
Abstract
PbMO3 (M = 3d transition metals) family shows systematic variations in charge distribution and intriguing physical properties due to its delicate energy balance between Pb 6s and transition metal 3d orbitals. However, the detailed structure and physical properties of PbFeO3 remain unclear. Herein, we reveal that PbFeO3 crystallizes into an unusual 2ap × 6ap × 2ap orthorhombic perovskite super unit cell with space group Cmcm. The distinctive crystal construction and valence distribution of Pb2+0.5Pb4+0.5FeO3 lead to a long range charge ordering of the -A-B-B- type of the layers with two different oxidation states of Pb (Pb2+ and Pb4+) in them. A weak ferromagnetic transition with canted antiferromagnetic spins along the a-axis is found to occur at 600 K. In addition, decreasing the temperature causes a spin reorientation transition towards a collinear antiferromagnetic structure with spin moments along the b-axis near 418 K. Our theoretical investigations reveal that the peculiar charge ordering of Pb generates two Fe3+ magnetic sublattices with competing anisotropic energies, giving rise to the spin reorientation at such a high critical temperature.
Similar content being viewed by others

Introduction
Transition metal perovskite oxides (general formula: ABO3) display a variety of desirable electronic and magnetic properties, such as high-temperature superconductivity1, colossal magnetoresistance2,3, metal–insulator transition4, multiferroicity5,6,7, and electrocatalysis8. AMO3 (A = Pb/Bi) perovskite oxides are typical examples of charge degrees of freedom at the A site depending on 6s0 (Pb4+, Bi5+) and 6s2 (Pb2+, Bi3+) electron configurations for the prohibition of the 6s1 configuration9,10,11. For example, in Bi-based systems, BiCrO3 through BiCoO3 are all Bi3+M3+O3; however, BiNiO3 has an unusual valence state Bi3+0.5Bi5+0.5Ni2+O3 with ordered Bi3+ and Bi5+ charge states9,10,11,12,13,14,15.
In Pb-based systems, as the d level of the transition metal becomes deeper, different crystal structures and systematic charge distribution changes are observed16,17,18,19,20,21,22. Divalent lead appears in tetragonal PbTiO3 and PbVO317. However, the disordered coexistence of Pb2+ and Pb4+ states (charge glass) occurs in PbCrO3 (Pb3+Cr3+O3 on average), where a simultaneous insulator-to-metal transition and a large volume collapse arise from the melting of Pb charge glass and Pb–Cr charge transfer upon pressurizing to 2.5 GPa18. More interestingly, a 1:3 ordered Pb2+ and Pb4+ and a 1:1 ordered Co2+ and Co3+ have been observed in PbCoO3 with the charge format of Pb2+Pb4+3Co2+2Co3+2O12 (Pb3.5+Co2.5+O3 on average)19,20. Moreover, a pressure-induced spin-state transition and Pb–Co intermetallic charge transfer have been discovered in this compound19,20. For PbNiO3, the electronic configuration is Pb4+Ni2+O3 in the presence of a single-valence Pb4+ state21. Currently, in PbMO3 systems, only PbMnO3 and PbFeO3 remain elusive. Herein, we focus on PbFeO3, which has been reported by Tsuchiya et al. in 200722. However, the difficulties in the synthesis of samples and in resolving the crystal structure have inhibited investigation of its structure and physical properties.
Alongside the intriguing charge properties at the Pb site, the spin degree of freedom at the M-site in PbMO3 systems has also received significant attention. For example, a pressure-induced high-spin to low-spin-state transition of Co2+ accompanied by an unusual increase in the resistance in PbCoO3 has been reported recently19. The perovskite family of RFeO3 (R = rare earth) exhibits various spin-related properties such as multiferroicity23,24,25, laser-induced ultrafast spin reorientation (SR)26,27, and ultrafast photomagnetic recording28, and it is anticipated that the evolution of magnetism and spin structure of PbFeO3 as a function of temperature, magnetic field or pressure will be similarly rich. A characteristic feature of RFeO3 is the presence of two magnetic sublattices, i.e., the R3+ sublattice and Fe3+ sublattice29,30,31,32, which generate three types of competitive exchange interactions (Fe–Fe, R–Fe, and R–R). The strongest Fe–Fe interaction induces a canted antiferromagnetic (AFM) ordering of Fe3+ (S = 5/2) spins below the Néel temperature TN = 600–740 K with a Néel vector Gx and a weak ferromagnetic (FM) vector Fz. This type of spin structure is energetically preferred for Fe3+ moments and occurs in almost all Fe3+O6 perovskite-type frameworks25,30,33,34,35. Furthermore, weak R–R interactions can result in AFM ordering in the R sublattice at a low temperature (<10 K). One of the most interesting phenomena in RFeO3 is the SR induced by temperature and/or magnetic field, where the alignment of Fe3+ spin moments changes from one crystal direction to another. As the Fe spin reorientation is regarded to be closely related to anisotropic R–Fe magnetic exchange interactions, the current PbFeO3 with nonmagnetic A-site Pb ions may provide a new avenue for understanding the distinct underlying mechanism of SR transition.
In this study, comprehensive investigations based on synchrotron X-ray diffraction (SXRD), neutron powder diffraction (NPD), electron diffraction (ED), hard X-ray photoemission spectroscopy (HAXPES), soft X-ray absorption spectroscopy (XAS), Mössbauer spectroscopy, characterization of magnetic and electrical properties, and density functional theory (DFT) calculations were performed to examine the structure, charge state, and magnetic properties of PbFeO3. We discovered that PbFeO3 crystallized into a Cmcm space group with a new charge ordering of a unique -A-B-B- type, whereby in the direction of the layers stacking, one layer composed by Pb2+ is interleaved by two layers built up from a mixture of Pb4+ and Pb2+ in a 3:1 ratio. Moreover, a high-temperature weak FM transition and subsequently a spin reorientation transition occurred at ~600 and 418 K, respectively. Related mechanisms are proposed to explain the SR of PbFeO3.
