
Abstract
Haematopoietic stem cells (HSCs) are characterized by their self-renewal potential associated to dormancy. Here we identify the cell surface receptor neogenin-1 as specifically expressed in dormant HSCs. Loss of neogenin-1 initially leads to increased HSC expansion but subsequently to loss of self-renewal and premature exhaustion in vivo. Its ligand netrin-1 induces Egr1 expression and maintains quiescence and function of cultured HSCs in a Neo1 dependent manner. Produced by arteriolar endothelial and periarteriolar stromal cells, conditional netrin-1 deletion in the bone marrow niche reduces HSC numbers, quiescence and self-renewal, while overexpression increases quiescence in vivo. Ageing associated bone marrow remodelling leads to the decline of netrin-1 expression in niches and a compensatory but reversible upregulation of neogenin-1 on HSCs. Our study suggests that niche produced netrin-1 preserves HSC quiescence and self-renewal via neogenin-1 function. Decline of netrin-1 production during ageing leads to the gradual decrease of Neo1 mediated HSC self-renewal.
Similar content being viewed by others

Introduction
Haematopoietic stem cells (HSCs) are highly quiescent and give rise to cycling multipotent progenitors (MPPs), which are in turn responsible for maintaining steady-state hematopoiesis1,2,3,4,5. Upon transplantation, HSCs harbour multilineage and serial long-term engraftment potential6,7,8,9. The CD34− HSC compartment is heterogeneous and consists of both dormant HSCs (dHSCs) and active HSCs (aHSCs) with dHSCs showing superior serial engraftment potential10,11. dHSCs can be identified via label retention approaches10,11,12,13 or by employing Gprc5c-GFP reporter mice11. All dHSCs reside in a transcriptionally and metabolically rather inactive state and rest in the G0 cell cycle phase.
Upon ageing the number of immunophenotypic HSCs increases, but their self-renewal capability diminishes and a myeloid differentiation bias emerges14,15,16,17,18,19. Various HSC intrinsic hallmarks of ageing, such as the disruption of cellular polarity, and epigenetic instability have been identified20,21,22. Concomitantly, it has become clear that the bone marrow (BM) microenvironment undergoes remodelling upon ageing and contributes to functional decline of HSCs23,24,25. Still, the crosstalk between extrinsic niche-derived and HSC intrinsic factors mediating stem cell maintenance and quiescence, particularly in the context of ageing, remains elusive26,27. Based on this, we hypothesize that changes in interactions maintaining quiescence in young BM may contribute to the functional decline of HSCs.
A number of cell surface receptors, activated by niche-derived ligands such as THPO-MPL, DARC-CD82, or Histamine-H2R, have been described to directly modulate HSC behaviour28,29,30,31. Interestingly, some of these, including CXCR4-CXCL12 (C-X-C chemokine receptor type 4/C-X-C motif chemokine 12) and SCF-c-Kit (stem cell factor/c-Kit), also seems to play a key role during neural development32,33. Neogenin-1 (Neo1), a cell surface receptor first identified as a regulator of axon guidance, has been implicated in a wide variety of functions ranging from cell migration and survival to angiogenesis34. Its role has recently also been studied in the innate and adaptive immune systems35,36,37. It shares almost 50% amino acid homology with DCC (deleted in colorectal cancer)38,39. The extracellular domain of Neo1 has been described to bind members of both the “repulsive guidance molecule” (RGM-a–c) and netrin (Ntn) families34,39. Neo1 can modulate cytoskeletal activities and can function as a co-receptor for bone morphogenetic proteins (BMPs)40,41. However, the functional role of Neo1 or its ligands such as Ntn1 in HSC biology remains uncertain1,42. Here, we identify Ntn1–Neo1 signalling as an important regulator of HSC quiescence.
Results
Neo1 is specifically expressed in the most quiescent HSCs
Neo1 expression in HSCs has previously been reported by us and others1,42,43,44. To further characterize Neo1 expression within the hematopoietic stem and progenitor cell (HSPC) compartment, we isolated various HSPC populations (Fig. 1a and Figure S1a) and found Neo1 to be exclusively expressed in HSCs (Fig. 1b). This HSC-specific expression pattern of NEO1 was also apparent at the protein level (Fig. 1c and Figure S1b). NEO1 levels in HSCs were heterogeneous as ~20% of HSCs expressed particularly high levels on the surface (Fig. 1c). Next, we studied whether this subset of NEO1 high-expressing HSCs corresponds to HSCs (dHSCs) by conducting label-retaining assays using SCL-tTA;H2B-GFP mice10 (Figure S1c). After 150 days of doxycycline chase, we found Neo1 transcripts and protein to be expressed at higher levels in dHSCs compared to aHSCs and MPP1s, suggesting that Neo1 is associated with dormancy (Fig. 1d, e). As expected, dHSCs specifically expressed the dHSC marker Gprc5c11 (Figure S1d). To independently validate increased Neo1 expression in dHSCs, we employed Gprc5c-GFP reporter mice and isolated dormant GFPpos and active GFPneg HSCs (Figure S1e). In agreement, we found higher Neo1 RNA and protein levels in Gprc5c-GFPpos vs. Gprc5c-GFPneg HSCs (Figure S1f, g). As HSCs are a highly quiescent population during steady state, we next addressed whether Neo1 levels not only rapidly diminished during hematopoietic differentiation, but also upon HSC activation. Therefore, we treated mice with either poly-I:C (pIC) mimicking viral, or lipopolysaccharide (LPS) mimicking bacterial infection45,46. HSCs showed a robust, reversible loss of Neo1 expression in response to either stimulus (Fig. 1f, g). Collectively, these data strongly link Neo1 expression to dormancy in HSCs.

a Overview of hematopoietic stem and progenitor cells (HSPCs) and their immunophenotypes. b Relative expression of Neo1 in HSPCs from 3-month-old mice; n = 4–7 (HSC-MPPs) and 9 (CMP/MEP/GMP), two independent experiments. c MFI of NEO1 in HSPCs from 3-month-old mice; n = 90 (MPP2), 118 (MPP34), 126 (MPP1) and 145 (HSC), two independent experiments. d Relative expression of Neo1 in dHSC and aHSC from SCL-tTA; H2B-GFP mice, chase for 5 months; n = 3. e MFI of NEO1 in dHSCs and aHSCs from SCL-tTA; H2B-GFP mice, chase for 5 months; n = 30 (aHSC)–47 (dHSC). f Relative expression of Neo1 in HSCs, 16 h, 5 and 7 days after PBS or poly-I:C injections; n = 3–5 (PBS16h). g Relative expression of Neo1 in HSCs, 16 h after PBS or LPS injections; n = 3 (LPS)–5 (PBS). For all panels, ±SD is shown. n indicates biological replicates. Scale bars in IF images are 5 μm. P value was determined by two-tailed t test. Source data are provided as a Source Data file.
Neo1-mutant mice reveal a competitive advantage upon transplantation
Considering the HSC-specific expression pattern of Neo1, we set out to study the function of Neo1 in the hematopoietic system. Unfortunately, in our hands, no commercial antibody allowed the robust and reproducible isolation of viable Neo1+ cells by flow cytometry when using Neo1-mutant cells as controls42. Thus, we employed a Neo1 gene-trapped mouse model to genetically address the functional role of Neo1 in HSC biology (Neo1gt/gt)38,47,48. Although Neo1 expression in the BM of mutant mice was diminished by >90% (Fig. 2a), the hematopoietic compartment did not exhibit altered HSPC or mature cell frequencies in 5–6-week-old animals (Figure S1h). To analyse Neo1-deficient hematopoiesis, we performed reconstitution analysis with BM cells derived from 5- to 6-week-old Neo1-mutant animals (Fig. 2b). First, we non-competitively transplanted total BM derived from Neo1-mutant or control littermates (CD45.2) into CD45.1 recipients and assessed HSC numbers 4 months after primary or secondary transplantation (Fig. 2c). We observed that the frequency of HSCs, while similar at 4 months after transplantation, increased in Neo1-mutant chimeras upon secondary transplantation. To further investigate this expansion of HSCs, we performed competitive transplantations of Neo1-mutant or control BM cells (Fig. 2d). We found that Neo1-mutant BM cells showed a competitive advantage compared to control counterparts as evident by peripheral blood leucocyte contribution in secondary recipients and in BM HSC contribution in primary and secondary transplantations (Fig. 2e, f). As HSC frequencies in both transplantation assays increased over time, we also investigated primary chimeras 8 months after transplantation and again found an increase in HSC numbers in Neo1-mutant chimeras (Fig. 2g, h). We observed no difference in HSC homing (Fig. 2i, j), suggesting that self-renewal and output of Neo1-mutant HSCs are altered.

