to such species with chiral phosphoric acid (CPA) catalysis, providing efficient access to enantioenriched triarylmethanes and allenes, respectively39,40. Inspired by these studies and in continuation of our interests in remote stereocontrol, we envisioned the possibility of achieving further remote stereocontrol with this system. When the thiophene benzylic cation is adorned with a 2-indolyl group, this cation is stabilized in the form of highly conjugated indole imine methide IP-b′ (Fig. 1d). Further nucleophilic conjugate addition is expected to take place at the tenth position based on our preliminary results, thereby representing not only thiophene dearomatization but also remote 1,10-conjugate addition. Notably, such remote stereocontrol has been rarely observed41,42,43. In addition, this dearomative 1,10-addition entails additional selectivity control, such as regioselectivity (1,6- vs. 1,10-addition), double bond Z/E selectivity, and axial chirality in some cases, none of which was a major problem in previous precedents of 1,6- and 1,8-addition of indole imine-methides. With the powerful bifunctional activation with CPA catalysis for remote control44,45,46,47,48,49,50,51,52,53,54,55, herein we have realized such an efficient asymmetric process.
Results and discussion
Reaction development
Our initial studies took advantage of QM intermediates for the asymmetric conjugate addition (see the SI for details)50,51,52,53,54,55. Unfortunately, these proved to be rather difficult to control regarding enantioselectivity and/or Z/E ratio. Subsequently, we employed indole-substituted tertiary alcohol 1a as the substrate and 2-phenylpyrrole 2a as the nucleophile (Table 1). With different CPA catalysts, the reaction in DCM proceeded successfully at room temperature to form the desired dearomatization product 3a (entries 1–10). Among these catalysts, the [H8]BINOL-derived (R)-B2 was identified as the best, resulting in 91% yield, 85% e.e., and >20:1 E/Z ratio. While comparable results could be obtained with catalysts A3, A4, and B1 (entries 3–5), B2 was used for further optimization on other parameters, which indicated that PhCl served as the superior solvent (entry 13). Finally, when the temperature was decreased to −40 oC, 3a was obtained in 99% yield, 96% e.e., and >20:1 E/Z (entry 14).
Substrates scope exploration
Having identified the suitable directing group and optimal conditions, next we examined the substrate scope of this asymmetric process (Table 2). A wide range of 2-thiophenyl tertiary alcohols 1 and pyrroles 2 reacted to form the dearomatization products. In most cases, good yields E/Z ratios, and enantioselectivities were obtained. Electronically different phenyl substituents bearing various functional groups at the ortho, meta, and para positions showed good performance (3a–h, 87–97% yields, 92–99% e.e., and >20/1 E/Z). Moreover, polycyclic and heterocyclic aryl substituents were also well tolerated (3i–k). Notably, substrates 1j and 1k have two thiophenyl groups, but only one thiophene ring was dearomatized with high efficiency and chemoselectivity. Particularly, the case of 1k also represented an efficient enantioselective desymmetrization. An alkyl-substituent could also be engaged in this process with excellent stereoselectivity, but in low yield. The low efficiency was caused by a competitive 1,6-addition pathway leading to chiral triarylethane 3l′ due to lower steric hindrance of this pathway (see the SI for details). Next, alcohols bearing differently-substituted indolyl groups were examined. All these reactions produced the desired products 3m–r with satisfactory results, including the 3-unsubstituted indolyl one (3r). Substitution on the thiophene ring also led to the desired product 3s, albeit with compromised efficiency and stereoselectivity. Finally, various pyrroles successfully served as nucleophiles, giving the corresponding enantioenriched sulfur heterocycles with respectable efficiency and selectivity (3t–w). Other electron-rich arenes, such as indole, naphthol, furan, thiophene, and 1,3,5-trimethoxybenzene, were also examined. Among them, indole reacted to form the desired product in good yield, but with a low Z/E selectivity and enantioselectivity. Other nucleophiles were not successful (see the SI for details).
We also applied this strategy to the asymmetric dearomatization of other heteroarenes, such as selenophenes (Table 3). After slight modification of the conditions (see the SI for details), the analogous selenophene substrates 4 reacted efficiently to form the corresponding chiral Se-heterocycles 5 with excellent stereoselectivity. The free alcohol functional group was also compatible with this catalytic system.