Results
Crystal structure of PbFeO3
According to a previous report22, the sample quality of PbFeO3 is extremely sensitive to the synthesis conditions. We carefully optimized the synthesis pressure and temperature, and finally obtained a nearly single phase by synthesizing at 8 GPa and 1423 K for 30 min. The SXRD pattern of PbFeO3 is shown in Fig. 1a. Except for a tiny Fe2O3 impurity phase, all the diffraction peaks can be indexed based on an unusual 2ap × 6ap × 2ap orthorhombic perovskite super unit cell, where ap refers to the pseudolattice parameter of a cubic ABO3 perovskite subcell. To identify the reflection conditions as well as possible space groups, we performed an ED experiment. ED patterns indicate that PbFeO3 has an orthorhombic cell with a 2ap × 6ap × 2ap superlattice (see Fig. 1b and Supplementary Fig. S1). The reflection conditions are 0kl (k = 2n), h0l (h, l = 2n), hk0 (h + k = 2n), h00 (h = 2n), 0k0 (k = 2n), and 00 l (l = 2n), which are consistent with the space groups of Cmc21 (No. 36), C2cm (No. 40), and Cmcm (No. 63). We determined the final crystal structure using a Rietveld refinement of the SXRD pattern based on the primary structural models suggested by the ED. In comparison, we discovered that the most reliable structure model was Cmcm (see Fig. 1a), as reducing the symmetry cannot improve the quality of the fit any more. Since X-rays are not sensitive to the light element of oxygen, we perform NPD to precisely determine the oxygen position. The Rietveld refinement results of NPD data collected at 300 K are illustrated in Fig. 1c. We observed a G-type AFM structure at 300 K, which will be discussed later. In the crystal symmetry of Cmcm, Pb atoms occupied six special Wyckoff positions 4c (0, y, 0.25), Fe atoms occupied two special positions 8d (0.25, 0.25, 0) and 16h (x, y, z), whereas O atoms occupied one 16 h (x, y, z) site, three different 8f (0, y, z) sites, three different 8g (x, y, 0.25) sites, and one 8e (x, 0, 0) site. Table 1 and Supplementary Table S1 list the refined structure parameters, including the detailed lattice constants and atomic positions. The bond lengths and the results of bond valence sum (BVS) calculations for the NPD and SXRD data are listed in Table 2 and Supplementary Table S2, respectively. The obtained lattice parameters were a = 7.89945(15) Å, b = 23.46820(45) Å, and c = 7.73406(15) Å (corresponding to perovskite cell parameters ap of 3.94973, 3.91137, and 3.86703 Å along with directions a, b, and c, respectively). As shown in Fig. 1d, each Fe atom is coordinated by six ligand O atoms with Fe–O distances varying from 1.95 to 2.14 Å, forming a perovskite-type FeO6 octahedron framework (see Fig. 1d). Moreover, the structure refinement results demonstrate that the Pb layers perpendicular to the b axis show a –shorter–longer–shorter– stacking with an interlayer distance of 3.8078 Å, 4.1186 Å, and 3.8078 Å. Figure 1e shows the high-angle annular dark-field (HAADF) image of PbFeO3. It is clear that the distances for the bright spots, which are the locations of the Pb layers, show a modulation similar to the –shorter–longer–shorter– pattern mentioned above. This result further confirms the reliability of the crystal structure determined from the ED, SXRD, and NPD data.

a Rietveld refinement for SXRD pattern recorded at room temperature for PbFeO3. Observed (black circles), calculated (red line), and difference (gray line) values are shown. The allowed Bragg positions in Cmcm symmetry are indicated by ticks (dark cyan). b ED patterns along [010] pseudocubic zone axis at RT. c Rieveld refinement for NPD pattern at 300 K for PbFeO3. Observed (black circles), calculated (red line), and difference (blue) values are shown. Bragg positions of PbFeO3 are indicated by ticks (dark cyan), which correspond to the allowed nuclear (magenta) and magnetic (dark cyan) Bragg peaks of PbFeO3; and allowed nuclear (navy) and magnetic (violet) Bragg reflections of the impurity phase Fe2O3 (~5 wt%). d Crystal structure of PbFeO3. e Left part: illustration of Pb modulations; Right part: HAADF image along [001] pseudocubic zone axis of PbFeO3. Distances for the bright spots, which are the locations of Pb, indicate a modulation with a shorter–longer–shorter pattern. f Sketch of unique -A-B-B- type of charge ordering of PbFeO3 composed of two types of differently charged layers.
Charge-order structure of PbFeO3
The BVS calculations (see Table 2) reveal that the lead atoms, located at the three positions (Pb1, Pb2, and Pb3), show a valence state close to +2, but the other three (Pb4, Pb5, and Pb6) provide valence sums corresponding to a valence state close to +4. However, Fe always shows a +3 state regardless of its atomic positions. These features indicate that the valence distribution of PbFeO3 should be Pb2+0.5Pb4+0.5Fe3+O3. Based on the charge distribution in the ac plane with different y-axis values, Pb2+ and Pb4+ show a unique long-range charge ordering, as shown in Fig. 1f. Specifically, there exist two types of layers with respect to the estimated valence states of Pb. Within each unit cell repetition period along the crystallographic b axis, two identically charged layers consisting of Pb1 and Pb2 atoms characterized by a Pb2+ charge state are located at y ≈ 0 and 1/2. In addition, four differently charged layers, each built up by three Pb4+ atoms: Pb4, Pb5, and Pb6 and just one Pb2+ charged Pb3 atom located at y ≈1/6, 1/3, 2/3, and 5/6, respectively. This 3:1 ratio of the Pb4+ and Pb2+ ions provided an average oxidation state +3.5 for Pb atoms in these layers. Therefore, a peculiar -A-B-B- charge ordering, i.e., one layer with exclusive Pb2+ oxidation state (A) is followed by two layers (B) with an average oxidation state of Pb3.5+, was realized in PbFeO3. This unprecedented charge ordering renders PbFeO3 unique in all the reported charge-order perovskite oxides36,37, resulting in the formation of a 2ap × 6ap × 2ap perovskite supercell. Neither the melting of Pb charge ordering nor the valence and spin-state changes of Fe were observed when the sample was heated to temperatures near its decomposition temperature of approximately 730 K (Supplementary Fig. S2) or cooled to a low temperature down to 10 K, as confirmed by temperature-dependent XAS (see Supplementary Fig. S3). Other charge-order compounds in AMO3 perovskite systems, such as BiNiO3, PbCrO3, and PbCoO3 as mentioned above16,18,19, remained unchanged upon cooling or heating at ambient pressure; however, they exhibited pressure-induced intersite charge transfer transitions. Moreover, BiNiO3 with R, Pb, and Sb substitution for Bi or Fe substitution for Ni indicated a temperature-induced charge transfer accompanied by negative thermal expansion15,38,39,40,41,42,43. The investigation of current PbFeO3 under pressure and doping effects will be performed in our future study.