a Relative expression of Neo1 in the total bone marrow of Wt and Neo1gt/gt mice; n = 6, three independent experiments. b Workflow: generation of full chimeras. c Absolute frequencies of bone marrow CD45.2+ HSCs in full Wt and Neo1gt/gt chimeras 4 months after first and second transplantation; n = 5 (2nd Tx)–8 (Ctrl 1st Tx) and 9 (Neo1 1st TX), two independent experiments. d Workflow: competitive transplantations. e Peripheral blood CD45.2+ chimerism during 1° and 2° competitive transplantations of Wt and Neo1gt/gt bone marrow; n = 13–17 (for exact n/timepoint please see Source data file), three independent experiments, Analysis with two-way ANOVA, multiple comparison with LSD Fisher’s test. f CD45.2+ chimerism of HSCs at endpoints of 1° and 2° competitive transplantations of Wt and Neo1gt//gt bone marrow; n = 11 (2nd TX), 12 (Ctrl 1st Tx), 14(Neo1 1st TX), three independent experiments. Whiskers are min–max, box is 25–75th percentile and line is mean. g Workflow: full chimeras studied in (h). h Absolute frequencies of bone marrow CD45.2+ HSCs in full Wt and Neo1gt//gt chimeras after 8 months; n = 8 (Ctrl)–9 (Neo1), three independent experiments. i Workflow: Homing assay in (j). j Absolute frequencies of CD45.2+ bone marrow LSK cells 48 h after transplantation of 10,000 sorted Wt and Neo1gt//gt LSK; n = 5 (Ctrl)–6 (Neo1). For all panels, ±SD is shown. n indicates biological replicates. P value was determined by two-tailed t test unless stated otherwise. Source data are provided as a Source Data file.
Aged Neo1-mutant HSCs display features of premature exhaustion
Next, we addressed whether the HSC expansion observed in Neo1-mutant chimeras would lead to malignant transformation or HSC exhaustion over time (Fig. 3a). Interestingly, 15 months after the generation of primary chimeras, the initial expansion of the Neo1-mutant HSC pool reverted and both HSC and MPP1 frequencies decreased (Fig. 3b). When we compared absolute blood counts in aged Neo1-mutant chimeras to controls, we found reduced absolute lymphocyte and neutrophil counts, as well as reduced haemoglobin levels indicative of hematopoietic malfunction (Fig. 3c). As expected, chimeras displayed increased myeloid differentiation upon ageing, and this effect was exacerbated in Neo1-mutant chimeras over time (Fig. 3d). To address whether this decline in mature cell output was caused by an HSC defect, we re-transplanted 100 CD45.2+ HSCs derived from either aged Neo1-mutant or control chimeras (Fig. 3e). Four months after transplantation, Neo1-mutant HSCs had generated significantly less progeny then controls (Fig. 3f). To validate functional exhaustion, we re-transplanted BM of aged chimeras into secondary and tertiary recipients (Fig. 3g). In these mice, aged Neo1-mutant BM exhibited a pronounced failure to engraft and depletion of HSCs and all MPP populations was observed, suggesting that the original Neo1-mutant HSCs from the aged chimeras had a decreased self-renewal potential (Fig. 3h, i). Meanwhile, we observed no increase in malignancies arising in Neo1-mutant chimeras. Next, we analysed cell cycle behaviour of Neo1-mutant HSCs. We found less HSCs residing in the G0 phase in 4–5-week-old Neo1-mutant mice compared to their control littermates (Figure S1i). This decrease in G0-HSCs was also apparent in full chimeras both 4 and 8 months after transplantation (Fig. 3j, k and Figure S1j) and Neo1-mutant HSCs expressed higher levels of the cell cycle activation marker CDK6. In addition, increased incorporation of bromodeoxyuridine (BrdU) above the expected injection-induced activation was observed in Neo1-mutant HSCs (Fig. 3l, m and Figure S1k). Altogether, Neo1-mutant HSCs harbour diminished long-term repopulation potential, associated with a loss of quiescence and increased proliferation.

a Workflow: aged chimeras, analysed in (b–d). b Absolute frequencies of bone marrow CD45.2+ HSPCs in full Wt and Neo1gt//gt chimeras after 15 months; n = 7 (Ctrl)–11 (Neo1), two independent experiments. c Absolute blood counts of full Wt and Neo1gt//gt chimeras after 15 months; n = 7 (Ctrl)–11 (Neo1), two independent experiments, for HB: 4 (Ctrl)–7 (Neo1). d Frequencies of B cells and myeloid cells of C45.2+ cells in peripheral blood of Wt and Neo1gt//gt chimeras after 15 months; n = 5–13 (for exact n/timepoint please see Source data file), two independent experiments. Analysis with two-way-ANOVA, multiple comparisons with LSD Fisher’s test. e Workflow: assessment of HSC potency derived from 15 months (aged) chimeras. f Frequency of CD45.2+ vs. competitor HSCs 16 weeks transplantation of 100 or 500 HSCs from of aged Wt and Neo1gt//gt chimeras; n = 6 (Ctrl + 500 HSC Neo1)–7(100 HSC, Neo1), two independent experiments. g Workflow: secondary and tertiary transplantations of 15 months (aged) chimeras. h Absolute frequencies of bone marrow CD45.2+ HSPCs in 2° transplantations of aged Wt and Neo1gt//gt chimeras after 4 months; n = 7 (Ctrl)–8 (Neo1), two independent experiments. i Absolute frequencies of bone marrow CD45.2+ HSPCs in 3° transplantations of aged Wt and Neo1gt//gt chimeras after 4 months; n = 6, two independent experiments. j Workflow: generation of full chimeras used in (k–m). k Cell cycle phase of CD45.2+ HSCs derived from Wt and Neo1gt//gt chimeras after 4 months; n = 4 (Ctrl)–6 (Neo1), two independent experiments. l MFI of CDK6 in CD45.2+ HSC derived from Wt and Neo1gt//gt chimeras after 4 months; n = 23 (Neo1)–29 (Ctrl). m Frequency of BrdU+ CD45.2+ HSC derived from Wt and Neo1gt//gt chimeras after 4 months, 48 h post BrdU injection; n = 6, two independent experiments. For all panels, ±SD is shown. n indicates biological replicates. P value was determined by two-tailed t test unless stated otherwise. Scale bars in IF images are 5 μm. Source data are provided as a Source Data file.
Molecular signatures of activation and HSC dysfunction are enriched in Neo1-mutant HSCs
To understand the molecular basis for the disruption of long-term self-renewal caused by loss of Neo1, we performed RNA-sequencing (RNA-seq) analysis of Neo1gt/gt and wild-type (Wt) CD45.2+ HSCs 4 months (expanding Neo1-mutant HSCs) and 15 months (exhausted Neo1-mutant HSCs) after transplantation (Fig. 4a and Figure S2a). The principal component analysis showed the main mode of transcriptional variation to be attributable to age. The molecular consequences of mutant Neo1 were recapitulated by PC-2 and the difference increased upon ageing (Fig. 4a and Figure S2b). As expected, Neo1 expression itself was diminished in Neo1-mutant HSCs, but interestingly strongly upregulated in aged compared to young Wt HSCs (Figure S2c). Analysis of shared functional differences between young and old Neo1-mutant HSCs compared to controls using Gene Set Enrichment Analysis (GSEA), we revealed cell cycle-associated gene sets like Hallmark(HM)_Mitotic_Spindle and HM_G2M_Checkpoint to be enriched in Neo1 mutants (Fig. 4b) validating the functional data. This pattern of increased activation in Neo1-mutant HSCs was also observed employing HSC-specific cell cycle signatures49 (Fig. 4b). In line with these data, the signature for aHSCs was enriched in Neo1-mutant HSCs, in turn the signature for dHSCs was enriched in Wt HSCs11 (Fig. 4b). Reflecting the observed functional deficits of Neo1-mutant HSCs, the MoIO50 signature associated with superior HSC function was overrepresented in Wt HSCs, while the NoMO signature50, enriching for less quiescent, functionally inferior HSCs were enriched in Neo1-mutant HSCs (Fig. 4b). Analysis of differentially expressed genes (DEGs) identified genes associated with differentiation such as Itga2b and Gata150,51,52, as well as cell cycle regulators such as Cdk653 (Fig. 4c and Figure S2f) or Mki67 (Figure S2d) to be upregulated in Neo1-mutant HSCs. In contrast, genes known to regulate HSC self-renewal or quiescence, such as Egr154,55, Zfp3656 and c-Fos57 were downregulated (Fig. 4c and Figure S2f). Interestingly, Cdk6 has been shown to suppress Egr1 expression during HSC activation, which was suggested to promote HSC quiescence based on genetic data and thus is a likely downstream target of Neo154. No other Ntn1 receptors were differentially expressed (Figure S2e). Therefore, the molecular data support the functional findings by revealing footprints of both loss of quiescence and diminished expression of HSC self-renewal related genes in Neo1-mutant HSCs. In addition, we found that HSC ageing signatures20 were enriched in Neo1-mutant HSCs reflecting the observed functional decline (Fig. 4d). In line, Klf6, which has been proposed to maintain features of young HSCs in human, was downregulated in Neo1-mutant HSCs58 (Fig. 4e and Figure S2f). Finally, we report gene sets associated with nuclear factor-κB (NF-κB) signalling, as well as signalling of the NEO1 ligand netrin-1 (Ntn1) to be depleted in Neo1-mutant HSCs, suggesting that these signalling pathways may be downstream of NEO1 activation (Fig. 4f).

