
Abstract
In addition to the serotonin 5-HT2A receptor (5-HT2AR), the dopamine D2 receptor (D2R) is a key therapeutic target of antipsychotics for the treatment of schizophrenia. The inactive state structures of D2R have been described in complex with the inverse agonists risperidone (D2Rris) and haloperidol (D2Rhal). Here we describe the structure of human D2R in complex with spiperone (D2Rspi). In D2Rspi, the conformation of the extracellular loop (ECL) 2, which composes the ligand-binding pocket, was substantially different from those in D2Rris and D2Rhal, demonstrating that ECL2 in D2R is highly dynamic. Moreover, D2Rspi exhibited an extended binding pocket to accommodate spiperone’s phenyl ring, which probably contributes to the selectivity of spiperone to D2R and 5-HT2AR. Together with D2Rris and D2Rhal, the structural information of D2Rspi should be of value for designing novel antipsychotics with improved safety and efficacy.
Similar content being viewed by others

Introduction
Dopamine is a neurotransmitter that controls numerous physiologic functions in the brain and peripheral nervous system via dopamine receptors of the G-protein-coupled receptor (GPCR) superfamily. In humans, five dopamine receptors (D1R–D5R) have been identified and have been classified according to their sequence, intracellular signaling, pharmacology, and localization as D1-class receptors (D1R and D5R) or D2-class receptors (D2R, D3R, and D4R)1,2,3. Thus, D2R is similar to both D3R and D4R, with 80 and 54% sequence identities, respectively, in their transmembrane helices4,5,6. D2R is highly distributed in the striatum, nucleus accumbens, and olfactory tubercle7,8, and it plays important pharmacologic roles in numerous human disorders related to dopaminergic dysfunction, including schizophrenia9,10,11 and Parkinson’s disease12,13.
D2R antagonists have been developed as antipsychotics to block dopaminergic transmission for the treatment of schizophrenia14. Antipsychotics are either typical or atypical; typical antipsychotics generally antagonize D2R, whereas atypical antipsychotics antagonize both D2R and the serotonin 5-HT2A receptor (5-HT2AR). Both of these groups at least improve the positive symptoms of schizophrenia15. However, they also are associated with a wide range of severe side effects, such as extrapyramidal symptoms, weight gain, metabolic disorders, and constipation16,17. Extrapyramidal symptoms are caused by excessive inhibition of D2R in the nigrostriatal pathway. Other side effects are primarily due to the undesired binding of antipsychotics to other aminergic receptors, such as the serotonin 5-HT2C receptor (5-HT2CR), which exhibits 46% sequence identity with 5-HT2AR.
GPCR structures have been successfully utilized for the structure-guided discovery of new ligands18,19. In D2-class receptors, D3R and D4R structures were determined in complex with the benzamide antipsychotics eticlopride and nemonapride (D3Reti and D4Rnem), respectively20,21. The inactive conformations of D2R have been described in complexes with risperidone (D2Rris), a pyridopyrimidine antipsychotic22, and with haloperidol (D2Rhal), a butyrophenone antipsychotic23. D2Rris and D2Rhal are practically identical, as shown by the RMSD values of Cα atoms between them (Supplementary Table 1). A G-protein-bound active conformation of D2R also was reported in complex with an agonist, bromocriptine (D2Rbro)24. Interestingly, the conformation of the extracellular loop (ECL) 2 in D2Rris and D2Rhal is entirely different from those of D3Reti, D4Rnem, and D2Rbro (Supplementary Fig. 1). Additionally, while the conformation of ECL1 is relatively conserved among the structures of D2-class receptors, Trp10023.50 on ECL1 of D2Rris is uniquely directed to the binding pocket (Supplementary Fig 1).
In this study, we describe the structure of D2R in complex with spiperone (D2Rspi), a butyrophenone typical antipsychotic that binds with high affinity to D2R, D3R, D4R, and 5-HT2AR25. We also present a structural comparison of D2Rspi with other D2R structures, D3Reti, and D4Rnem, in addition to 5-HT2AR complexed with risperidone (5-HT2ARris)26 and 5-HT2CR with ritanserin (5-HT2CRrit)27. The structure of D2Rspi given herein provides valuable information for the rational design of antipsychotics with improved receptor selectivity.