The distinctive valence distribution of PbFeO3 can be further confirmed by Fe L2,3-edges XAS, Pb L3-edge XAS, and Pb-4f XPS. The sharp multiple spectral features at the 3d transition element L2,3-edges are extremely sensitive to the valence state44,45 and local environment46,47. The Fe L2,3-edge results of PbFeO3 are shown in Fig. 2a, together with Fe2O3 as a Fe3+ reference. Both of them show similar peak energy positions and spectral features, indicating the formation of the Fe3+ (3d5) state in PbFeO3, consistent with the BVS result. Considering that the Pb L3-edge spectral profile obtained from the high-resolution partial fluorescence yield (PFY) model is an effective method to identify the valence state of Pb, we performed Pb L3-edge XAS using the PFY model20,44,48. As shown in Fig. 2b, a lower energy shoulder appeared at ~13,030 eV, which is assigned to the dipole-allowed transition from the 2p3/2 core level to the unoccupied 6s state. Typically, this pre-edge can be observed in Pb4+ ions with two 6s holes but is absent in Pb2+ ions with fully occupied 6s states49. Therefore, we can confirm that a Pb4+ component exists in PbFeO3. Meanwhile, we observed that the pre-edge peak height of PbFeO3 was lower than that of PbNiO3, which possesses a pure Pb4+ state. After subtracting the background originating from the edge jump and the Pb 5d states, the spectral intensity of the pre-edge peak (see the green curve in Fig. 2b) in PbFeO3 is approximately half of that in PbNiO3, indicating that the average valence state of Pb is +3. Moreover, the valence state of Pb can be further confirmed by HAXPES measurements. Figure 2c shows the HAXPES results for PbFeO3 and other PbMO3 compounds with M = Ti, Cr, Co, and Ni used as standard references. Two components appeared in both the Pb 4f7/2 and Pb 4f5/2 peaks for PbCrO3 (Pb2+0.5Pb4+0.5CrO3), PbCoO3 (Pb2+0.25Pb4+0.75CoO3), and PbFeO3. Each peak can be deconvoluted into two Gaussians, as reported previously18,19. The 6s0 electronic configuration in Pb resulted in lower binding energy than that of 6s2 because of a strong screening effect19; hence, the components at lower binding energies are attributable to Pb4+ ions. The peak energies of PbFeO3 were close to those of Pb2+0.5Pb4+0.5Cr3+O3, indicating the coexistence of Pb2+ and Pb4+ ions. We estimated the fractions of Pb2+ and Pb4+ from the area ratios of Pb2+ and Pb4+ using PbCrO3 data as the standard for Pb2+0.5Pb4+0.5. As shown in Fig. 2d and Supplementary Table S3, the fitting demonstrates a nearly equal ratio between Pb2+ and Pb4+ in PbFeO3. Based on the BVS, XAS, and HAXPES results, we confirm that the charge configuration of PbFeO3 is Pb2+0.5Pb4+0.5Fe3+O3 with novel ordered Pb2+ and Pb4+ distribution.

a XAS of Fe L2,3-edge of PbFeO3, and reference Fe2O3; b Pb L3-edge XANES of PbFeO3 (black circles) and PbNiO3 (blue curve) for comparison. The green curve in (b) shows the pre-edge peak 6s state of PbFeO3 after subtracting the background (dashed gray curve). c Pb-4f HAXPES results for PbTiO3, PbCrO3, PbFeO3, PbCoO3, and PbNiO3 at RT. Predominant of Pb4+ and Pb2+ are evident in the spectrum for PbFeO3. d Average Pb valence state calculated from area ratios of Pb2+ and Pb4+ components. PbTiO3, PbCrO3, and PbNiO3 are standard for Pb2+, Pb3+ (Pb2+0.5Pb4+0.5), and Pb4+, respectively.
Electrical transport property of PbFeO3
The electrical properties of PbFeO3 are shown in Fig. 3a. A strong insulating feature was observed below 350 K (resistivity ρ > 107 Ω ∙ cm), and a temperature-dependent behavior appeared with the values reduced by four to five orders of magnitude in the temperature range of 350–630 K. The data can be described by a three-dimensional (3D) variable-range-hopping (VRH) regime with the function ρ(T) = ρ0exp(T0/T)1/4, where T0 = (33/π)/kBξ2D(EF), kB is the Boltzmann constant, D(EF) the density of states (DOS) at the Fermi level, and ξ the localization length of a wave function for localized electrons [exp(-r/ξ)]50,51. The inset of Fig. 3a shows the plot of lnρ vs. T−1/4 for the resistivity data of PbFeO3, and the straight line of the plot is the fitting result obtained using the VRH model in the temperature range of 520–630 K. The fitting results yielded T0 = 5414 K, and ρ0 = 2.0 × 10−4 Ω ∙ cm. This implies that in this system the electrical transport is dominated by the hopping of localized charge carriers. This feature is consistent with the intrinsic insulating behavior of the Fe3+O6 perovskite framework, such as LaFeO3 and BiFeO3, owing to the strong electronic correlation effects47,52. Moreover, the valence band-edge positions obtained from HAXPES indicate that the valence bond is away from the Fermi energy and the binding energy is ~1 eV (see Fig. 3b), consistent with the insulating nature of PbFeO3. It is noteworthy that no electrical anomaly was observed at the magnetic transition temperatures as shown later.

a Temperature dependence of resistivity measured above RT for PbFeO3. Inset shows 3D VRH model fitting between 520 and 630 K. b HAXPES spectrum near valence band for PbFeO3. The energy bandgap is evaluated to be ~1 eV.
Electronic structures and charge-order analysis by DFT
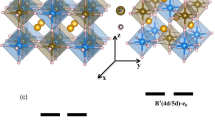
The crystal and electronic structures of the extraordinary charge-ordered states of Pb2+ and Pb4+ were further investigated by employing the DFT calculations for PbFeO3. To gain a deeper insight, we conducted a comparative analysis of the experimentally observed charge-order structure and other typically known A-site charge-ordered phases. We optimized the crystal structure of various Pb2+/Pb4+ 1:1 charge-ordered phases (see Supplementary Fig. S4) considering several collinear FM and AFM arrangements of Fe spins. The corresponding results are summarized in Fig. 4. We discovered that the experimentally determined Cmcm Pb2+/Pb4+ charge-order phase corresponded to the lowest energy structure and was 56 meV/f.u., which was lower compared with the nonpolar columnar-ordered Pmmn structure (see Figs. 4a and 4b). All the polar phases (Cm, Pc, and Pm shown in Supplementary Fig. S4), which were considered in this study, had higher energies compared with their nonpolar counterparts. The existence of two different types of lead ions in the Cmcm structure was reflected in our analysis of the calculated density of states (see Fig. 4c). The conduction bands within the energy range of the Fermi level to 2 eV comprised Pb 6s orbitals that were strongly hybridized with O 2p orbitals. This implies that within the Cmcm structure, half of the Pb ions were highly valent (+4), similar to several other Pb-based materials16,53. The other half of the Pb ions formed a 6s2 electronic structure, which corresponds to the +2 valence state. Consequently, the Fe ions exhibited half-filled t2g3eg2 electronic structures, thereby resulting in a strong AFM interaction between the Fe spins, which stabilized the G-type AFM structure (see Fig. 4b). The G-type AFM phase was insulating and indicated a bandgap of ~0.8 eV, agreeing with the experimentally determined value.