a, Left: workflow for RNA-seq of CD45.2+ HSCs from Wt and Neo1gt//gt chimeras after 4 and 15 months. Right: sparse PCA; n = 2 (WT old/young, Neo1 young)–3 (Neo1 old). b GSEA for cell cycle and HSC potency of Wt vs. Neo1gt//gt HSCs. FDR < 0.05, NOM p value <0.05. c Normalized read counts of DEG in HSCs from young and old Wt and Neo1gt//gt chimeras, n = 4 (Ctrl)–5 (Neo1). d GSEA for HSC ageing signatures in Wt vs. Neo1gt//gt HSCs. FDR < 0.05, NOM p value <0.05. e Normalized read counts of Klf6 in HSCs from young and old Wt and Neo1gt//gt chimeras, n = 4 (Ctrl)–5 (Neo1). f GSEA for signalling pathways in Wt vs. Neo1gt//gt HSCs. FDR < 0.05, NOM p value <0.05. For all panels, ±SD is shown. n indicates biological replicates. Scale bars in IF images are 4 μm. P value was determined by two-tailed t test unless stated otherwise. Source data are provided as a Source Data file.
Interestingly, when we tested enrichment for the Reactome_Netrin-1_Signalling gene set on RNA-seq data of a recent study of HSPC1, it was enriched in HSCs compared to all MPP populations, suggesting that Ntn1 signalling is physiologically active in homeostatic HSCs (Figure S2g). In summary, we discover molecular features of both loss of quiescence and loss of self-renewal in Neo1-mutant HSCs, paralleling functional results.
NTN1 maintains HSC engraftment potential and quiescence via NEO1 signalling
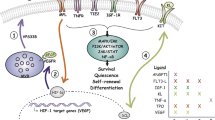
Next, we assessed whether the NEO1 ligands NTN1, RGM-a and RGM-b alone or in combination with their co-ligand BMP-2 were able to affect HSC behaviour. Because neither RGMs and Ntn1 nor additional Ntn1 receptors were expressed in HSCs (Figure S2h)1, we sorted and cultured HSCs in the presence of NTN1, RGM-A and RGM-B with or without BMP-2 (Fig. 5a). To assess active NEO1 signalling, we monitored Egr1 expression, which was downregulated in Neo1-mutant HSCs (Fig. 5b). After 48 h of stimulation, only NTN1, but none of the other ligands, induced expression of Egr1 (Fig. 5b). This induction was absent in Neo1-mutant HSCs (Fig. 5b). In addition, we detected a Neo1-dependent decrease in G2–S–M and an increase in G0-phase HSCs as well as diminished CDK6 protein levels (Fig. 5c, d), paralleling the data from Neo1-mutant HSCs in vivo (Fig. 4c, g). We further confirmed the induction of quiescence by NTN1 with HSCs isolated from FUCCI59 and c-Myc-GFP mice60 reporter mice (Figure S2i, j). Gene sets associated with NF-κB signalling were downregulated in Neo1-mutant HSCs. Since NF-κB is essential for HSC maintenance and known to protect HSCs from premature differentiation upon stress61, we hypothesized that NTN1 may induce NF-κB signalling. To test this hypothesis, we isolated HSCs from p65-GFP mice, cultured them +/− NTN1 or +/− the p65 nuclear translocation inhibitor JSH-23 (Fig. 5e). We observed increased nuclear p65 levels upon NTN1 treatment, which was blocked by JSH-23 (Fig. 5e), suggesting that NTN1 maintains the canonical NF-κB pathway. We next assessed whether in vitro NTN1 stimulation translates into improved HSC engraftment in vivo. For this purpose, we stimulated 500 HSCs derived from either CD45.2 or CD45.1/2 mice with or without Ntn1 for 48 h, mixed treated with untreated congenically distinct HSCs and transplanted them into lethally irradiated recipients (Fig. 5f). Four months after transplantation, we found increased engraftment of HSCs cultured with NTN1 in the BM, independent of genotype (Fig. 5g). This showed that ex vivo treatment with NTN1 robustly improved the in vivo function of cultured HSCs. This effect of NTN1 was dependent on the presence of NEO1 since it was absent in Neo1-mutant HSCs (Figure S2k). Collectively, these data suggest that the NTN1–NEO1 axis preserves NF-κB activity, quiescence and in vivo function of cultured HSCs.

a Workflow: In vitro stimulation of sorted HSCs used in (b–d), analysis after 48 h. b Relative expression of Egr1 in Wt HSCs; n = 3 (other), 4 (RGM-a + b), 16 (Ctrl/Neo1), for ctrl/Ntn1, four independent experiments. c Representative cell cycle plots pre-gated on HSCs and quantification with or without Ntn1 treatment; n = 3 (Neo1), 11 (Wt-Ctrl), 12 (Wt-Ntn1), three independent experiments for ctrl HSC. d MFI of CDK6 in Wt HSCs 48 h after Ntn1 treatment, quantification of MFI per cell; n = 114 (Ctrl) and 134 (Ntn1). e Workflow: representative images and quantification of total cell/nuclear MFI of p65-GFP HSC 48 h after treatment with Ntn1 or Ntn1 + JSH-23; n = 8 (JSH-23), 78 (Ctrl), 91 (Ntn1), two independent experiments. f Workflow: competitive transplantation of Ntn1 stimulated CD45.2 and CD45.1/2 HSCs. g Chimerism of bone marrow LSK-SLAM cells 4 months after competitive transplantation of Control vs. Ntn1-treated HSCs; n = 6 (CD45.1/2), 7 (CD45.2), two independent experiments. For all panels, ±SD is shown. n indicates biological replicates. Scale bars in IF images are 4 μm. P value was determined by two-tailed t test unless stated otherwise. Source data are provided as a Source Data file.
Conditional Ntn1 deletion depletes HSCs and leads to activation and differentiation in vivo
Next, we addressed the role of Ntn1 in hematopoiesis in vivo. Mice homozygous for an Ntn1-null allele (Ntn1β-geo/β-geo) die perinatally due to defects in cerebral development62 and heterozygous mice display no hematopoietic phenotype (Figure S3a). Therefore, we generated CAGGS:CreERT2; Ntn1flox/flox mice63,64, which allows tamoxifen (Tam)-inducible ubiquitous deletion of Ntn1 (Figure S3h). We induced deletion of Ntn1 at 6 weeks after birth (Ntn1ΔCAGGSCre/ΔCAGGSCre) and analysed mice 8 weeks later (Fig. 6a). Ntn1 deletion caused an increase in the relative frequencies of myeloid cells, especially neutrophils in both peripheral blood and BM (Figure S3b–d). Strikingly, the frequency of HSCs in Ntn1ΔCAGGSCre/ΔCAGGSCre BM was significantly reduced, while simultaneously the frequency of both MPP2 and MPP3/4 cells expanded (Fig. 6b, c and Figure S3e, f). In response to the induced Ntn1 deletion, HSCs entered a more proliferative, less quiescent state, represented by an increase of HSCs in G2–S–M and a reduction in G0 (Fig. 6d). After Ntn1 deletion, HSCs also expressed reduced levels of Egr1, while expression of Cdk6, as well as the differentiation associated genes Gata1 and Itga2b increased (Fig. 6e). Finally, Neo1 expression was upregulated in Ntn1ΔCAGGSCre/ΔCAGGSCre HSCs, suggesting a compensatory upregulation in response to the absence of its ligand (Fig. 6e).

a Workflow: analysis of Ntn1flox/flox and CAGGS:CreERT2; Ntn1flox/flox mice 8 weeks after Cre induction for (b–e). b Representative flow cytometry plots of the LSK population of Ntn1flox/flox and Ntn1ΔCAGGS/ΔCAGGS mice. c Frequencies of bone marrow HSCs in Ntn1flox/flox and Ntn1ΔCAGGS/ΔCAGGS mice; n = 7 (flox)–10 (∆CAGGS), two independent experiments. d Cell cycle phase of HSCs derived from Ntn1flox/flox and Ntn1ΔCAGGS/ΔCAGGS mice; n = 8 (flox) and 10 (∆CAGGS), two independent experiments. e Relative expression of quiescence and activation related genes in HSCs derived from Ntn1flox/flox and Ntn1ΔCAGGS/ΔCAGGS mice; n = 6 (flox)–9 (∆CAGGS), two independent experiments. f Frequencies of bone marrow HSCs in Ntn1flox/flox and Ntn1ΔCAGGS/ΔCAGGS mice 5 months after Cre induction; n = 8 (∆CAGGS) and 12 (flox), three independent experiments. g Cell cycle phase of HSCs derived from Ntn1+/LSL-Rosa26-Ntn1 and Ntn1-OE mice; n = 8 (∆CAGGS) and 12 (flox), three independent experiments. h Workflow: competitive transplantation of CAGGS:CreERT2, Ntn1ΔCAGGS/ΔCAGGS and Ntn1-OE mice 5 months after Cre induction, analysed in (i–k). i Representative FACS plots of peripheral blood leucocytes pre-gated on CD45+ cells at 16 weeks after transplantation. j Peripheral blood CD45.2+ chimerism during competitive transplantations; n = 13 (OE)–14 (Cre/∆CAGGS), two independent experiments. Analysis was done with two-way-ANOVA, multiple comparison with LSD Fisher’s test. k Bone marrow HSC CD45.2+ chimerism after 16 weeks of competitive transplantation; n = 12 (Cre)–13 (∆CAGGS/OE), two independent experiments. l Workflow: transplantation of 200 HSCs sorted from CAGGS:CreERT2 and Ntn1ΔCAGGS/ΔCAGGS mice at 5 months after Cre induction. m Frequencies of bone marrow HSCs 8 weeks transplantation; n = 6. For all panels, ±SD is shown. n indicates biological replicates. P value was determined by two-tailed t test unless stated otherwise. Source data are provided as a Source Data file.