Mechanistic studies and synthetic applications
This process could also be directly extended to the dearomatization of the furan ring. At −40 oC, the reaction between furan carbinol 6 with pyrrole proceeded smoothly to form the desired product 7 in 92% yield with 92% e.e. and 6.7:1 E/Z ratio (Fig. 2, Eq. 1). The practicality was further demonstrated by a 1-mmol-scale reaction of 1a and 2a, resulting in comparably excellent results to the small-scale one (Eq. 2). Moreover, the double bond in the sulfur heterocycle of product 3t could be further reduced to form enantioenriched tetrahydrothiophene 8 (Eq. 3). It is worth noting that enantioenriched tetrahydrothiophenes and tetrahydroselenophenes are useful molecules in organic synthesis and medicinal chemistry56,57,58,59,60,61,62. For example, they are known chiral ligands or organocatalysts for asymmetric synthesis 56,57,58,59.

a Reaction extension to asymmetric dearomatization of the furan ring. b Mmol-scale synthesis. c Selective reduction of C-C double bond. d Control experiments with N-substituted substrates. e Reaction extension to N-methyl indole as the nucleophile.
Control experiments were carried out to gain some insights into the mechanism (see the SI for details). First, we examined the N-methylated pyrroles 2a′ and 2b′, which are more nucleophilic than 2a and 2b, respectively. However, their reactivity was found to be lower, requiring a much higher temperature (RT vs. −40 oC under the standard conditions). The corresponding products 3a′ and 3t′ were obtained in only moderate yields with slightly decreased enantioselectivities (Eq. 4). The results indicated that hydrogen bonding with N−H motif of the nucleophile is not necessary for good stereocontrol, but this interaction might help reduce the barrier of the nucleophilic addition. In contrast, N-methylation in the substrate indole motif (1a′) led to dramatic changes in both reactivity and stereoselectivity. At room temperature, the reaction with 2b produced a mixture of diastereomers in 2:1 dr and 1:1 E/Z (Eq. 5). The presence of so many isomers made the enantioselectivity determination difficult. This observation strongly suggested that the N−H motif in the indole unit plays a crucial role in stereocontrol, which is likely to facilitate the key imine methide intermediate generation and allow subsequent hydrogen-bonding interaction with the catalyst. Notably, other nucleophiles were also examined (Supplementary Table 12), and it was found that N-methyl indole performed well in this system to furnish the desired product 10 in moderate yield and excellent stereoselectivity (44% yield, 95% e.e., E/Z >20:1, Eq. 6).
Density functional theory studies
To better understand the mechanism and origins of selectivity, density functional theory (DFT) calculations were conducted on the reaction of tertiary alcohol 1a and 2-phenylpyrrole 2a by dimethyl phosphoric acid (Fig. 3) or chiral phosphoric acid (R)-B2 (Fig. 4) using Gaussian 1663. Geometry optimizations were performed at B3LYP-D3BJ/6-31 G(d) the level of theory64,65, and single-point energies were computed with M06-2X/6-311+G(d,p)-CPCM(chlorobenzene)66,67,68,69. The calculated potential energy profile is given in Fig. 3. The binding of substrate CP2 to catalyst CP1 affords relatively stable complex CP3. Then intermediate CP3 undergoes acid-catalyzed dehydration via transition states TS1-E and TS1-Z to obtain the imine methide intermediates CP4-E and CP4-Z, with activation free energies of 18.5 and 18.3 kcal/mol, respectively. Subsequently, the nucleophilic attack of 2-phenylpyrrole CP5 to the Z-isomer CP4-Z via TS2-Z-Z and TS2-Z-E lead to CP6-Z and CP6-E. The difference in energy between these two transition states is 5.1 kcal/mol, showing great stereoselectivity (>20/1, E/Z). The subsequent intermolecular hydrogen shift forms the final product CP7 and regenerates the free catalyst via TS3-E and TS3-Z. However, the formation of the preferred product CP7-E through the nucleophile 2-phenylpyrrole CP5 attacks the E-isomer CP4-E via TS2-E-E and TS2-E-Z is very difficult, and has an overall barrier of 20.7 and 19.6 kcal/mol with respect to the stable intermediate CP3, respectively. The enantio-determining step for this reaction is the nucleophilic attack of 2-phenylpyrrole on the indole imine methide CP4-Z, TS2.