a Description of Pb2+/Pb4+-ordered optimized Cmcm structure. b Calculated energies of various Pb2+/Pb4+-ordered structures considering four collinear arrangements between Fe spins. Relative energy (\({\Delta}E\)) is shown with respect to undistorted ferromagnetic phase. c Calculated density of states of lowest energy charge-ordered Cmcm structure in G-type antiferromagnetic phase.
Various lattice distortions contributed to the formation of the charge-order Cmcm structure. As shown in Fig. 4a, the Pb charge-ordered patterns lead to the formation of layered-like components (denoted as L1 and L2) made up of Fe2 ions. A layer of Fe1 ions remains sandwiched between every such L1 and L2 layers. We can categorize these distortions that contribute to the nonpolar Cmcm structure as follows. First, based on the oxygen octahedral rotations around the crystallographic axes, we have identified, (a) in-phase oxygen octahedral rotations around the crystallographic c axis, which correspond to the \(M_3^ +\) symmetry of the undistorted and charge disordered cubic \(Pm\overline 3 m\) structure, (b) out-of-phase oxygen octahedral rotations around the b axis (corresponding to the \(R_4^ +\) symmetry of the cubic \(Pm\overline 3 m\) structure), and (c) in-phase oxygen octahedral rotations around the a axis around the Fe1 ions (corresponding to the Z4 [1/3, 1/2, 0] symmetry of the cubic \(Pm\overline 3 m\) structure), which can transform the cubic \(Pm\overline 3 m\) structure to \(Pmma\) structure. Next, we categorize the distortions that have been observed to occur exclusively around the Fe2 ions in the layered-like components (L1 and L2) of the charge-order Cmcm structure in response to the non-trivial arrangement of the Pb2+ and Pb4+ cations (as illustrated in Supplementary Fig. S9). This category of distortions can be further classified as, (a) strong anti-ferro-distortive (AFD) movements of the oxygen and Fe2 ions in response to the difference in the charge states of the PbO layers along the b axis which correspond to the DT1 [0, 1/3, 0] symmetry (transform \(Pm\overline 3 m \to P4/mmm\)), (b) weak buckling in the O-Fe2-O connections along with the a and c axes (which follows the DT2 [0, 1/3, 0] symmetry transforming \(Pm\overline 3 m \to Pmmm\)), (c) AFD movements of the oxygens and Fe2 ions along the a and c axes which transform as Z4 [1/6, 1/2, 0] (transform \(Pm\overline 3 m \to Cmmm\), hereafter denoted as Z4-I) and Z4 [1/3, 1/2, 0] (transform \(Pm\overline 3 m \to Pmma\), hereafter denoted as Z4-II) symmetries. The third category of distortions is the weak AFD movements of the Pb and oxygens ions following \(R_5^ +\), \(X_5^ +\), and \(T3\) symmetries. The theoretically determined Fe–O bond lengths, as listed in Fig. 4a, agree well with their experimentally determined values (see Table 2). Furthermore, similar AFD phenomena were also exhibited by other layered and layered-like charge-order structures that were taken into consideration (Supplementary Fig. S4) in this study.
Magnetic transitions and spin structures of PbFeO3
The zero-field cooling (ZFC) magnetic susceptibility curve is illustrated in Fig. 5a. As the temperature decreased to TN ≈600 K, the magnetic susceptibility increased significantly, indicating a long-range magnetic transition. Upon further cooling to TSR ≈418 K, an abrupt decrease occurred, suggesting the presence of an SR transition. Based on the isothermal magnetization curves measured at different temperatures (see Fig. 5b), a linear magnetization behavior can be observed at 630 K, in accordance with paramagnetism. However, between TN and TSR, considerable magnetic hysteresis occurred. For example, at 450 K, the observed coercive field was ~0.5 T. Moreover, there was a small amount of residual magnetization (0.01 μB/Fe). These features indicate that a weak FM transition originating from the canted AFM spins would occur at TN. Below 300 K, the magnetization almost resumed to a linear behavior with negligible coercive field and residual magnetization, illustrating that the AFM structure did not comprise a weak ferromagnetic component. These behaviors are similar to those observed in RFeO3 perovskite oxides with a spin reorientation transition33,34,35. The Mössbauer spectrum at 300 K is shown in Supplementary Fig. S5. The spectrum can be fitted with three sets of magnetically split sextets with area ratios of 60, 34, and 6%. These components are attributed to Fe at the 16 h site, 8d site, and at α-Fe2O3 impurities. The values of the isomer shift and magnetic hyperfine field for the 16 h component (0.42 mm/s, 48.8 T) and those for the 8d component (0.38 mm/s, 42.0 T) indicate that Fe in PbFeO3 is trivalent and in a magnetically ordered state at RT.

a Temperature dependence of magnetic susceptibility of PbFeO3 measured at 0.01 T. b Isothermal magnetization loops measured at various temperatures. c Magnetic structures of PbFeO3 between TSR and TN (Γ4) as well as below 300 K (Γ1). Red arrows depict Fe3+ moments. d Magnetic-phase diagram for PbFeO3. Red and blue spots denote magnetic moments determined from neutron diffraction. The error bars of magnetic moments are within the symbols. Green line provides visual guidance. CAFM collinear antiferromagnetism, WFM weak ferromagnetism, PM paramagnetism.
To further determine the spin ordering of PbFeO3, we performed temperature-dependent NPD measurements from 2 to 625 K, as shown in Supplementary Figs. S6, S7, and S8. Supplementary Fig. S6 shows the diffraction patterns together with the Rietveld refinement results at 100 and 475 K. The Rietveld analysis for magnetic diffraction revealed a commensurate magnetic order with wave vector k = (0, 0, 0). Furthermore, a canted G-type AFM structure described by Γ4 (Gx, Ay, Fz) (Bertaut’s notation54) with antiferromagnetically coupled spins along the a axis and an allowed net magnetization along the c axis was determined between TSR and TN. The weak FM component had likely contributed to the magnetic hysteresis observed in the M–H measurement at 450 K; however, it was extremely small to be determined by the refinement. When the temperature was below TSR, a collinear G-type AFM structure described by Γ1 (Ax, Gy, Cz) with spin along the b axis was also confirmed by the refinement, consistent with the linear feature of the magnetization shown in Fig. 5b. It is apparent that the symmetry of Γ1 (Ax, Gy, Cz) forbids a FM component, which is also consistent with the absence of magnetic hysteresis below TSR. Supplementary Fig. S7 shows the NPD fitting for several characteristic peaks with the spin direction along a axis (Γ4), b axis (Γ1), and c axis (Γ2) for comparison. It is evident that the Γ4 and Γ1 spin models can provide the most reliable fitting for the higher and lower temperature magnetic phases, respectively. Therefore, our studies confirmed a temperature-induced SR from Γ4 (Gx, Ay, Fz) to Γ1 (Ax, Gy, Cz) in PbFeO3. Figure 5c shows the two different types of spin configurations. The refined magnetic moments at T = 475 and 100 K in units of µB were Ma = 2.57 (2) and Mb = 3.78 (1), respectively. Figure 5d shows the magnetic-phase diagram of PbFeO3 based on the current results. With decreasing temperature, the compound changed from paramagnetic to canted AFM with a small amount of net FM moment at the critical temperature of TN ≈600 K. Upon further cooling to TSR ≈418 K, a continuous SR transition occurred, changing the weak FM phase to a collinear AFM phase. The evaluated temperature range for the SR was between 300 and 418 K. Temperature-dependent magnetic moments for various phases are also shown in the phase diagram. Based on the refinement, the magnetic moment at T = 2 K in units of µB was Mb = 3.8 (1), which was somewhat lower than the expected value (5 µB) for an Fe3+ ion probably due to the considerable covalence/hybridization effects between Fe and O atoms (see Fig. 5c).