The observed reduced numbers of HSCs were even more pronounced at 5 months post Ntn1 deletion, suggesting a progressive loss of HSCs after Ntn1 deletion (Fig. 6f and Figure S3g, i). To test whether increased levels of NTN1 could alter HSC behaviour in vivo, we generated CAGGS:CreERT2; LSL-Rosa26-Ntn1 mice (Ntn1-OE) and induced Cre expression in 6-week-old animals, leading to a 30-fold increase of Ntn1 levels in BM endothelial cells after 5 months (Figure S3h). While we found no difference in HSPC frequencies (Figure S3j), quiescent G0-HSCs increased, suggesting that Ntn1 overexpression in the BM microenvironment leads to increased HSC quiescence in vivo (Fig. 6g and Figure S3k). In addition, the frequency of cycling HSCs 5 months after Ntn1 deletion was significantly increased, reproducing the 2-month timepoint (Fig. 6d, g and Figure S3k). In summary, Ntn1 mediates HSC quiescence not only in culture but also in vivo and loss of Ntn1 activates and progressively depletes quiescent, functional HSCs.
Conditional Ntn1 deletion impairs HSC function
To study, whether the Ntn1-mediated increase (Ntn1-OE) or reduction in HSC quiescence and frequency (Ntn1 deletion) is associated with functional consequences, we competitively transplanted total BM of Ntn1-OE, Ntn1ΔCAGGSCre/ΔCAGGSCre or control (CAGGS-CreERT2) mice 5 months after Tam induction (Fig. 6h). Upon Ntn1-OE, we neither observed any differences in peripheral blood leucocytes nor in HSC frequencies 4 months after transplantation (Fig. 6i, k). In contrast, Ntn1 deletion led to a reduced contribution of CD45.2+ donor cells to peripheral blood leucocytes (Fig. 6i, j) accompanied with a strong reduction of HSC numbers 4 months after transplantation (Fig. 6k). Next, we addressed the engraftment potential of 200 purified HSCs (LSK, CD150+, CD48−, CD34−) isolated either from a microenvironment, in which Ntn1 was deleted for 5 months (Ntn1ΔCAGGSCre/ΔCAGGSCre) or expressed on normal levels (Fig. 6l). Two months post transplantation, the HSC frequency was significantly reduced compared to control HSC, which have developed in an Ntn1-proficient environment (Fig. 6m). These data show that HSCs derived from an Ntn1-deficient BM become functionally impaired and this self-renewal defect is not reversed by transplanting them back into an Ntn1-proficient recipient microenvironment.
Ntn1 expressed by arterioles maintains HSCs
We next investigated which niche cells express Ntn1. By screening published datasets, we found that Ntn1 is expressed at low levels in sinusoidal (SEC: CD45−, CD31+, Sca-1medium, Pdpn+) and at higher levels in arteriolar endothelial cells (AEC: CD45−, CD31+, Sca-1high, Pdpn−)65. In addition, Ntn1 expression has been reported in periarteriolar smooth muscle cells (SMCs)66. To examine Ntn1 expression within the BM niche, we isolated AECs, SECs, CD45+ hematopoietic and RFP+ cells derived from Sma-RFP (smooth muscle actin-RFP) reporter mice marking SMCs67 (Figure S4a, b). While we found no expression in CD45+ hematopoietic cells, we detected the highest Ntn1 levels in AECs and SMCs (Fig. 7a). To investigate whether periarteriolar smooth muscle-derived Ntn1 regulates HSCs, we generated Sma-CreERT2; Ntn1flox/flox mice, injected adult mice with Tam and studied HSCs 8 weeks after Cre induction. In line with depletion of HSCs upon global Ntn1 deletion, we detected a decrease in HSCs in Ntn1ΔSmaCre/ΔSmaCre animals compared to controls (Fig. 7b). This reduction was, however, not as strong as we observed upon global Ntn1 deletion using CAGGS-Cre (Fig. 6), suggesting additional Ntn1 sources like AECs. As BM arterioles deteriorate upon ageing, leading to the loss of HSC maintaining SCF23,24, we isolated SECs and AECs from young and old Wt mice and found diminished Ntn1 expression specifically in old AECs (Fig. 7c). When we investigated Neo1 in aged HSCs, we found expression was still restricted to HSCs, but levels were significantly increased (Figure S4c), in line with our RNA-seq data from aged Wt chimeras (Figure S2d). To further confirm this, we performed RNA-seq of young and old LSK-SLAM cells. We found membrane-associated processes and receptors to be upregulated upon ageing (Figure S4d). Specifically, Neo1 expression increased robustly on RNA and protein level in old HSCs (Fig. 7d, e). Several studies have previously compared transcriptional profiles of young vs. old HSCs (using different marker combinations). However, the studies showed a wide variety of DEGs with little consistency (Figure S4e). To identify consistently changed DEGs upon HSC ageing, we added 12 previously published transcriptome datasets of aged HSCs to our own study and performed a meta-analysis (Figure S4e). In these 13 datasets, not a single DEG was shared among ten or more studies, again highlighting the heterogeneity. Nevertheless, 13 genes were consistently differentially expressed in eight to nine datasets (Fig. 7f). Seven of these were receptors and one of these was Neo1, suggesting that Neo1 is one of the most consistently upregulated genes found upon HSC ageing.

a Relative expression of Ntn1 in CD45+ cells, SEC, AEC and RFP+ SMC derived from Sma-RFP mice; n = 4 (CD45/AEC), 6 (SEC) and 7 (SMA-RFP), two independent experiments. b Frequencies of HSCs in bone marrow of in Ntn1flox/flox and Ntn1ΔSma/ΔSma mice; n = 8 (flox) and 10 (∆SMA), three independent experiments. c Relative expression of Ntn1 in SEC, AEC and CD45+ cells derived from young and old Wt mice; n = 3 (yCD45/oAEC), 4 (oSEC), 6 (ySEC) and 7 (yAEC), three independent experiments. d Normalized read counts of Neo1 in young, and old LSK-SLAM cells; n = 5 (young) and 7 (old), FDR < 0.0001. e MFI of NEO1 in sorted 6 or 24 months LSK-SLAM cells; n = 592 (young)–593 (old). f Most abundant common DEGs in published ageing studies and own data, additional details in the “Methods” section. g Relative expression of Neo1 in LSK-SLAM cells isolated from either denervated or healthy legs of individual mice; n = 8, two independent experiments. h Relative expression of Neo1 in HSCs of aged mice, before and after 2 months post transplantation; n = 6 (before) and 8 (after), two independent experiments. i Model of Neo1/Ntn1 axis in young and old mice. For all panels, ±SD is shown. n indicates biological replicates. Scale bars in IF images are 5 μm. P value determined by two-tailed t test unless stated otherwise. Source data are provided as a Source Data file.
It has recently been established that surgical BM denervation mirrors the phenotype of arteriolar degeneration upon ageing and thereby induces premature HSC ageing24. Therefore, we tested whether the observed Neo1 upregulation during HSC ageing (Fig. 7d) or as a consequence of Ntn1 deficiency (Fig. 6e) was recapitulated upon denervation-mediated induction of premature marrow ageing. One hind limb per Wt mouse was surgically denervated and LSK-SLAM cells 4 months after surgery were analysed. We found an increase in Neo1 expression in HSCs of seven out of eight denervated femurs compared to sham-operated nerve-intact contralateral femurs of the same mice (Fig. 7g). The Neo1 upregulation is consistent with a model that the normal or accelerated ageing process leads to a decrease in Ntn1 expression in the microenvironment, mediating a compensatory Neo1 upregulation to maintain signalling when its ligand Ntn1 becomes limiting.
Finally, we investigated whether the niche mediated upregulation of Neo1 in HSCs of 30-month-old mice (NTN1low environment) can be reversed by transplanting them into 2-month-old young mice (NTN1high environment). Indeed, Neo1 expression in HSCs significantly decreased again in young mice (Fig. 7h). These data further support the link between the level NTN1 production in the BM microenvironment and expression of its receptor Neo1 on HSCs in young and old mice (Fig. 7i). However, the compensatory upregulation of Neo1 expression due to age-dependent ligand deprivation is not sufficient to maintain NEO1 function, since ablation of either Ntn1 or Neo1 leads to proliferation and decreased self-renewal of HSCs, a hallmark of aged HSCs.
Discussion
Here, we identify arteriolar niche-derived NTN1 ligand and its cognate HSC-specific receptor NEO1 as a novel ligand-receptor signalling axis regulating HSC quiescence and long-term self-renewal. This axis is deregulated upon ageing and loss of either of its components leads to functional HSC impairment. NTN1–NEO1 represents a novel intercellular and non-cell autonomous signalling network by which NTN1 produced by perivascular niches binds to HSCs to fine-tune HSC dynamics, in particular cell cycle activity and long-term self-renewal.