Results
Overall structure of D2Rspi
Because wild-type D2R is not expressed in Spodoptera frugiperda (Sf9) insect cells, we prepared a stable construct for crystallization trials. D2R was stabilized by the truncation of 34 N-terminal residues and the replacement of the intracellular loop (ICL) 3 with the thermostabilized apocytochrome b562RIL28 (D2R-bRIL). D2R-bRIL was further stabilized by the mutations S121K3.39 and L123W3.41 (here, superscripts indicate residue numbers according to the Ballesteros–Weinstein scheme15) and the replacement of bRIL with mbIIG, the loop-modified cytochrome b562IIG29 (D2R-mbIIG S121K3.39/L123W3.41, see Methods). The use of mbIIG instead of bRIL was essential to obtain D2R crystals. S121K3.39 is a mutation of the allosteric sodium ion binding site of class A GPCRs that mimics the presence of the sodium ion, therefore stabilizing the inactive state30,31. The S121K3.39, L123W3.41, and S121K3.39/L123W3.41 mutants, in addition to the stabilized construct, showed similar affinities for spiperone to the wild-type human D2R (Supplementary Table 2 and Supplementary Fig. 2), suggesting that these mutations did not substantially affect the binding of spiperone. Additionally, the L123W3.41 mutant showed similar antagonist activity to that of the wild-type human D2R by a transforming growth factor alpha (TGFα) shedding assay32, which measured the antagonist activities of wild-type and mutant D2R for spiperone against a fixed concentration of dopamine (Supplementary Table 3 and Supplementary Fig. 3). However, because the S121K3.39 mutation stabilizes the inactive state of D2R, the antagonist activities of the mutants with S121K3.39 could not be determined (Supplementary Table 3 and Supplementary Fig. 3). Eticlopride and sulpiride enhance the affinity for D2R in the presence of the sodium ion, whereas spiperone does not33. This enhancement is ascribed to an interaction network from the bound sodium ion in the allosteric binding site33. The S121K3.39 mutation decreased the affinity for eticlopride (Supplementary Table 2), suggesting that the side chain of Lys1213.39 does not sufficiently mimic the sodium ion in the allosteric binding site for the binding of eticlopride.
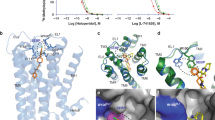
For crystallization, we generated an antibody recognizing the D2R structure (IgG3089) and prepared a novel Fab fragment (Fab3089) of IgG3089. Using the stabilized construct and Fab3089, we successfully obtained crystals of D2R (Supplementary Fig. 4a). The structure of D2R was determined in complex with spiperone at 3.1 Å resolution using an X-ray free-electron laser (Fig. 1, Table 1, and Supplementary Fig. 4). D2Rspi bound to Fab3089 at the extracellular region (Fig. 1a and Supplementary Fig. 4c), exhibiting a canonical GPCR fold with seven transmembrane helices (TM1–7) and an intracellular amphipathic helix 8 (H8) (Fig. 1). The D2Rspi structure demonstrated the inactive conformation because the seven helical bundle structure and the conformations of the four activation microswitches are more similar to those of the inactive state conformation in D2Rris than those of the active state conformation in D2Rbro (Supplementary Fig. 5 and Supplementary Table 1). We also compared the PIF motif of D2Rspi with those of the inactive state and the active state structures of the β2-adrenergic receptor to confirm D2Rspi demonstrated the inactive conformation, because D2Rris contains the I122A3.40 mutation in the motif.

a Overall structure of D2Rspi-Fab 3089 complex. Extracellular (b) and side (c) views of D2Rspi. Spiperone, D2R, Fab3089, and mbIIG are indicated in orange, green, cyan, and gray, respectively. The disulfide bond and the C-terminal segment of ECL2 are shown in yellow and magenta, respectively, in (b) and (c). Ext, extracellular; Int, intracellular.
There were no secondary structures in the ECLs and ICL1; ICL2 in D2Rspi was disordered. Spiperone was bound to the orthosteric binding site (Fig. 1 and Supplementary Fig. 4d). Like other class A GPCRs, the ligand-binding pocket was covered by the C-terminal segment of ECL2, which is stabilized by a disulfide bond between Cys1073.25 on TM3 and Cys18245.50 on ECL234 (Fig. 1).