Possible origins of spin reorientation in PbFeO3
In Fe-based perovskite oxides RFeO333,34,55, the anisotropic R–Fe magnetic interactions are generally identified as driving factors behind the SR transitions. In the PbFeO3 system under investigation, the nonmagnetic nature of the A-site Pb2+ and Pb4+ ions negates the possibility of magnetic interactions between A- and B-site cations. However, PbFeO3 still exhibits a SR transition at a much higher critical temperature than those observed in most RFeO3 perovskites. This indicates that distinct mechanisms of SR transition are operational in PbFeO3. We further explored this phenomenon by the application of first-principles electronic structure calculations and finite temperature Monte Carlo simulations. Our investigations are based on the spin Hamiltonian as described below Eq. (1),
Here, the first, second, and third terms denote the symmetric exchange interactions between individual Fe spins (\({\mathbf{S}}^1\) and \({\mathbf{S}}^2\) denote Fe1 and Fe2 spin, respectively). The longitudinal and transverse anisotropy for the Fe1 and Fe2 ions are represented as (\(D^1\) and \(E^1\)) and (\(D^2\) and \(E^2\)), respectively. The magnetic interaction strengths estimated on the basis of the crystal structure experimentally determined at 300 K (denoted as SExpt) and the first-principles optimized structure (denoted as SOpt) are listed in Supplementary Fig. S10. The interactions between Fe spins, corresponding to either of these structures, are both strong and antiferromagnetic in nature leading to G-type AFM order. We also found that, while the Fe1 ions energetically favor the orientation of the spins along the b axis, the Fe2 spins, in contrast, favor the a-axis spin orientation. Moreover, the magnetic anisotropy of the Fe1 magnetic sublattice is stronger than that of the Fe2 sublattice. Our results of total energy calculations identified the Gy phase with spins parallel to the b axis to be the lowest energy phase for both the crystal structures, with energy of ~0.020 meV/Fe lower than that of the Gx phase with spins along the a axis. This is consistent with experimental observations. The Monte Carlo simulated magnetic transition temperature is around ~582 K (see Supplementary Fig. S11), which is also in good agreement with the experimentally observed TN. Below 582 K, both Fe1 and Fe2 sublattices exhibit G-type AFM ordering with their spins oriented along the b axis (see Supplementary Fig. S11). The spin Hamiltonian premised on SOpt exhibits similar behavior (see Supplementary Fig. S11). Here, the anti-symmetric and anisotropic–symmetric exchange interactions between the Fe spins that lead to the spin canting were not taken into consideration, as these interactions, being weak for Fe3+ ions, are least likely to cause a spin orientation transition within a temperature interval of ~180 K.
The peculiar arrangement of the Pb ions leads to the creation of the two magnetic Fe1 and Fe2 sublattices with mutually competing magnetic anisotropic energies. We, therefore, explored the possible origin of SR transition through the nature of modulation of single-ion anisotropic energies. We calculated the total energies of the magnetic phases (Gx, Gy, and Gz) as functions of the modulation of the structural distortions mentioned earlier. Among the various distortions in SExpt, only the symmetric distortions which owe their origin to the special arrangements of Pb cations, and eventually are transformed following DT1, DT2, and Z4 symmetries (see Supplementary Fig. S9), were taken into consideration. Our results (as shown in Fig. 6) indicate that an increase in the DT1 distortion results in the relative shortening of the average Fe1–O bond length along the b axis, which in turn enhances the magnetic anisotropy energies of Fe1 ions, contributing to the enhancement of stability of the Gy phase. This stability of the Gy phase is maintained within a range of ~0.8 Å modulation of the amplitude of this distortion with respect to SExpt. An increase in the DT2 and Z4-I distortions, which modulates the Fe2–O–Fe2 bond lengths and angles in the ac plane, also increases the stability of the Gy phase. The decrease in DT2 and Z4-I distortions, however, gives rise to the magnetic-phase transition from Gy to Gx phase (as shown in Fig. 6b, c). The same phase transition (i.e., from Gy to Gx phase) is also observed by increasing the AFD that follows the Z4-II symmetry along the c axis (see Fig. 6d). Interestingly, a decrease in the amplitude of this distortion below ~0.4 Å indicates a transition from the Gy to Gz phase. Our results are summarized as follows. (1) The Gz phase, in most cases, is higher in energy in comparison to the Gx and Gy phases. (2) DT2 and Z4-I are the two distortions that have been identified to contribute weakly to the formation of the Cmcm structure and significantly in the process of magnetic-phase transitions. Notably, the transformation of these distortions from zero to any finite value has no bearing on the overall symmetry of the structure. (3) Modulation of these distortions with respect to temperature can result in the transition from Gx to Gy magnetic phase. Notably, there is an exact match of the results between total energy calculations and Monte Carlo simulations based on estimated parameters of the Spin Hamiltonian as a function of DT2 and Z4-I structural distortions (as presented in Supplementary Fig. S12).

Calculated relative energies of Gx, Gy, and Gz magnetic phases as functions of the modulation of the structural distortions corresponding to the DT1 (transforms \(Pm\overline 3 m \to P4/mmm\), a), DT2 (transforms \(Pm\overline 3 m \to Pmmm\), b), Z4 (transforms \(Pm\overline 3 m \to Cmmm\), c) and Z4 (transforms \(Pm\overline 3 m \to Pmma\), d) symmetries with respect to the crystal structure experimentally determined at 300 K (SExpt).