In agreement with Neo1 being specifically expressed by dHSCs (Fig. 1), Neo1 is part of the MoIO signature marking functionally superior HSCs50. Expression of Neo1 is also highest in Vwf+ HSCs residing on the top of the hematopoietic hierarchy68 and NEO1+ cells have recently been reported as a subpopulation within Hoxb5+ HSCs42. Intriguingly, Dnmt3a mutant HSCs show increased quiescence, as well as a robust upregulation of Neo1 expression69,70,71, suggesting it as a potential target for Dnmt3a mutant hematopoietic disorders.
When characterizing Neo1-mutant hematopoiesis, we observed an initial increase in HSC numbers associated with loss of quiescence and subsequently loss of HSC self-renewal over time that correlated with decreased expression of Egr1 and increased expression of Cdk6. Similarly, hematopoietic loss of Egr1 leads to increased cycling and initial HSC expansion followed by a loss of engraftment potential upon serial transplantation55. Since we analysed a hypomorphic Neo1 mouse model with severely decreased (>90%) but remaining minor expression38,47,48, our results possibly underestimate the biological relevance of Neo1 in HSCs. It has been reported, that ≈80% of Neo1gt/gt mice die prenatally. The ones born develop hydrocephalus of varying degree, with around one in five displaying severe phenotypes with macroscopically visible “dome-shaped” skulls48. Since this was reproducible in our analysis, we used only Neo1gt/gt mice without macroscopic features of hydrocephalus, which showed normal, healthy behaviour. In these Neo1gt/gt mice, the HSC numbers were unchanged at the time of analysis. Nevertheless, we cannot formally exclude that additional factors such as neuronal stress may contribute to some extent to the described HSC phenotype in the primary Neo1gt/gt mutants.
NEO1 can bind multiple neural guidance molecules, which mediate context-dependent effects. As an example, RGMs are known to inhibit neuronal migration72, while NTN1 acts as a chemoattractant for commissural axons63. In HSCs, we found NTN1, but neither RGMs nor BMP-2, to modulate HSC behaviour. This is intriguing because, in the developing bone, NEO1 modulates cartilage growth via canonical BMP signalling73. However, the relevance of BMP signalling for adult HSCs remains uncertain74.
Over the past years, the role of netrins in neurobiology, originally established using gene-trapped mice62, has been challenged by novel conditional Ntn1 alleles63,75,76. When we repurposed these to investigate hematopoiesis, we found increased activation and progressive loss of HSC numbers as well as self-renewal potential after global deletion of Ntn1, mimicking the Neo1-mutant phenotype. Further, in vivo overexpression and in vitro stimulation with NTN1 enhanced HSC quiescence and increased engraftment potential of cultured HSCs upon transplantation, respectively. These results are in line with studies showing quiescence-inducing compounds that maintain HSC engraftment potential in vitro11,29,77 as well as studies that associate loss of self-renewal capability in vivo with divisional history13,78,79. Altogether, the data strongly suggest that NTN1 acts as a paracrine NEO1 ligand modulating HSC behaviour. Furthermore, Ntn1 has been described to support immature states of iPSCs and cancer stem cells80,81, suggesting that it maintains stemness in various settings. Here, we demonstrate that NTN1/NEO1 signalling increase NF-κB activation in HSCs, a pathway known to protect HSCs from exhaustion during stress, while the loss of p65 leads to hematopoietic failure61,82.
Within the BM niche, we found Ntn1 to be expressed in AECs and SMC in line with the previous studies65,83,84. These as well as other perivascular cells secrete multiple molecules that support HSCs including SCF and CXCL1226,27,65,83,85,86. Upon ageing, BM arterioles are remodelled leading to a depletion of periarteriolar stromal cells and SCF, affecting hematopoiesis23,24,25. In line, NTN1 secretion by SMCs is known to guide axons of the sympathetic nervous system during arteriolar growth66. The connection between the sympathetic nervous system and arterioles is intriguing, as denervation disrupts BM arterioles and mediates accelerated HSC ageing24.
Our data strongly support the link between NTN1 production in the BM microenvironment and expression of its receptor Neo1 on HSCs. Loss of Ntn1 expression in the niches during: (a) physiological ageing, (b) accelerated ageing by surgical denervation or (c) by genetic ablation results in compensatory upregulation of Neo1 expression due to ligand deprivation, which, however, is not sufficient to maintain Neo1 function. Such a mechanism has also been observed for the Ntn1 receptors DCC and NEO1 upon loss of Ntn1 during development76.
Collectively, our data suggest that NTN1 produced mainly by arteriolar niches preserves quiescence and self-renewal of HSCs via NEO1, while ageing-associated decline of Ntn1 leads to the gradual decrease of Neo1-mediated HSC self-renewal.
Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andreas Trumpp a.trumpp@dkfz.de. Certain materials are shared with research organizations for research and educational purposes only under an MTA to be discussed in good faith with the recipient.
Experimental mouse models
SCL-tTA; H2B-GFP mice
This transgenic mouse line expresses the fusion protein histone H2B-GFP under the tetracycline-responsive regulatory element and the tTA-S2 transactivator from the endogenous Scl locus10. Doxycycline was supplemented in drinking water of 8–16-week-old mice for 150 as previously described10. To set the gates for GFP+ cells, age-matched H2B-GFP littermates were used. SCL-tTA; H2B-GFP mice were backcrossed to C57BL/6J.
C57BL/6J (CD45.2, CD45.1 or CD45.2/CD45.1) mice were either purchased from Envigo (the Netherlands) or Janvier Labs (France) or bred in-house.
Gprc5c-GFP mice (Tg(Gprc5c-EGFP)JU90Gsat)
This transgenic mouse line was previously generated by inserting an EGFP gene into a BAC clone at the initiating ATG codon of the first coding exon of the Gprc5c gene and this BAC clone was subsequently used to generate transgenic reporter mice87. Analysed mice were backcrossed to C57BL/6J.
Myc-eGFP mice
This transgenic mouse line expresses a fusion protein of c-Myc and eGFP60.
FUCCI mice (B6-Tg(Gt(ROSA)26Sor-Fucci2)#Sia)
This transgenic mouse line allows the identification of cell cycle phase via fluorescent fusion proteins, mice were sacrificed after 8–16 weeks59.
Neo1 gt/gt mice (B6.129P2-Neo1 Gt(KST265)Byg/Mmmh)
These mice harbour a gene-trapped Neo1 allele that leads to a strong reduction of Neo1 expression38. For transplantation experiments, male and female animals 4–6 weeks of age were used. Control transplantations were always performed using gender-matched, wild-type littermates. For competitive transplantations, competitor BM was also age- and gender-matched.
Ntn1 β-geo/+ mice (Ntn1 Gt(ST629)Byg)
These mice harbour a gene-trapped Ntn1 allele that leads to a strong reduction of Ntn1. Heterozygous mice can be used as reporter mice employing the β-gal reporter in the gene-trapped vector62.
Ntn1 fl/fl mice
This transgenic mouse line contains loxP sites flanking coding sequences containing both the principal ATG (based on Ntn1 complementary DNA (cDNA) sequence NM_008744) and the cryptic ATG (based on Ntn1 cDNA: BC141294) and the alternative promoter described in intron 363. To generate global Ntn1 deletion, we crossed Ntn1fl/fl mice to CAGGS-CreERT2 mice (Jackson Laboratories). For smooth muscle-specific deletion, Ntn1fl/fl mice were crossed to Sma-CreERT2 mice. For 8 weeks endpoints, Ntnfl/fl crossings only female, and for 5 months endpoints, only male mice were analysed to reduce variability.
+/LSL-Rosa26-Ntn1 mice
This transgenic mouse line was generated for this study. The human NETRIN-1 was cloned in Rosa26-lox-stop-lox plasmid (Soriano). Mice were generated by SEAT CNRS Gustave Roussy Phenomin. We crossed these mice to CAGGS:CreERT2 mice (Jackson Laboratories), inducing global overexpression of Ntn1. To reduce variability, only male animals were analysed at 5 months after Cre induction.
Sma-RFP mice (C.Cg-Tg(aSMA-RFP)#Rkl
The mouse line harbours an RFP reporter for Sma and thereby allows identification of SMCs67. Sma-RFP mice are on a BALB/C background.
All other mouse models are on a B6J background.
All mice were bred in-house in the animal facilities of DKFZ, University Medical Center Groningen, INSERM or Albert-Einstein College of medicine under specific pathogen-free conditions in individually ventilated cages at 24°, a humidity of 80% with fixed day/night cycles of 12 h. According to German, French, American or Dutch guidelines, mice were euthanized by cervical dislocation and all animal procedures were performed according to protocols approved by the Regierungspräsidium Karlsruhe, Animal Care and Use Committee of Albert-Einstein College of Medicine, the Instantie voor Dierenwelzijn committee, Universitair Medisch Centrum Groningen/Rijksuniversiteit Groningen or University of Lyon local Animal Ethic Evaluation Committee. To reduce animal numbers, remaining BM/cDNA samples generated in this and previous studies were used whenever possible.
Method details
pIC- or LPS-induced inflammatory stress
Mice were injected intraperitoneally with pIC (100 μg/mouse in 0.1 ml phosphate-buffered saline (PBS)), LPS (5 μg/mouse in 0.1 ml PBS) or PBS alone. Sixteen hours (LPS/pIC/PBS), 5 or 8 days (pIC/PBS) later, mice were sacrificed and BM cells were used for subsequent analysis.