Binding mode of spiperone
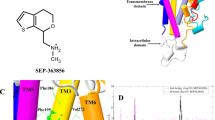
In D2Rspi, spiperone was surrounded by residues from TM2, 3, 5, 6, and 7 and ECL2 (Figs. 1 and 2a, b). The binding mode of spiperone was consistent with the results of the TGFα shedding assay (Supplementary Table 3) and of the ligand-binding assay for spiperone using the mutants35,36,37,38.

a Close-up view of the ligand-binding pocket of D2Rspi. Spiperone and D2R are indicated in orange and green, respectively. The side chains of the contact residues within 4.5 Å of spiperone are shown as green sticks. The side chain of W10023.50 is indicated in magenta. The side chains of the residues in the PIF motif are shown as blue sticks. b Diagram of the interactions between D2R and spiperone. c Fab3089 binding site. The side chains of I18345.51 of D2Rspi and Y55 of Fab3089 are shown as green and cyan sticks, respectively. d Vertical cross section of (a). Red and blue circles indicate the EBP and the bottom hydrophobic cleft, respectively.
The tertiary amine in the triazaspiro ring formed a salt bridge with Asp1143.32; this is strictly conserved in aminergic receptor structures (Fig. 2a, b). In D2R, the mutant of Asp1143.32 loses its affinities to both agonists and antagonists22,36. This interaction may be stabilized by the conserved hydrogen bond between Asp1143.32 and Tyr4167.43 39. On the opposite side of spiperone from Asp1143.32, there is hydrophobic contact between spiperone and Phe3896.51. This contact is likely essential for spiperone binding, given that F389A6.51 showed a 34-fold reduction in affinity for spiperone compared to that of wild-type D2R35. Additionally, there was contact between the triazaspiro ring and Ile18345.51, Ile18445.52, and Cys18245.50 on ECL2 (Fig. 2a, b). The loss of this contact with the I184A45.52 mutation significantly reduced the antagonist activity for spiperone (Supplementary Table 3), suggesting that the contact with Ile18445.52 is crucial for the antagonist activity of spiperone. The contact between Ile18345.51 and spiperone was influenced by the binding of Fab3089, because the side-chain conformation of Ile18345.51 was stabilized by Tyr55 of Fab3089 (Fig. 2c). The I183A45.51 mutant slightly increased the antagonist activity for spiperone (Supplementary Table 3).
On the extracellular side of the salt bridge, the phenyl ring of spiperone was bound in an extended binding pocket (EBP) between TM2 and TM3 (Fig. 2a, d). The EBP was formed by residues Val872.57, Trp902.60, Val912.61, Leu942.64, Trp10023.50, Phe1103.28, Val1113.29, and Cys18245.50. The drastic reduction of the antagonist activity of W100A23.50 for spiperone indicates that Trp10023.50 is crucial for maintaining EBP conformation (Supplementary Table 3). Because of the triazaspiro ring rigidity and the direction of the conserved salt bridge between the tertiary amine and Asp1143.32, the EBP is likely essential for spiperone binding. In the EBP of D2Rspi, spiperone’s phenyl ring forms hydrophobic contacts with Trp902.60, Val912.61, Leu942.64, Phe1103.28, and Val1113.29 (Fig. 2a, b). Mutations of most of these residues resulted in considerably decreased antagonist activity for spiperone (Supplementary Table 3).
On the intracellular side of the salt bridge, spiperone’s fluorophenyl ring penetrated deeply into the ligand-binding pocket, binding in the bottom hydrophobic cleft (Fig. 2a, b, d). Similar bottom hydrophobic clefts have been observed in the structures of the histamine H1 receptor40, 5-HT2AR26, and 5-HT2CR27. In the cleft, the fluorophenyl ring formed a CH–π interaction with Cys1183.36; hydrophobic interactions with Thr1193.37, Ile1223.40, Ser1975.46, Phe1985.47, and Phe3826.44; and edge-to-face π interactions with Trp3866.48 and Phe3906.52 (Fig. 2a, b). Trp3866.48 is a microswitch in the CWxP motif. Indeed, W386L6.48 affected activation by dopamine and showed no antagonist activity for spiperone (Supplementary Table 3 and Supplementary Fig. 3). The interaction with Phe3906.52 is essential for the binding of spiperone, because F390A6.52 drastically decreases the affinity for spiperone35. By contrast, contact with Phe1985.47 is not essential for the binding of spiperone, given that F198A5.47 showed a similar affinity for spiperone to that of wild-type D2R35. The S197A5.46 mutant also showed an affinity for spiperone similar to that of the wild-type D2R36,37,38, thus strengthening contact between spiperone and the Cβ atom of Ser1975.46.