Discussion
In summary, the crystal structure and physical properties of PbFeO3 were investigated in detail through SXRD, ED, XAS, HAXPES, NPD, and theoretical calculations. It was discovered that PbFeO3 possessed an unusual 2ap × 6ap × 2ap orthorhombic superlattice with space group Cmcm. The BVS, XAS, HAXPES, and DFT calculation results revealed that the electronic configuration was Pb2+0.5Pb4+0.5Fe3+O3. A unique long-range charge ordering of the -A–B–B-type of the layers of Pb atoms with different oxidation states occurred. In the direction of the layers’ stacking, one layer with exclusively Pb2+ oxidation state (A) was followed by the two layers (B) with an average oxidation state of Pb3.5+, which in turn was a result of mixing 3×Pb4+ and 1×Pb2+ atoms in them. The single Pb2+ state formed two identically charge layers (A) at y ≈ 0 and 0.5, whereas the layers comprised of 3:1 ordered Pb4+ and Pb2+ atoms contributed to four other charge layers at the positions y ≈1/6, 1/3, 2/3, and 5/6. When the temperature decreased to 600 K, PbFeO3 experienced a canted AFM phase transition with a weak net FM moment. Further cooling induced a spin reorientation transition at ~418 K, thereby changing the canted AFM structure to a collinear one; the magnetic Fe moments were aligned along the a and b axes above and below the SR temperatures, respectively. The PbFeO3 system under investigation exhibits a unique phenomenon where two magnetic Fe1 and Fe2 sublattices are created as a result of the peculiar arrangement of Pb2+ and Pb4+ ions. The mutually competing magnetic anisotropic energies of these two sublattices is a plausible contributing factor for the spin reorientation in PbFeO3 at a higher critical temperature of 418 K. This is different from the case of RFeO3 perovskites where the presence of a magnetic rare-earth ion plays the most important role in the observed SR transitions. Since the magnetic sublattices owe their origin to a certain arrangement of Pb2+/Pb4+ ions, this may introduce a unique opportunity of inducing magnetic phase transition (\(M = 0 \leftrightarrow M \ne 0\), where \(M\) denotes net magnetization in the system) by driving a redistribution of Pb ions via an external electric field and/or strain. This work provides a new avenue for studying the charge ordering phase and distinctive spin orientation transition with potential applications in advanced spintronic devices due to the high transition temperature and possible tuning.
Methods
Materials synthesis
Polycrystalline samples of PbFeO3 were prepared using a high-pressure and high-temperature method. Starting materials of high purity (>99.9%) PbO, PbO2, and Fe2O3 with a 1:1:1 molar ratio were thoroughly mixed in an agate mortar within an argon-filled glovebox, and then sealed into gold capsules of diameter 3 mm and height 3.5 mm. The capsule was treated at 8 GPa and 1423 K for 30 min in a cubic-anvil-type high-pressure apparatus and quenched to room temperature prior to the slow release of pressure. The temperature window was narrow. Many impurities were introduced when the temperature increased or decreased by 50 K.
Characterizations
The SXRD data for PbFeO3 were collected using a large Debye–Scherrer camera installed at the BL02B2 beamlines of SPring-8 with wavelengths of 0.41965 Å. The structure was refined using the Rietveld method via the GSAS program56. Temperature-dependent NPD at high temperatures (>300 K) was measured at HB-2A at the High Flux Isotope Reactor of the Oak Ridge National Laboratory57 with a wavelength of 2.41 Å. NPD patterns were obtained at ≤300 K using a high-resolution diffractometer HRPT58 at the Swiss Spallation Neutron Source of the Paul Scherrer Institute with a wavelength of 1.886 Å, and a vanadium container measuring 6 mm in diameter was used. The NPD data were analyzed using the Rietveld package, Fullprof59. The ED patterns and high-angle annular dark-field (HAADF) images at room temperature (RT) were obtained using a JEOL JEM-ARM200F (scanning) transmission electron microscope. The field dependence of the isothermal magnetization (M) and temperature-dependent magnetic susceptibility (χ) below 400 K were measured using a Quantum Design superconducting quantum interference device magnetometer. High-temperature magnetic susceptibility data at 400–650 K were collected using a MicroSense vibrating sample magnetometer. The resistivity at high temperatures was measured using the standard four-probe method. The size of the sample was ~2 × 1 × 1 mm for the electrical measurements. The valence states of PbFeO3 were determined via soft X-ray absorption spectroscopy and hard X-ray absorption near-edge spectroscopy (XANES). The XAS of Fe-L2,3 was collected at beamline BL11A of the National Synchrotron Radiation Research Center in Taiwan using the total-electron-yield mode. The spectrum of the single-crystal Fe2O3 sample was measured simultaneously to serve as an absolute energy reference. The high-resolution partial fluorescence yield Pb-L3 XAS spectra were measured at Pb L1 emission line with an overall resolution of ∼2 eV at beamline ID 20 of the Synchrotron SOLEIL, France. HAXPES measurements for Pb 4f core levels and valence bands were performed at RT with E = 7930.1 eV to investigate the valence state using a hemispherical photoelectron analyzer (R4000, VG Scienta) installed at BL09XU of SPring-8. Powder samples of PbMO3 with M = Ti, Cr, Fe, and Ni were pasted onto a carbon tape. Carbon black powder was mixed with PbTiO3 before it was pasted onto a carbon tape to prevent charge-up due to the high insulation of the sample. The binding energy was calibrated using the Fermi edge of a gold film sample. Mössbauer spectroscopy was performed on a 57Fe-25%-enriched sample at RT using the conventional absorption method.
First-principles calculations
The crystal and electronic structures of various Pb2+/Pb4+ charge-ordered states were studied by employing the DFT+\(U\)60 approach using the projector-augmented plane-wave method61 as implemented in the VASP code62,63. The Perdew–Burke–Ernzerhof (PBE)64 exchange-correlation functional was used. We constructed the initial structure of various A-site charge-ordered phases by freezing the anti-ferrodistortive displacements of oxygen ions and the oxygen octahedra rotations (which consequently created an A-site oxygen polyhedral with variation in size and oxygen coordination number) within the cubic \(Pm\overline 3 m\) structure, which comprised eight chemical formula unit cell sizes. The corresponding structures were optimized considering several collinear arrangements of the Fe spins. We set the screened Coulomb interaction (\(U\)) to 4.5 eV and 1.0 eV for Hund’s coupling (\(J_H\)) on the Fe site. A kinetic-energy cutoff value of 500 eV and a Hellman–Feynman force convergence criterion of 0.01 eV Å−1 were used. A k-point mesh was considered based on the crystal symmetry. We further cross-checked the electronic structures of the charge-ordered states using the linearized augmented plane-wave (LAPW) method as implemented in the Wien2k code65 using the same values of \(U\) and \(J_H\) at the Fe site and k-point mesh, as used in the VASP code. We used the −7.5 Ry energy cutoff to separate the valence states from the core states. We calculated values of the symmetric exchange interactions between Fe spins and single-ion magnetic anisotropy energies of Fe1 (\(E^1\) and \(D^1\)) and Fe2 (\(E^2\) and \(D^2\)) ions by employing the linearized augmented plane-wave (LAPW) method. We estimated SIA parameters associated to Fe1 and Fe2 magnetic sublattices by modulating the lattice distortions which owe their origin to the special Pb2+/Pb4+ ordered pattern. The Monte Carlo simulations based on the first-principles parameterized spin Hamiltonian were conducted considering 8 × 4 × 8 (6144 magnetic ions) supercell size of the \(Cmcm\) structure and maximum sample number of 109.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Keimer, B., Kivelson, S. A., Norman, M. R., Uchida, S. & Zaanen, J. From quantum matter to high-temperature superconductivity in copper oxides. Nature 518, 179–186 (2015).