a Free energy profile of dimethyl phosphoric acid-catalyzed dearomatization of thiophenes. b Color-filled NCI isosurfaces of the transition state TS2-E-E, TS2-E-Z, TS2-Z-Z, and TS2-Z-E (blue, strong attraction; green, weak interaction; red, steric effect). The distances are given in Ångstroms.

The distances are given in Ångstroms, and energies are given in kcal/mol.
We first investigated the dearomative transformations from different configurations of imine methide CP4. As depicted in Fig. 3a, the transition states TS2-E-E and TS2-E-Z from the E-isomer are higher in energy than TS2-Z-E by 6.9 and 5.8 kcal/mol, respectively. To gain further insight into the energy difference, the non-covalent interaction (NCI) analysis was performed70,71. Figure 3b shows color-filled NCI isosurfaces for all non-covalent interactions in the transition states. The crucial difference between these competing transition structures is the location of the phenyl group of thiophene, which is orientated toward the catalyst in unfavored TS2-E-E and TS2-E-Z. To accommodate the phenyl group, it is hard for transition states TS2-E-E and TS2-E-Z to form additional stabilizing hydrogen-bonding interactions. In contrast, TS2-Z-E enjoys additional favorable C–H···O–P interaction (shown in light blue). Therefore, E-isomer CP4-E is less prone to dearomative transformations than Z-isomer CP4-Z, basically owing to the weaker hydrogen-bonding interactions.
To elucidate the origins of stereoselectivity, we explored the structures of dimethyl phosphoric acid-catalyzed transition states TS2-Z-Z and TS2-Z-E, which generate Z-selectivity product CP7-Z and E-selectivity product CP7-E, respectively. The 5.1 kcal/mol difference in energy between TS2-Z-Z and TS2-Z-E corresponds to the excellent stereoselectivity (>20:1 ratio). The primary difference between these two transitions states (Fig. 3b) is the orientation of the electron-deficient sulfur center of the thiophene relative to the electron-rich pyrrole ring. In minor TS2-Z-Z, the electron-deficient sulfur center is far away from the electron-rich pyrrole ring, while it points toward the center of the electron-rich pyrrole ring in major TS2-Z-E (shown in light blue), suggesting the presence of attractive chalcogen-bonding interaction in TS2-Z-E72,73. In addition, the π–π interactions between the phenyl rings in TS2-Z-Z are weaker than those in TS2-Z-E (shown as large green disks). Therefore, the favorable chalcogen-bonding and π–π interactions are the main contributions to the 5.1 kcal/mol preference for forming an E-selectivity product.
We also studied chiral phosphoric acid (R)-B2 catalyzed enantioselective addition of the 2-phenylpyrrole to the imine methide. The calculated si-face attack in TS2-R-E is found to be 2.2 kcal/mol lower in free energy than the re-face attack in TS2-S-E, which is consistent with the 96% e.e. observed experimentally. To better recognize the factors that impact the enantioselectivity, optimized structures of transition states TS2-R-E and TS2-S-E were compared (Fig. 4). Apparently, transition state TS2-R-E enjoys additional stabilizing C–H···O–P interaction compared to TS2-S-E. Such favorable hydrogen-bonding interactions decrease the energies of transition state TS2-R-E. In contrast, there is a lack of C–H···O–P interaction in unfavored TS2-S-E. Further calculations by removing the substituents of the catalyst with methyl groups, then computed the single-point ΔΔE‡ without optimization, show a 2.3 kcal/mol advance for the transition state TS2-R-E. The energetics provide reasonable agreement with experimental observations. Consequently, hydrogen-bonding interactions play a leading role in determining high enantioselectivity.