Ile1223.40 and Phe3826.44 belong to the PIF motif located at the bottom of the ligand-binding pocket in aminergic receptors41. When activated, the PIF motif conformationally rearranges, with the outward movement of the cytoplasmic side of TM6. The PIF motif conformation in D2Rspi is that of the inactive state (Supplementary Fig. 5). Thus, the direct interactions of spiperone with Ile1223.40 (3.7 Å distance) and Phe3826.44 (3.6 Å distance) in the PIF motif may block the conformational rearrangements of the PIF motif and help to stabilize the inactive conformation, as has been observed in the structural studies of 5-HT2AR26 and 5-HT2CR27. Of the 21 contact residues, 20 were conserved between D2R and D3R (Supplementary Table 4), reflecting the similarly high affinity of these receptors for spiperone42.
Comparison with D2Rris, D2Rhal, and D2Rbro
There are striking structural differences in the ligand-binding pocket of D2Rspi and the other inactive state structures of D2R: D2Rris and D2Rhal (Fig. 3a, b and Supplementary Table 1). In D2Rspi, the ligand-binding pocket was covered by the C-terminal segment of ECL2 (Fig. 3a–c), on which the side chains Ile18345.51 and Ile18445.52 pointed to the entrance and the bottom of the ligand-binding pocket, respectively. This conformation was conserved in the structures of other aminergic receptors, including D3R, D4R, and 5-HT2AR (Supplementary Fig. 6). In D2Rris and D2Rhal, however, ECL2 extended away from the top of the receptor core (Fig. 3a, b, d). In this conformation, Ile18345.51 was buried in the hydrophobic core outside the ligand-binding pocket, and Ile18445.52 reached the top of the ligand-binding pocket (Fig. 3a, b). Thus, Ile18445.52 did not contact with risperidone in D2Rris, while Ile18445.52 contacted with spiperone in D2Rspi. These findings were consistent with I184A45.52 showing similar antagonist activity for risperidone with the wild-type (Supplementary Table 3).

a Extracellular view of the superposition of D2Rspi and D2Rris. The side chains of disulfide bridge, I18345.51 and I18445.52 are shown as sticks. Red arrows indicate the shift of helices in D2Rris with the distance relative to D2Rspi. b Extracellular view of ECL1 and ECL2 of D2Rspi, D2Rris, D2Rhal and D2Rbro. The side chains of the disulfide bridge, W10023.50, I18345.51, and I18445.52 are shown as sticks. Surface representation of D2Rspi (c) and D2Rris (d) viewed from the extracellular side. ECL1 and ECL2 are pink and yellow, respectively. e The EBP of D2Rspi and the corresponding part of D2Rris, D2Rhal and D2Rbro. The side chains of W902.60 and F1103.28 are shown as sticks. f Side view of the superposition of D2Rspi, D2Rris, D2Rhal and D2Rbro around TM5 and ECL2. Red arrows indicate the shift of the extracellular half of TM5 in D2Rris and D2Rhal relative to D2Rspi. In (b), (e), and (f), D2Rspi (green), D2Rris (cyan), D2Rhal (purple), D2Rbro (olive), spiperone (orange), risperidone (magenta), haloperidol (ivory), and bromocriptine (lightblue) are shown.
On ECL1, D2R demonstrated the diverse side-chain conformation of Trp10023.50. In D2Rspi, Trp10023.50 interacted with the conserved disulfide bond (Fig. 3b). The conformation was highly conserved in the structures of class A GPCRs (Supplementary Fig. 6). The interaction between a disulfide bond and tryptophan is often observed in protein structures; this may contribute to protecting the disulfide bond and stabilizing the structure43,44. Trp10023.50 in D2Rhal exhibited similar side-chain conformation with that of D2Rspi, although it did not contact with the disulfide bond because of ELC2 flipping (Fig. 3b). In D2Rris, Trp10023.50 moves to the ligand-binding pocket and forms a T-stacking interaction with risperidone’s tetrahydropyridopyrimidinone ring22 (Fig. 3b). Trp10023.50 in D2Rris forms a hydrophobic patch with Leu942.64 and Ile18445.52, covering the ligand-binding pocket. Despite this, the ligand-binding pocket in D2Rris was more exposed to the extracellular solution compared with that in D2Rspi (Fig. 3c, d). W100A23.50 in D2R has been shown to reduce the residence times of several antipsychotics22. Based on these results, it was hypothesized that the hydrophobic patch in D2Rris contributes to the slow dissociation of antipsychotics22. However, the results were also consistent with the conformation observed in D2Rspi, in which Trp10023.50 stabilized the conformation of ECL2 and EBP.