Dagotto, E., Hotta, T. & Moreo, A. Colossal magnetoresistant materials: the key role of phase separation. Phys. Rep. 344, 1–153 (2001).
Salamon, M. B. & Jaime, M. The physics of manganites: structure and transport. Rev. Mod. Phys. 73, 583–628 (2001).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Tokura, Y., Seki, S. & Nagaosa, N. Multiferroics of spin origin. Rep. Prog. Phys. 77, 076501 (2014).
Wang, X. et al. Observation of magnetoelectric multiferroicity in a cubic perovskite system: LaMn3Cr4O12. Phys. Rev. Lett. 115, 087601 (2015).
Zhou, L. et al. Realization of large electric polarization and strong magnetoelectric coupling in BiMn3Cr4O12. Adv. Mater. 29, 1703435 (2017).
Seo, J. S. et al. A homochiral metal-organic porous material for enantioselective separation and catalysis. Nature 404, 982–986 (2000).
Niitaka, S. et al. Crystal structure and dielectric and magnetic properties of BiCrO3 as a ferroelectromagnet. Solid State Ion-. 172, 557–559 (2004).
Naka, M., Seo, H. & Motome, Y. Theory of valence transition in BiNiO3. Phys. Rev. Lett. 116, 056402 (2016).
Belik, A. A. et al. Neutron powder diffraction study on the crystal and magnetic structures of BiCoO3. Chem. Mater. 18, 798–803 (2006).
Harrison, W. A. Valence-skipping compounds as positive-U electronic systems. Phys. Rev. B 74, 245128 (2006).
Oka, K. et al. Pressure-induced spin-state transition in BiCoO3. J. Am. Chem. Soc. 132, 9438–9443 (2010).
Ishiwata, S. et al. High pressure synthesis, crystal structure and physical properties of a new Ni(II) perovskite BiNiO3. J. Mater. Chem. 12, 3733–3737 (2002).
Azuma, M. et al. Colossal negative thermal expansion in BiNiO3 induced by intermetallic charge transfer. Nat. Commun. 2, 347 (2011).
Azuma, M. et al. Pressure-induced intermetallic valence transition in BiNiO3. J. Am. Chem. Soc. 129, 14433–14436 (2007).
Belik, A. A., Azuma, M., Saito, T., Shimakawa, Y. & Takano, M. Crystallographic features and tetragonal phase stability of PbVO3, a new member of PbTiO3 family. Chem. Mater. 17, 269–273 (2005).
Yu, R. et al. Melting of Pb Charge glass and simultaneous Pb-Cr charge transfer in PbCrO3 as the origin of volume collapse. J. Am. Chem. Soc. 137, 12719–12728 (2015).
Sakai, Y. et al. A-site and B-site charge orderings in an s-d level controlled perovskite oxide PbCoO3. J. Am. Chem. Soc. 139, 4574–4581 (2017).
Liu, Z. et al. Sequential spin state transition and intermetallic charge transfer in PbCoO3. J. Am. Chem. Soc. 142, 5731–5741 (2020).
Inaguma, Y. et al. Synthesis, structural transformation, thermal stability, valence state, and magnetic and electronic properties of PbNiO3 with perovskite- and LiNbO3-type structures. J. Am. Chem. Soc. 133, 16920–16929 (2011).
Tsuchiya, T. et al. High-pressure synthesis of a novel PbFeO3. Mater. Res. Soc. Symp. Proc. 988, QQ09–QQ16 (2006).
Tokunaga, Y. et al. Composite domain walls in a multiferroic perovskite ferrite. Nat. Mater. 8, 558–562 (2009).
Tokunaga, Y., Iguchi, S., Arima, T. & Tokura, Y. Magnetic-field-induced ferroelectric state in DyFeO3. Phys. Rev. Lett. 101, 097205 (2008).
Tokunaga, Y., Taguchi, Y., Arima, T. & Tokura, Y. Magnetic biasing of a ferroelectric hysteresis loop in a multiferroic orthoferrite. Phys. Rev. Lett. 112, 037203 (2014).
Kimel, A. V., Kirilyuk, A., Tsvetkov, A., Pisarev, R. V. & Rasing, T. Laser-induced ultrafast spin reorientation in the antiferromagnet TmFeO3. Nature 429, 850–853 (2004).
de Jong, J. A., Kimel, A. V., Pisarev, R. V., Kirilyuk, A. & Rasing, T. Laser-induced ultrafast spin dynamics in ErFeO3. Phys. Rev. B 84, 104421 (2011).
Kimel, A. V. et al. Ultrafast non-thermal control of magnetization by instantaneous photomagnetic pulses. Nature 435, 655–657 (2005).
Yuan, S. J. et al. Spin switching and magnetization reversal in single-crystal NdFeO3. Phys. Rev. B 87, 184405 (2013).
Tsymbal, L. T. et al. Magnetic and structural properties of spin-reorientation transitions in orthoferrites. J. Appl. Phys. 101, 123919 (2007).
Hahn, S. E. et al. Inelastic neutron scattering studies of YFeO3. Phys. Rev. B 89, 014420 (2014).
Nikitin, S. E. et al. Decoupled spin dynamics in the rare-earth orthoferrite YbFeO3: evolution of magnetic excitations through the spin-reorientation transition. Phys. Rev. B 98, 064424 (2018).
White, R. L. Review of recent work on the magnetic and spectroscopic properties of the rare-earth orthoferrites. J. Appl. Phys. 40, 1061–1069 (1969).
Li, E. et al. Spin switching in single crystal PrFeO3 and spin configuration diagram of rare earth orthoferrites. J. Alloy. Compd. 811, 152043 (2019).
Zhao, H. J., Iniguez, J., Chen, X. M. & Bellaiche, L. Origin of the magnetization and compensation temperature in rare-earth orthoferrites and orthochromates. Phys. Rev. B 93, 014417 (2016).