In summary, we have developed an efficient organocatalytic enantioselective dearomatization of thiophenes in the context of asymmetric 1,10-conjugate addition. It is also a rare example of excellent remote stereocontrol. By suitable stabilization of 2-thiophenyl carbocation with an indole motif in the form of an extended indole imine methide, chiral phosphoric acid serves as a superior bifunctional catalyst to promote intermolecular C−C bond formation with excellent chemo-, regio- and enantioselectivity as well as the product double bond Z/E ratio. This protocol can be extended to the asymmetric dearomatization of selenophenes and furans. Control experiments and DFT calculations illustrated a possible pathway in which multiple hydrogen-bonding interactions play a crucial role in achieving excellent stereocontrol.
Methods
General procedure for the catalytic asymmetric dearomatization of thiophenes
Condition A
At −40 oC, to an oven-dried 8-mL vial charged with a solution of the tertiary alcohol 1 (0.2 mmol) and pyrrole 2 (0.24 mmol) in PhCl (3.6 mL) was slowly added a solution of catalyst (R)-B2 (20 mg, 0.015 mmol, 10 mol%) in PhCl (0.4 mL). The reaction mixture was stirred at the same temperature for 12 h. After that, triethylamine (two drops) was added to quench the reaction. The mixture was concentrated under reduced pressure and purified by silica gel (deactivated by triethylamine) flash chromatography to afford the desired product 3.
Condition B
At −20 oC, to an oven-dried 8-mL vial charged with a solution of the tertiary alcohol 1 (0.2 mmol) and pyrrole 2 (2.0 mmol) in toluene (3.6 mL) was slowly added a solution of catalyst (R)-B2 (20 mg, 0.015 mmol, 10 mol%) in toluene (0.4 mL). The reaction mixture was stirred at the same temperature for 24 h. After that, triethylamine (two drops) was added to quench the reaction. The mixture was concentrated under reduced pressure and purified by silica gel (deactivated by triethylamine) flash chromatography to afford the desired product 3.
Data availability
All data generated and analyzed during this study are included in this article and its Supplementary Information, or also available from the authors upon reasonable request. The X-ray crystallographic coordinate for structure 5a has been deposited at the Cambridge Crystallographic Data Centre under deposition numbers CCDC 2022226, respectively, and can be obtained free of charge from the CCDC via http://www.ccdc.cam.ac.uk/data_request/cif.
References
Pape, A. R., Kaliappan, K. P. & Kündig, E. P. Transition-metal-mediated dearomatization reactions. Chem. Rev. 100, 2917–2940 (2000).
Ortiz, F. L., Iglesias, M. J., Fernandez, I., Sanchez, C. M. A. & Gomez, G. R. Nucleophilic dearomatizing (DNAr) reactions of aromatic C, H-systems. a mature paradigm in organic synthesis. Chem. Rev. 107, 1580–1691 (2007).
Zhuo, C.-X., Zhang, W. & You, S.-L. Catalytic asymmetric dearomatization reactions. Angew. Chem. Int. Ed. 51, 12662–12686 (2012).
Dalpozzo, R. Strategies for the asymmetric functionalization of indoles: an update. Chem. Soc. Rev. 44, 742–778 (2015).
Wu, W.-T., Zhang, L. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 45, 1570–1580 (2016).
Wertjes, W. C., Southgate, E. H. & Sarlah, D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 47, 7996–8017 (2018).
Xia, Z.-L., Xu-Xu, Q.-F., Zheng, C. & You, S.-L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 49, 286–300 (2020).
Roche, S. P. & Porco, J. A. Jr. Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed. 50, 4068–4093 (2011).
Zheng, C. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 36, 1589–1605 (2019).
Huck, C. J. & Sarlah, D. Shaping molecular landscapes: recent advances, opportunities, and challenges in dearomatization. Chem 6, 1589–1603 (2020).
Charest, M. G., Lerner, C. D., Brubaker, J. D., Siege, D. R. & Myers, A. G. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 308, 395–398 (2005).
Sullivan, B., Carrera, I., Drouin, M. & Hudlicky, T. Symmetry-based design for the chemoenzymatic synthesis of oseltamivir (Tamiflu) from ethyl benzoate. Angew. Chem. Int. Ed. 48, 4229–4231 (2009).
Hernandez, L. W., Pospech, J., Klöckner, U., Bingham, T. W. & Sarlah, D. Synthesis of (+)-Pancratistatins via catalytic desymmetrization of benzene. J. Am. Chem. Soc. 139, 15656–15659 (2017).