Cell isolation and flow cytometry
Mouse BM cells were isolated, and HSCs and MPP1–4 progenitors defined by immune phenotype (lineage-negative (Lin−), Sca-1+, c-Kit+ (LSK), CD135−/+, CD150−/+, CD48−/+, CD34−/+, CD45.2 CD45.1/2, CD45.1) (see also Fig. 1a), or LSK-SLAM (Lin− c-Kit+ CD150+ CD48−) purified by FACS and subsequently subjected to RNA-seq, population quantitative real-time PCR (qPCR) analysis, in vitro experiments, reconstitution assays or cytological analysis. Briefly, BM was isolated from pooled femora, tibiae, ilia and vertebrae by gentle crushing in PBS using a mortar and pistil. If no depletion of lineage-positive (Lin+) cells was performed, lysis of erythrocytes was performed using ACK Lysing Buffer (Thermo Fisher Scientific). To deplete Lin+ cells we used the Dynabeads Untouched Mouse CD4 Cells Kit (Invitrogen). Briefly, total BM was stained for 30 min with 100 μl/mouse of the Lineage Cocktail provided in the Dynabeads Untouched Mouse CD4 Cells Kit (Invitrogen) in PBS. Labelled cells were then incubated for 20 min with 1.5 ml/mouse of washed polyclonal sheep anti-rat IgG-coated Dynabeads provided in the Kit. Cells were depleted using a magnet, enriching for the Lin− cell fraction. To purify HSC and MPP1–4, the Lin− fraction was stained for 30 min using the following monoclonal antibodies: anti-lineage [anti-CD4 (clone GK1.5), anti-CD8a (53-6.7), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-GR1 (RB6-8C5) and anti-TER119 (Ter-119)]; anti-CD117/c-Kit (2B8); anti-Ly6a/Sca-1 (D7); anti-CD34 (RAM34); anti-CD150 (TC15-12F12.2); anti-CD48 (HM48-1); anti-CD135 (A2F10); CD45.1 (A20); CD45.2 (104). The coupled fluorochromes used depended on the experiment to allow sorting of different fluorescent protein containing reporters or isolation of HSC from transplants. Monoclonal antibody conjugates were purchased from eBioscience, BD Bioscience or BioLegend. Cell sorting was then performed on a FACS Aria I, II and III, FACS Aria Fusion (Becton Dickinson) using BD FACSDiva v8.0.3 (BD Bioscience) or MoFlo Astrios or XDP cell sorters (Beckman Coulter). Sorted in Complete Stem Cell Medium (specified elsewhere) for in vitro culture, cytology and reconstitution experiments, or RNA lysis buffer (ARCTURUS PicoPure RNA Isolation Kit (Life Technologies, Invitrogen) for population RNA-seq or qPCR and stored at −80 °C.
For Figure S4c, long-term HSCs were characterized as Lin−Sca-1+c-Kit+CD48−CD150+, ST-HSCs as Lin−Sca-1+c-Kit+CD48−CD150− and MPPs as Lin−Sca-1+c-Kit+CD48+CD150−. For isolation of committed progenitor subsets, the following markers were used for isolation: CLP (Lin−CD127+Sca-1loc-Kitlo), CMP (Lin−CD127−Sca-1−c-Kit−CD34+CD16/CD32hi), GMP (Lin−CD127−Sca-1-c-Kit+CD16/CD32hi) and MEP (Lin−CD127−Sca-1−c-Kit+CD34−CD16/CD32+). Data were analysed using FlowJo, versions 6-10.5.3.
CDK6/NEO1 staining
For analyses of NEO1 expression, BM cells were isolated from mice and stained for HSC/MPP markers and sorted as described. Cells were plated on poly-l-lysine-coated slides and then fixed with BD Cytofix/Cytoperm Buffer (Beckton Dickinson). Subsequently, Cdk6 (Abcam) or Neo1 (Abcam) staining was performed in 0.1% Triton (Sigma) and 5% bovine serum albumin (BSA). Secondary antibodies coupled to AF-488 were used. After washing, slides were embedded in anti-fade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and imaging was performed employing a Zeiss LSM 700 or LSM 710 confocal microscope using ZEN blue v2.5 (Zeiss international). Experimental replicates were always performed side by side and imaged in one session without change of laser intensities or gain to avoid technical bias and allow the comparability. DAPI signal was not used for quantification or normalization. The analysis was performed with FIJI v.2.0.
NEO1 staining (Fig. 7e)
A total of 4000–6000 long-term HSCs were seeded in spots of an immunofluorescent adhesion slide (VWR) and allowed to settle for 20 min. Cells were then fixed and permeabilized with Fixation and Permeabilization Solution (BD Biosciences) for 20 min on ice. Cells were then blocked with 4% BSA for 30 min at room temperature and then stained with 1:100 mouse Neo1 biotinylated antibody (R&D) at 4 °C. Cells were washed three times in 0.1% Triton X-100 PBS solution and stained with 1:500 secondary antibody streptavidin Alexa-647 for 1 h at 4 °C. After washing, coverslips were mounted in anti-fade reagent with DAPI (Invitrogen).
p65-GFP staining
After 48 h of the culture of 2000 HSCs/well, cells were plated on poly-l-lysine-coated slides and then fixed with BD Cytofix/Cytoperm Buffer (Beckton Dickinson). Subsequently, anti-GFP-488 (Abcam) staining was performed in 0.1% Triton (Sigma) 5% BSA for 1 h to increase the signal. After washing, coverslips were mounted in anti-fade reagent with DAPI (Invitrogen).
Cell cycle analysis
HSC/MPP surface staining (LSK, CD150, CD48, CD34) was performed on BM cells or in vitro treated HSPCs. Cells were fixed with BD Cytofix/Cytoperm Buffer (Beckton Dickinson). Subsequently, intracellular Ki-67 (BD Biosciences) staining was performed using PermWash solution (Beckton Dickinson). Prior to flow cytometry analysis, cells were stained with Hoechst 33342 (Invitrogen) or DAPI (Thermo Fisher).
Population RNA-seq
For Fig. 4e–j and Figure S2c–e, population RNA-seq data were generated as previously described1. Briefly, total RNA isolation was performed using ARCTURUS PicoPure RNA Isolation Kit (Life Technologies, Invitrogen) according to the manufacturer’s instructions. Total RNA was used for quality controls and for normalization of starting material. cDNA libraries were generated using 1 ng of total RNA for Neo1-deficient/Wt HSCs using the SMARTer Ultra Low RNA Kit for Illumina Sequencing (Clontech) according to the manufacturer’s indications. Sequencing was performed using the HiSeq2000 device (Illumina).
RNA-seq: young and old LSK-SLAM for Fig. 7d and Figure S4d
RNA was isolated from 15,000 LSK-SLAM cells (Lin−Sca-1+c-Kit+CD48−CD150+) using the Nucleospin XS Kit (Macherey Nagel) and quantified on Bionalyzer using RNA Pico 6000 Kit (Agilent). Ribosomal depletion was performed using a modified version of RiboZero Kit (Illumina). In all, 300 pg ribosomal-depleted RNA was used as input into TotalScript RNA-Seq Kit (Epicentre). Libraries were pooled and sequenced to 30–50 million reads on HiSeq 2500.
qPCR analysis
For qPCR, total RNA of 2000–10,000 cells were isolated as described above or using Nucleospin RNA XS Kit (Machery-Nagel) and reverse-transcribed using SuperScript VILO cDNA Synthesis Kit (Invitrogen) according to the manufacturer’s guidelines. For qPCR analysis, Fast SYBR Green Master Mix (Thermo Scientific) or LightCycler SYBR Green I (Roche) was used on a ViiA 7 Real-Time PCR System (Applied Biosystems) or a LightCycler 480 Instrument (Roche). RNA expression was normalized to Oaz1, Act2b, or Hprt housekeeping gene expression and presented as relative quantification (ratio = 2−DDCT). Primers were designed using the Universal ProbeLibrary Assay Design Center (Roche) or NCBI Primer-BLAST (NCBI) and ordered from Sigma. Primer sequences are available in the Supplementary Information file.
Reconstitution experiments
For the generation of full chimeras, 3 × 106 total BM cells from 4-week-old Wt CD45.2 or Neo1gt/gt CD45.2 mice were injected per recipient mouse.
For the generation of 50/50 chimeras, 1.5 × 106 total BM cells from 4-week-old Wt CD45.2 or Neo1gt/gt CD45.2 mice were mixed with 1.5 × 106 total BM cells derived from CD45.1/2 mice so that a total of 3 × 106 BM cells was injected per recipient mouse.
For transplantation after in vitro treatment, 500 sorted HSCs of CD45.1/2 or CD45.2 derived from 8- to 12-week-old animals were cultured for 48 h in respective conditions. Then, a well of the progeny of CD45.1/.2 was mixed to a well of the progeny of CD45.2 and then transplanted. A similar setup was used for the assessment of Neo1 dependency of in vitro Ntn1 treatment: 500 Neo1-deficient CD45.2+ HSCs were sorted after 8 months from old Neo1 full chimeras and then incubated with or without Ntn1. These Neo1-deficient HSCs were mixed with 500 CD45.1/2 HSC incubated without Ntn1, derived from 8-week-old CD45.1/2 animals and then transplanted into individual recipients.