The EBP is uniquely observed in D2Rspi among the inactive state structures of D2R. In D2Rspi, the side chain of Phe1103.28 that creates the EBP was flipped compared with those of D2Rris and D2Rhal (Fig. 3e), D3Reti, and the 5-HT2 receptors. In D2Rspi, the flipped Phe1103.28 side chain formed a stacking interaction with Trp902.60 (Fig. 3e).
D2Rris and D2Rhal also possessed the bottom hydrophobic cleft (Fig. 3f). However, the conformation of this cleft in D2Rris and D2Rhal was altered by the shift of the extracellular half of TM5 in the ligand-binding pocket relative to that in D2Rspi (Fig. 3a, f). Resultantly, Phe1895.38 and Ser1935.42 contacted risperidone in D2Rris, though these residues did not contact haloperidol in D2Rhal (Fig. 3f). The shift of TM5 observed in D2Rris and D2Rhal is likely inhibited in D2Rspi by steric contact between the extracellular end of TM5 and ECL2. Indeed, Phe1895.38 in D2Rris and D2Rhal occupied a similar position with Ile18445.52 in D2Rspi in the ligand-binding pocket (Fig. 3f). The shift can also be affected by the I122A3.40 mutation introduced to stabilize the receptor in D2Rris and D2Rhal22,23. In D2Rris and D2Rhal, the Cβ atom of Ala1223.40 was in contact (3.8 Å distance) with the carbonyl oxygen atom of Ser1975.46 (Supplementary Fig. 7a); when the side chain of Ala1223.40 was replaced by isoleucine using Coot45, the resulting side chain formed steric contacts (less than 3.0 Å distance) with the surrounding residues, including Ser1975.46 and Pro2015.50 on TM5, and with risperidone or haloperidol in any of the seven allowed side-chain rotamers for isoleucine (Supplementary Fig. 7b).
Unlike inactive state structures, D2Rbro showed a typical active state conformation in the microswitches and the seven helical bundles (Supplementary Fig. 5). Conformations of ECL2 and the extracellular end of TM5 in D2Rbro were more similar to those of D2Rspi than those of D2Rris and D2Rhal. (Fig. 3b, f). Trp10023.50 of D2Rbro existed at a similar position with those in D2Rspi and D2Rhal but with a different side-chain rotamer (Fig. 3b). EBP in D2Rspi was not observed in D2Rbro (Fig. 3e).
Comparison with 5-HT2ARris and 5-HT2CRrit
The conformations of the extracellular end of TM5, the conserved Trp23.50 on ECL1, the C-terminal segment of ECL2, and the disulfide bridge between ECL2 and TM3 in 5-HT2ARris and 5-HT2CRrit were similar to those of D2Rspi (Fig. 4a, b and Supplementary Fig. 6). On the C-terminal segment of ECL2, the Leu22845.51 and Leu22945.52 residues of 5-HT2AR contacted risperidone and zotepine, respectively26, and the Val20845.52 residue of 5-HT2CR interacted with ritanserin27. The high conservation of the residues in the ligand-binding pocket (Supplementary Table 4) and the structural similarity of 5-HT2ARris, 5-HT2CRrit, and D2Rspi explain why antipsychotics often bind to D2R, 5-HT2AR, and 5-HT2CR with high affinity.

Extracellular view of the superpositions of D2Rspi and either 5-HT2ARris (a) or 5-HT2CRrit (b). c The EBP of D2Rspi and the corresponding part of 5-HT2ARris. d The EBP of D2Rspi and the corresponding part of 5-HT2CRrit. e Vertical cross sections of D2Rspi and 5-HT2ARris. In this figure, D2Rspi (green), 5-HT2ARris (blue), 5-HT2CRrit (brown), spiperone (orange), risperidone (yellow), and ritanserin (gray) are shown.