Joule, J. A. & Mills, K. Heterocyclic Chemistry 5th edn (Wiley, 2010).
Delafuente, D. A., Myers, W. H., Sabat, M. & Harman, W. D. Tungsten(0) η2-thiophene complexes: dearomatization of thiophene and its facile oxidation, protonation, and hydrogenation. Organometallics 24, 1876–1885 (2005).
Sperry, J. B., Ghiviriga, I. & Wright, D. L. Electrochemical annulation of five-membered rings through dearomatization of furans and thiophenes. Chem. Commun. 42, 194–196 (2006).
Nguyen, S. T., Ding, X. & Peet, N. P. A remarkable reductive dearomatization of thiophene and furan rings. Synthesis 45, 1904–1908 (2013).
Komatsuda, M., Kato, H., Muto, K. & Yamaguchi, J. Pd-catalyzed dearomative three-component reaction of bromoarenes with diazo compounds and allylborates. ACS Catal. 9, 8991–8995 (2019).
Urban, S., Beiring, B., Ortega, N., Paul, D. & Glorius, F. Asymmetric hydrogenation of thiophenes and benzothiophenes. J. Am. Chem. Soc. 134, 15241–15244 (2012).
Ullah, A. et al. Palladium-catalyzed propargylative and allenylativedearomatization of 2-(chloromethyl)thiophenes: remarkable effect of solvent. Tetrahedron 72, 170–175 (2016).
Xu, J. et al. Catalyst-controlled chemodivergent modification of indoles with 2-furylcarbinols: Piancatelli reaction vs cross-dehydrative coupling reaction. J. Org. Chem. 82, 3561–3570 (2017).
Guo, Q.-X. et al. Highly enantioselective alkylation reaction of enamides by Brønsted-acid catalysis. Org. Lett. 11, 4620–4623 (2009).
Qi, S., Liu, C.-Y., Ding, J.-Y. & Han, F.-S. Chiral phosphoramide-catalyzed enantioselective synthesis of 2,3-diindolylarylmethanes from indol-2-yl carbinols and indoles. Chem. Commun. 50, 8605–8608 (2014).
Zhu, Z.-Q., Shen, Y., Liu, J.-X., Tao, J.-Y. & Shi, F. Enantioselective direct α-arylation of pyrazol-5-ones with 2-indolylmethanols via organo-metal cooperative catalysis. Org. Lett. 19, 1542–1545 (2017).
Zhang, H.-H. et al. Design and enantioselective construction of axially chiral naphthyl-indole skeletons. Angew. Chem. Int. Ed. 56, 116–121 (2017).
Xu, M.-M. et al. Cooperative catalysis-enabled asymmetric α-arylation of aldehydes using 2‑indolylmethanols as arylation reagents. J. Org. Chem. 83, 5027–5034 (2018).
Wang, H.-Q. et al. Application of 7-indolylmethanols in catalytic asymmetric arylations with tryptamines: enantioselective synthesis of 7-indolylmethanes. Adv. Syn. Catal. 360, 1850–1860 (2018).
Bertuzzi, G. et al. γ-Regioselective functionalization of 3-alkenylindoles via 1,6-addition to extended alkylideneindolenine intermediates. Adv. Synth. Catal. 360, 1296‒1302 (2018).
Li, X. et al. Catalytic enantioselective synthesis of chiral tetraarylmethanes. Nat. Catal. 3, 1010–1019 (2020).
Bera, K. & Schneider, C. Brønsted acid catalyzed [3+2]-cycloaddition of cyclic enamides with in situ generated 2-methide-2H-indoles: enantioselective synthesis of Indolo[1,2-a]indoles. Org. Lett. 18, 5660–5663 (2016).
Bera, K. & Schneider, C. Brønsted acid catalyzed [3+2]-cycloaddition of 2-vinylindoles with in situ generated 2-methide-2H-indoles: highly enantioselective synthesis of pyrrolo[1,2-a]indoles. Chem. Eur. J. 22, 7074–7078 (2016).