For secondary transplantations, 3 × 106 total BM cells were isolated and transplanted.
For potency assessment of 100/500 CD45.2 Wt or Neo1-mutant HSCs were sorted from straight chimeras 15 months after transplantation, mixed with 1 × 105 total BM cells from 8- to 12-week-old CD45.1/.2 mice and transplanted.
For assessment of HSC homing, 10 × 103 LSK cells derived from CD45.2 Wt or Neo1-deficient mice were sorted and transplanted.
For the generation of 50/50 chimeras, 1.5 × 106 total BM cells from 6.5-month-old Ntn1-OE/Ntn1ΔCAGGSCre/ΔCAGGSCre or CAGGS-Cre mice, were mixed with 1.5 × 106 total BM cells derived from CD45.1/2 mice so that a total of 3 × 106 BM cells was injected per recipient mouse.
For transplantations of sorted HSC, 200 HSCs from 6.5-month-old Ntn1ΔCAGGSCre/ΔCAGGSCre or CAGGS-Cre were mixed with 1 × 106 total spleen cells from CD45.1/2 mice and injected into the recipient.
For transplantation of a 30-month-old HSC, 3 × 106 total BM cells were isolated and injected into recipients.
For all experiments, cells were transplanted into fully irradiated (2 × 5 Gy) B6J mice (CD45.1). Contribution of CD45.2 or CD45.1/.2 donor cells was monitored in peripheral blood approximately every 4 weeks post transplantation in all transplantations using either LSRII, LSR Fortessa.
Outcome was addressed by absolute blood counts or flow cytometry using the following monoclonal antibodies: anti-CD45.1 (A20); anti-CD45.2 (104); anti-CD4 (clone GK1.5), anti-CD8a (53-6.7), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-GR1 (RB6-8C5). The applied fluorochromes depended on the experiment. For endpoint analysis of chimera animals, BM stainings were performed as following: anti-lineage [anti-CD4 (clone GK1.5), anti-CD8a (53-6.7), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-GR1 (RB6-8C5) and anti-TER119 (Ter-119)]; anti-CD117/c-Kit (2B8); anti-Ly6a/Sca-1 (D7; anti-CD34 (RAM34); anti-CD150 (TC15-12F12.2−; anti-CD48 (HM48-1); anti-CD45.1 (A20); anti-CD45.2 (104)). Monoclonal antibody conjugates were purchased from eBioscience, BD Bioscience or BioLegend.
Tam induction schema
For Sma:CreERT2; Ntn1fl/fl, CAGGS:CreERT2; Ntn1fl/fl Ntn1fl/fl and CAGGS:CreERT2; LSL-Rosa26-Ntn1 mice 3 Tam (Sigma-Aldrich) injections in 1 week were performed 4–6-week-old animals. At 8 weeks or 5 months after Cre induction mice were sacrificed and analysed.
BrdU analysis
Full chimeras of Neo1-mutant or Wt CD45.2 BM were injected intraperitoneally with 0.2 ml BrdU (1.8 mg/ml; Sigma) 4 months after transplantation and sacrificed 48 h after injection. Then, HSC surface staining was performed as described above, cells were processed and then staining with anti-BrdU (BD Bioscience) antibody (1:30) was performed.
HSPCs plating and in vitro treatment
In all, 5000 LSK-SLAM cells for RNA analysis, or 5000–10,000 HSCs for imaging or cell cycle analysis were sorted into and then cultured in Complete Stem Cell Medium (StemPro-34 SFM, Life Technologies containing 50 ng/ml SCF and 25 ng/ml TPO (all Preprotech), 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, StemPro-34 supplement as recommended). Cells were cultured in 96-well ultralow attachment plates (Corning) and were treated with either recombinant 1 μl/ml Ntn1 (R&D), 1 μl/ml Rgm-a (R&D), 1 μl/ml Rgm-b (R&D), or 200 ng/ml Bmp-2 (R&D) in addition to the standard cytokines. The concentration of JSH-23 was 6 μM. At 48 h after plating, cells were used for downstream procedures such as RNA isolation, imaging or flow cytometry as described in its respective chapter.
BM denervation by the transaction of femoral and sciatic nerves
Denervation of the femoral and tibial BM was done as previously described24. The femoral nerve was localized after its exit from the vertebral column deep in the psoas muscle. This was accomplished with a midline abdominal incision; the intestines were gently moved aside to visualize the psoas muscle. An incision was made in the psoas to visualize the femoral nerve, and a 1 cm section of the nerve was excised. Deep to the psoas (through the incision), the sciatic nerve was visualized in close proximity to the iliac crest of the pelvis, and a 1 cm section of the nerve was excised. For sham operation, both femoral and sciatic nerves were exposed by surgery, but were left intact.
BM digestion for stromal cell isolation
For isolation of stromal cells, we thoroughly cleaned dissected bones, crushed and digested them for 1 h in RPMI with 2% fetal calf serum as well as 0.25% collagenase Type I (Gibco). After digestion was stopped with 10% fetal calf serum-containing medium, red cell lyses, lineage depletion, staining and cell sorting were performed as described above.
Quantification, statistics and reproducibility
Standard quantifications, display and experimental design
Statistical analysis was performed with two-tailed paired Student’s t test or two-way analysis of variance using Fisher’s least significant difference for multiple comparisons as indicated in the respective figure legend. All data are presented as mean ± SD. For box and whiskers plots, error bars depict min to max values, the box is defined at 25–75th percentile and the median is marked with an additional line. Please see figure legends for detailed information. GraphPad Prism 7/8 was used for statistical analysis. The number of independent experiments is indicated in the respective figure legends. Sample exclusion was only done as a result of premature mouse death, infection or clear mistakes in sample processing.
RNA-seq analysis
For RNA-seq of Neo1-mutant and control HSCs, the following pipeline was used: sequenced read fragments were mapped to the mouse reference genome GRCm38 using STAR (STAR_2.6.1a)88. Expression counts estimates were generated using HTSeq (htseq-0.9.1)89. DESeq2 (DESeq2_1.20.0)90 was used to test for differential expression; results were considered significant at a p.adj. value <0.05. The analysis was performed in R-studio v3.5.2 (www.r-project.org).
For Fig. 7d, f and Figure S4d, differential expression was calculated using egdeR (3.24.3).
Downstream analysis
GSEA was performed using the GSEA software (3.0) on pre-ranked differential expression lists.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-seq data have been deposited in online repositories: data linked to Fig. 4: MTAB-7902, data linked to Fig. 7: GSE128050. Expression data from young and old Neo1-mutant or control HSC can be found in Supplementary Data 1. Expression data from the analysis of young and old LSK-SLAM cells can be found in Supplementary Data 2. Source data for all seven figures and four Supplementary Figures are available in Supplementary Data 3. Nucleotide sequences and additional source data are available upon reasonable request to the corresponding author.
References
Cabezas-Wallscheid, N. et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522 (2014).
Kiel, M. J. et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121 (2005).
Pietras, E. M. et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46 (2015).
Busch, K. et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546 (2015).
Rodriguez-Fraticelli, A. E. et al. Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216 (2018).
Benz, C. et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 10, 273–283 (2012).
Purton, L. E. & Scadden, D. T. Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell 1, 263–270 (2007).
Till, J. E. & McCulloch, E. A. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 14, 213–222 (1961).
Yamamoto, R. et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126 (2013).
Wilson, A. et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129 (2008).
Cabezas-Wallscheid, N. et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell 169, 807–823.e819 (2017).
Foudi, A. et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 27, 84–90 (2009).
Bernitz, J. M., Kim, H. S., MacArthur, B., Sieburg, H. & Moore, K. Hematopoietic stem cells count and remember self-renewal divisions. Cell 167, 1296–1309 e1210 (2016).
Sudo, K., Ema, H., Morita, Y. & Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 192, 1273–1280 (2000).
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M. & de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703 (2011).
Verovskaya, E. et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 122, 523–532 (2013).
Sawen, P. et al. Murine HSCs contribute actively to native hematopoiesis but with reduced differentiation capacity upon aging. Elife 7, https://doi.org/10.7554/eLife.41258 (2018).
Yamamoto, R. et al. Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell 22, 600–607 e604 (2018).
Geiger, H., de Haan, G. & Florian, M. C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 13, 376–389 (2013).
Sun, D. et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688 (2014).
Beerman, I. et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425 (2013).
Florian, M. C. et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 10, 520–530 (2012).
Kusumbe, A. P. et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532, 380–384 (2016).
Maryanovich, M. et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24, 782–791 (2018).
Ho, Y. H. et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell 25, 407–418 e406 (2019).
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014).
Pinho, S. & Frenette, P. S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 20, 303–320 (2019).
Chen, X. et al. Bone marrow myeloid cells regulate myeloid-biased hematopoietic stem cells via a histamine-dependent feedback loop. Cell Stem Cell 21, 747–760 e747 (2017).
Hur, J. et al. CD82/KAI1 maintains the dormancy of long-term hematopoietic stem cells through interaction with DARC-expressing macrophages. Cell Stem Cell 18, 508–521 (2016).
Duchene, J. et al. Atypical chemokine receptor 1 on nucleated erythroid cells regulates hematopoiesis. Nat. Immunol. 18, 753–761 (2017).