Wollan, E. O. & Koehler, W. C. Neutron diffraction study of the magnetic properties of the series of perovskite-type compounds [(1-x)La, xCa]MnO3. Phys. Rev. 100, 545–563 (1955).
Azuma, M. et al. Systematic charge distribution changes in Bi- and Pb-3d transition metal perovskites. Dalton Trans. 47, 1371–1377 (2018).
Oka, K. et al. Tuning negative thermal expansion in Bi1-xLnxNiO3 (Ln = La, Nd, Eu, Dy). Appl. Phys. Lett. 103, 061909 (2013).
Nakano, K. et al. Glassy distribution of Bi3+/Bi5+ in Bi1-xPbxNiO3 and negative thermal expansion induced by intermetallic charge transfer. Chem. Mater. 28, 6062–6067 (2016).
Sakai, Y. et al. Polar-nonpolar phase transition accompanied by negative thermal expansion in perovskite-type Bi1-xPbxNiO3. Chem. Mater. 31, 4748–4758 (2019).
Nishikubo, T. et al. Optimized negative thermal expansion induced by gradual intermetallic charge transfer in Bi1-xSbxNiO3. Appl. Phys. Express 11, 061102 (2018).
Nabetani, K. et al. Suppression of temperature hysteresis in negative thermal expansion compound BiNi1-xFexO3 and zero-thermal expansion composite. Appl. Phys. Lett. 106, 061912 (2015).
Nishikubo, T. et al. Enhanced negative thermal expansion induced by simultaneous charge transfer and polar-nonpolar transitions. J. Am. Chem. Soc. 141, 19397–19403 (2019).
Burnus, T. et al. X-ray absorption and X-ray magnetic dichroism study on Ca3CoRhO6 and Ca3FeRhO6. Phys. Rev. B 77, 205111 (2008).
Chang, C. F. et al. Dynamic atomic reconstruction: how Fe3O4 thin films evade polar catastrophe for epitaxy. Phys. Rev. X 6, 041011 (2016).
Hollmann, N. et al. Electronic and magnetic properties of the kagome systems YBaCo4O7 and YBaCo3MO7 (M=Al, Fe). Phys. Rev. B 80, 085111 (2009).
Kuo, C. Y. et al. k=0 magnetic structure and absence of ferroelectricity in SmFeO3. Phys. Rev. Lett. 113, 217203 (2014).
Chen, C. T. et al. Electronic states in La2-xSrxCuO4+δ probed by soft-X-ray absorption. Phys. Rev. Lett. 66, 104–107 (1991).
Chen, K. et al. Valence state of Pb in transition metal perovskites PbTMO3 (TM=Ti, Ni) determined from X-ray absorption near-edge spectroscopy. Phys. Status Solidi B 255, 1800014 (2018).
Mott, N. F. & Davis, E. A. Electronic Processes in Non-crystalline Materials, 2nd edn. (Clarendon, 1979).
Mott, N. F. Conduction in Non-crystalline Materials (Clarendon, 1987).
Wang, J. et al. Epitaxial BiFeO3 multiferroic thin film heterostructures. Science 299, 1719–1722 (2003).
Payne, D. J. et al. Nature of electronic states at the Fermi level of metallic beta-PbO2 revealed by hard X-ray photoemission spectroscopy. Phys. Rev. B 75, 153102 (2007).
Bertaut, E. F. in Magnetism III. (eds Rado, G. T. & Suhl, H.) 149–209 (Academic Press Inc., 1963).
Cao, S. et al. Tuning the weak ferromagnetic states in dysprosium orthoferrite. Sci. Rep. 6, 37529 (2016).
Larson, A. C. & Dreele, R. B. V. General Structure Analysis System (GSAS). Los Alamos National Laboratory Report LAUR, 86–784 (2004).
Calder, S. et al. A suite-level review of the neutron powder diffraction instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum. 89, 092701 (2018).
Fischer, P. et al. High-resolution powder diffractometer HRPT for thermal neutrons at SINQ. Phys. B 276-278, 146–147 (2000).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B 192, 55–69 (1993).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Blochl, P. E. Projector augumented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. Ab-Initio molecular-dynamics for liquid-metals. J. Non-Cryst. Solids 193, 222–229 (1995).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blaha, P. et al. WIEN2k: an APW+lo program for calculating the properties of solids. J. Chem. Phys. 152, 074101 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant nos. 11934017, 51772324, 11921004, 11904392), the National Key R&D Program of China (grant nos. 2018YFE0103200, 2018YFA0305700), the Beijing Nature Science Foundation (grant nos. Z200007, 2202059), the Chinese Academy of Sciences (grant nos. XDB33010200, QYZDB–SSW–SLH013), and Grants-in-Aid for Scientific Research, JP18H05208, JP19K05246, and JP19H05625 from the Japan Society for the Promotion of Science (JSPS). The theoretical investigation was supported by the Grants-in-Aid for Scientific Research 19K05246 from the JSPS and TSUBAME supercomputing facility. The synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (2018A1667, 2018B1636, and 2019B1896). This work is partly based on experiments carried out at the Swiss Spallation Neutron Source SINQ (grant no. 200021_165855), Paul Scherrer Institute, Villigen, Switzerland. We acknowledge the support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials. This research used resources at the High Flux Isotope Reactor, a DOE Office of Science User Facility operated by ORNL. The Mössbauer spectroscopic measurement was performed at Nagoya Institute of Technology under the Nanotechnology Platform Program of MEXT, Japan.
Author information
Authors and Affiliations
Contributions
Y.W.L., R.Y., and M.A. conceived the study; X.Y., J.Z., L.C., and C.J. prepared the sample, X.Y., Z.L., and L.Z. performed resistance measurements; D.S., J.Y., and M.K. measured LT neutron diffraction; X.W., Z.H., H.L., C.T.C., C.S., A.E., and L.T. measured XES and XAS; Y.S., T.N., and M.A. measured SXRD and HAXPES; H.C. and S.C. measured HT-NPD. K.M. measured Mössbauer spectrum. S.W. and H.D. performed DFT calculations. C.D., H.H., X.Y., and J.Z. analyzed crystal structure. H.H. performed ED measurements. All the authors discussed the results; Y.W.L., X.Y., R.Y., D.S., H.D., and M.A. wrote the paper with comments from all the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, X., Zhao, J., Das, H. et al. Observation of novel charge ordering and spin reorientation in perovskite oxide PbFeO3. Nat Commun 12, 1917 (2021). https://doi.org/10.1038/s41467-021-22064-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-22064-9
This article is cited by
-
Elastic, optoelectronic and photocatalytic properties of semiconducting CsNbO3: first principles insights
Scientific Reports (2023)
-
Influence of La3+ substitution on the structural, magnetic and electrical characteristics of Sr2FeTiO6 double perovskites
Journal of Materials Science: Materials in Electronics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