Sun, X.-X. et al. Catalytic asymmetric [3+3] cycloaddition of azomethine ylides with C3-substituted 2-indolylmethanols. Adv. Syn. Catal. 359, 2660–2670 (2017).
Sun, M. et al. Catalytic asymmetric (4+3) cyclizations of in situ generated ortho-quinone methides with 2-indolylmethanols. Angew. Chem. Int. Ed. 58, 8703–8708 (2019).
Zhang, Y.-C., Jiang, F. & Shi, F. Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: strategies, reactions, and outreach. Acc. Chem. Res. 53, 425–446 (2020).
Palmieri, A., Petrini, M. & Shaikh, R. R. Synthesis of 3-substituted indoles via reactive alkylideneindolenine intermediates. Org. Biomol. Chem. 8, 1259–1270 (2010).
Bartoli, G., Bencivenni, G. & Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 39, 4449–4465 (2010).
Wang, L., Chen & Xiao, Y. J. Alkylideneindoleninium ions and alkylideneindolenines: key intermediates for the asymmetric synthesis of 3-indolyl derivatives. Asian J. Org. Chem. 3, 1036–1052 (2014).
Palmieri, A. & Petrini, M. Sulfonyl azoles in the synthesis of 3-functionalized azole derivatives. Chem. Rec. 16, 1353–1379 (2016).
Yue, C., Na, F., Fang, X., Cao, Y. & Antilla, J. C. Chiral phosphoric acid catalyzed asymmetric synthesis of heterotriarylmethanes from racemic indolyl alcohols. Angew. Chem. Int. Ed. 57, 11004–11008 (2018).
Li, X. & Sun, J. Organocatalytic enantioselective synthesis of chiral allenes: remote asymmetric 1,8-addition of indole imine methides. Angew. Chem. Int. Ed. 59, 17049–17054 (2020).
Krause, N. & Thorand, S. Copper-mediated 1,6-, 1,8-, 1,10- and 1,12-addition and 1,5-substitution reactions in organic synthesis. Inorg. Chim. Acta 296, 1–11 (1999).
Uraguchi, D., Yoshioka, K., Ueki, Y. & Ooi, T. Highly regio‑, diastereo‑, and enantioselective 1,6- and 1,8- additions of azlactones to di- and trienyl N‑acylpyrroles. J. Am. Chem. Soc. 134, 19370–19373 (2012).
den Hartog, T. et al. On the mechanism of Cu-catalyzed enantioselective extended conjugate additions: A structure-based approach. ACS Catal. 5, 560–574 (2015).
Wang, Z. et al. Organocatalytic asymmetric synthesis of 1,1-diarylethanes by transfer hydrogenation. J. Am. Chem. Soc. 137, 383–389 (2015).
Wang, Z., Wong, Y. F. & Sun, J. Catalytic asymmetric 1,6-conjugate addition of para-quinone methides: formation of all-carbon quaternary stereocenters. Angew. Chem. Int. Ed. 54, 13711–13714 (2015).
Dong, N., Zhang, Z.-P., Xue, X.-S., Li, X. & Cheng, J.-P. Phosphoric acid catalyzed asymmetric 1,6-conjugate addition of thioacetic acid to para-quinone methides. Angew. Chem. Int. Ed. 55, 1460–1464 (2016).
Qian, D., Wu, L., Lin, Z. & Sun, J. Organocatalytic synthesis of chiral tetrasubstituted allenes from racemic propargylic alcohols. Nat. Commun. 8, 567 (2017).
Chen, M. & Sun, J. Catalytic asymmetric N-alkylation of indoles and carbazoles through 1,6-conjugate addition of aza-para-quinone methides. Angew. Chem. Int. Ed. 56, 4583–4587 (2017).
Zhang, L. et al. Organocatalytic remote stereocontrolled 1,8-additions of thiazolones to propargylic Aza‑p‑quinone methides. Org. Lett. 21, 7415–7419 (2019).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective Mannich‐type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed. 43, 1566–1568 (2004).
Uraguchi, D. & Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 126, 5356–5357 (2004).
Nakashima, D. & Yamamoto, H. Design of chiral N-triflyl phosphoramide as a strong chiral Brønsted acid and its application to asymmetric Diels−Alder reaction. J. Am. Chem. Soc. 128, 9626–9627 (2006).