Yoshihara, H. et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 1, 685–697 (2007).
Zou, Y. R., Kottmann, A. H., Kuroda, M., Taniuchi, I. & Littman, D. R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393, 595–599 (1998).
Molineux, G., Migdalska, A., Szmitkowski, M., Zsebo, K. & Dexter, T. M. The effects on hematopoiesis of recombinant stem-cell factor (Ligand for C-Kit) administered invivo to mice either alone or in combination with granulocyte colony-stimulating factor. Blood 78, 961–966 (1991).
Siebold, C., Yamashita, T., Monnier, P. P., Mueller, B. K. & Pasterkamp, R. J. RGMs: structural insights, molecular regulation, and downstream signaling. Trends Cell Biol. 27, 365–378 (2017).
Mirakaj, V., Jennewein, C., Konig, K., Granja, T. & Rosenberger, P. The guidance receptor neogenin promotes pulmonary inflammation during lung injury. FASEB J. 26, 1549–1558 (2012).
Schlegel, M. et al. Inhibition of neogenin fosters resolution of inflammation and tissue regeneration. J. Clin. Invest. 128, 4711–4726 (2018).
Muramatsu, R. et al. RGMa modulates T cell responses and is involved in autoimmune encephalomyelitis. Nat. Med. 17, 488–494 (2011).
Leighton, P. A. et al. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 410, 174–179 (2001).
Xu, K. et al. Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science 344, 1275–1279 (2014).
Healey, E. G. et al. Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein. Nat. Struct. Mol. Biol. 22, 458–465 (2015).
Lee, N. K. et al. Neogenin recruitment of the WAVE regulatory complex maintains adherens junction stability and tension. Nat. Commun. 7, 11082 (2016).
Gulati, G. S. et al. Neogenin-1 distinguishes between myeloid-biased and balanced Hoxb5 (+) mouse long-term hematopoietic stem cells. Proc. Natl Acad. Sci. USA 116, 25115–25125 (2019).
Forsberg, E. C. et al. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 1, e28 (2005).
Nestorowa, S. et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31 (2016).
Essers, M. A. et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908 (2009).
Baldridge, M. T., King, K. Y., Boles, N. C., Weksberg, D. C. & Goodell, M. A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010).
Bae, G. U. et al. Neogenin regulates skeletal myofiber size and focal adhesion kinase and extracellular signal-regulated kinase activities in vivo and in vitro. Mol. Biol. Cell 20, 4920–4931 (2009).
O’Leary, C. J. et al. Neogenin recruitment of the WAVE regulatory complex to ependymal and radial progenitor adherens junctions prevents hydrocephalus. Cell Rep. 20, 370–383 (2017).
Venezia, T. A. et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2, e301 (2004).
Wilson, NicolaK. et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell 16, 712–724 (2015).
Haas, S. et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell 17, 422–434 (2015).
Zhao, M. et al. N-cadherin-expressing bone and marrow stromal progenitor cells maintain reserve hematopoietic stem cells. Cell Rep. 26, 652–669 e656 (2019).
Laurenti, E. et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell 16, 302–313 (2015).
Scheicher, R. et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 125, 90–101 (2015).
Min, I. M. et al. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2, 380–391 (2008).
Galloway, A. et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 352, 453–459 (2016).
Deneault, E. et al. A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell 137, 369–379 (2009).
Adelman, E. R. et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 9, 1080–1101 (2019).
Sakaue-Sawano, A. et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498 (2008).
Huang, C. Y., Bredemeyer, A. L., Walker, L. M., Bassing, C. H. & Sleckman, B. P. Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur. J. Immunol. 38, 342–349 (2008).
Yamashita, M. & Passegue, E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 25, 357–372 e357 (2019).
Serafini, T. et al. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87, 1001–1014 (1996).
Dominici, C. et al. Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 545, 350–354 (2017).
Guo, C., Yang, W. & Lobe, C. G. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis 32, 8–18 (2002).
Xu, C. et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. 9, 2449 (2018).
Brunet, I. et al. Netrin-1 controls sympathetic arterial innervation. J. Clin. Invest. 124, 3230–3240 (2014).
LeBleu, V. S. et al. Identification of human epididymis protein-4 as a fibroblast-derived mediator of fibrosis. Nat. Med. 19, 227–231 (2013).
Sanjuan-Pla, A. et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236 (2013).
Challen, G. A. et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 44, 23–31 (2011).
Challen, G. A. et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15, 350–364 (2014).
Jeong, M. et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 23, 1–10 (2018).
Monnier, P. P. et al. RGM is a repulsive guidance molecule for retinal axons. Nature 419, 392–395 (2002).
Zhou, Z. et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev. Cell 19, 90–102 (2010).
Singbrant, S. et al. Canonical BMP signaling is dispensable for hematopoietic stem cell function in both adult and fetal liver hematopoiesis, but essential to preserve colon architecture. Blood 115, 4689–4698 (2010).
Yung, A. R., Nishitani, A. M. & Goodrich, L. V. Phenotypic analysis of mice completely lacking netrin 1. Development 142, 3686–3691 (2015).
Bin, J. M. et al. Complete loss of Netrin-1 results in embryonic lethality and severe axon guidance defects without increased neural cell death. Cell Rep. 12, 1099–1106 (2015).
Arai, F. et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118, 149–161 (2004).
Walter, D. et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 (2015).
Flach, J. et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014).
Ozmadenci, D. et al. Netrin-1 regulates somatic cell reprogramming and pluripotency maintenance. Nat. Commun. 6, 7398 (2015).
Sung, P. J. et al. Cancer-associated fibroblasts produce netrin-1 to control cancer cell plasticity. Cancer Res. 79, 3651–3661 (2019).
Stein, S. J. & Baldwin, A. S. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood 121, 5015–5024 (2013).
Asada, N. et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 19, 214–223 (2017).
Baccin, C. et al. Combined single-cell and spatial transcriptomics reveals the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 1, 38–48 (2020).
Ding, L., Saunders, T. L., Enikolopov, G. & Morrison, S. J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012).
Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013).
Gong, S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925 (2003).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I. et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We thank all technicians of the Trumpp, Mehlen and de Haan laboratories for technical assistance; S. Schmitt, M. Eich, K. Hexel and T. Rubner from the DKFZ Flow Cytometry Core, and W. Abdulahad, T. Bijma, G. Mesander and J. Teunis from UMCG Flow cytometry Unit facilities for their assistance; K. Reifenberg, P. Prückl, M. Schorpp-Kistner, A. Rathgeb and all members of the DKFZ and UMCG Laboratory Animal Core Facility for excellent animal welfare and husbandry. We also thank the DKFZ and ERIBA Genomics and Proteomics Core Facility for their assistance. This work was supported by the Heinrich F.C. Behr Foundation, Studienstiftung des deutschen Volkes (S.R.), by INCA, ERC, ANR, and Fondation Bettencourt (all to P.M.), Max Planck Society and the ERC-Stg-2017 (VitASTEM) (all to N.C.-W.), the FOR2033, FOR2674 and SFB873 funded by the Deutsche Forschungsgemeinschaft (DFG), the SyTASC consortium funded by the Deutsche Krebshilfe, the DKTK RiskY-AML Joint Funding program and the Dietmar Hopp Foundation (all to A.T). Studies in the Frenette laboratory were funded by the National Institutes of Health (DK056638, HL069438, HL116340 to P.S.F.). M.M. is a New York Stem Cell Foundation (NYSCF) Druckenmiller fellow. Studies in the de Haan laboratory were funded by the Netherlands Organization for Scientific Research/Mouse Clinic for Cancer and Ageing, the Landsteiner Foundation for Blood Transfusion Research (LSBR1703) and Marriage, a EU FP7 Marie Curie Initial Training Network (Contract number 316964).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.R., N.C.-W. and A.T. Methodology: S.R., A.F.S., J.P., N.R., P.S., L.L., S.L., M.M., B.G., A.R.R., D.Z., M.T., E.Z., B.D.-A., B.D., D.K., A.N., M.B., A.H.-W., K.S., C.K., P.Z., A.P., M.S. Investigation: S.R., A.F.S., J.P., M.M., S.L., N.R. Resources: M.H., P.M. Writing—original draft: S.R., A.F.S., N.C.-W. and A.T. Writing—review and editing: S.R., A.F.S., J.P., P.S., L.L., N.R., M.M., S.M.-F., P.S.F., M.H., L.B., P.M., G.d.H., N.C.-W. and A.T. Supervision: G.d.H., N.C.-W. and A.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Ayako Nakamura-Ishizu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Renders, S., Svendsen, A.F., Panten, J. et al. Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat Commun 12, 608 (2021). https://doi.org/10.1038/s41467-020-20801-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20801-0
This article is cited by
-
Depleting myeloid-biased haematopoietic stem cells rejuvenates aged immunity
Nature (2024)
-
The role of the haematopoietic stem cell niche in development and ageing
Nature Reviews Molecular Cell Biology (2024)
-
Implications of stress-induced gene expression for hematopoietic stem cell aging studies
Nature Aging (2024)
-
Clonal hematopoiesis and its impact on the aging osteo-hematopoietic niche
Leukemia (2024)
-
NSD2 E1099K drives relapse in pediatric acute lymphoblastic leukemia by disrupting 3D chromatin organization
Genome Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