Parmar, D., Sugiono, E., Raja, S. & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 114, 9047–9153 (2014).
Akiyama, T. & Mori, K. Stronger Brønsted acids: recent progress. Chem. Rev. 115, 9277–9306 (2015).
James, T., van Gemmeren, M. & List, B. Development and applications of disulfonimides in enantioselective organocatalysis. Chem. Rev. 115, 9388–9409 (2015).
McGarrigle, E. M. et al. Stronger Brønsted acids. Chem. Rev. 107, 5841–5883 (2007).
Chen, F., Tan, C. K. & Yeung, Y.-Y. C2-Symmetric cyclic selenium-catalyzed enantioselective bromoaminocyclization. J. Am. Chem. Soc. 135, 1232–1235 (2013).
Liu, X., An, R., Zhang, X., Luo, J. & Zhao, X. Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane‐based chiral sulfide. Angew. Chem. Int. Ed. 55, 5846–5850 (2016).
Illa, O. et al. Practical and highly selective sulfur ylide-mediated asymmetric epoxidations and aziridinations using a cheap and readily available chiral sulfide: extensive studies to map out scope, limitations, and rationalization of diastereo- and enantioselectivities. J. Am. Chem. Soc. 135, 11951–11966 (2013).
Zempleni, J., Wijeratne, S. S. K. & Hassan, Y. I. Biotin. Biofactors 35, 36–46 (2009).
Matsuda, H., Morikawa, T., Oda, M., Asao, Y. & Yoshikawa, M. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum. Bioorg. Med. Chem. Lett. 13, 4445–4449 (2003).
Jeong, L. S. et al. Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4’-thioadenosine derivatives without 4’-hydroxymethyl group as highly potent and selective antagonists. J. Med. Chem. 50, 3159–3162 (2007).
Frisch, M. J. et al. Gaussian 16, rev. A.03 (Gaussian Inc., 2016).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Zhao, Y. & Truhlar, D. G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167 (2008).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 120, 215–241 (2008).
Cossi, M., Rega, N., Scalmani, G. & Barone, V. Energies, structures, and electronic properties of molecules in solution with the C‐PCM solvation model. J. Comput. Chem. 24, 669–681 (2003).
Barone, V. & Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998).
Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Zhou, B., Haj, M. K., Jacobsen, E. N., Houk, K. N. & Xue, X. S. Mechanism and origins of chemo- and stereoselectivities of aryl iodide-catalyzed asymmetric difluorinations of β-substituted styrenes. J. Am. Chem. Soc. 140, 15206–15218 (2018).
Vogel, L., Wonner, P. & Huber, S. M. Chalcogen bonding: an overview. Angew. Chem. Int. Ed. 58, 1880–1891 (2019).
Acknowledgements
Financial support was provided by the Research Grants Council of Hong Kong (16302617 and 16302318), Innovation and Technology Commission (ITC-CNERC14SC01), the National Natural Science Foundation of China (91956114), Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002), and the National Science Foundation (CHE-1764328 to K.N.H). Computational work was supported by the Center for Computational Science and Engineering at Southern University of Science and Technology, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575). We also thank Dr. Herman H. Y. Sung for help with structure elucidation and Guannan Wang for help in experiments.
Author information
Authors and Affiliations
Contributions
X.L. conceived the project, performed the experiments, and wrote the paper. M.D. and P.Y. performed DFT calculations. K.N.H. directed the DFT calculations and mechanism analysis. J.S. conceived and directed the project and wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jun Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Duan, M., Yu, P. et al. Organocatalytic enantioselective dearomatization of thiophenes by 1,10-conjugate addition of indole imine methides. Nat Commun 12, 4881 (2021). https://doi.org/10.1038/s41467-021-25165-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25165-7
This article is cited by
-
Catalytic asymmetric (3 + 3) cycloaddition between different 2-indolylmethanols
Science China Chemistry (2024)
-
Remote copper-catalyzed enantioselective substitution of yne-thiophene carbonates
Science China Chemistry (2024)
-
Quinone methides and indole imine methides as intermediates in enantioselective catalysis
Nature Synthesis (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