Phe1103.28 and Trp902.60 forming EBP in D2Rspi corresponded with Trp1513.28 and Val1302.60 in 5-HT2AR and Trp1303.28 and Leu1092.60 in 5-HT2CR, respectively. In D2R, W90L2.60 reduced the antagonist activity more than F110W3.28 and similarly with W90L2.60/F110W3.28 (Supplementary Table 3). Spiperone shows a high affinity to D2R and 5-HT2AR but a low affinity to 5-HT2CR25. A potential explanation of this difference in affinity is that while Trp1513.28 in 5-HT2ARris can be flipped to form the EBP without any steric hindrance, Trp1303.28 in 5-HT2CRrit is challenging to flip because of the steric contact with Leu1092.60 (Fig. 4c, d). Thus, the EBP of D2Rspi and the putative EBP of 5-HT2ARris may contribute to spiperone’s higher selectivity for these receptors than for 5-HT2CR. The binding mode of spiperone in the EBP may be useful for designing selective D2R and 5-HT2AR antipsychotics.
A unique side-extended cavity was previously observed in the structure of 5-HT2AR between TM4 and TM5 that was suggested to contribute to the binding site of 5-HT2AR-selective drugs26. D2Rspi did not possess the side-extended cavity between TM4 and TM5 (Fig. 4e).
Comparison with D3Reti and D4Rnem
The conformation of D2Rspi was similar to that of D3Reti and D4Rnem, except for the extracellular half of TM6 (Fig. 5a, b). The conformation of the C-terminal segment of ECL2 in D2Rspi was almost identical to that of D3Reti and D4Rnem (Fig. 5a, b). On ECL2, Ile18345.52 in D3Reti and Leu18745.52 in D4Rnem contacted eticlopride20 and nemonapride21, respectively, similar to Ile18445.52 in D2Rspi, which contacts spiperone. A previous study showed that eticlopride and nemonapride bind just above the bottom hydrophobic cleft in D3Reti and D4Rnem22 (Supplementary Fig. 8a), but this is different from the binding of spiperone in D2Rspi, risperidone in D2Rris, and haloperidol in D2Rhal. To interact with these benzamide antipsychotics, the extracellular half of TM6 exhibited a greater tilt toward TM3 in D3Reti and D4Rnem than in D2Rspi, D2Rris, and D2Rhal (Fig. 5a, b and Supplementary Fig. 8b). Thus, His3496.55 in D3Reti and His4146.55 in D4Rnem interacted with eticlopride and nemonapride, respectively, whereas no contact was made between spiperone and His3936.55 in D2Rspi (Supplementary Fig. 8b). Due to the large tilt of TM6, the distances between Cys3.36 and Phe6.52 in D3Reti and D4Rnem were approximately 1.5 and 2.0 Å closer, respectively, than in D2Rspi (Fig. 5a, b and Supplementary Fig. 8b). Together, the side-chain flip of Cys1183.36 and the tilt of TM6 resulted in the closure of the bottom hydrophobic cleft in D3Reti and D4Rnem (Fig. 5a, b and Supplementary Fig. 8a, b). Conversely, the insertion of spiperone’s fluorophenyl ring between Cys3.36 and Phe6.52 created the bottom hydrophobic cleft and inhibited the large tilt of TM6 in D2Rspi. D2-class receptors show high affinities for eticlopride, nemonapride, spiperone, and risperidone25, and display conserved residues in the ligand-binding pocket. Therefore, it is likely that these receptors show a similar conformation with D3Reti and D4Rnem when they bind to benzamide antipsychotics and with D2Rspi, D2Rris, and D2Rhal when they bind to butyrophenone or pyridopyrimidine antipsychotics (Supplementary Fig. 8c).

Extracellular view of the superpositions of D2Rspi and either D3Reti (a) or D4Rnem (b). c The EBP of D2Rspi and the corresponding part of D3Reti. d Superposition of the EBP of D2Rspi and D4Rnem. In this figure, D2Rspi (green), D3Reti (yellow), D4Rnem (pink), spiperone (orange), eticlopride (blue), and nemonapride (red) are shown.
Spiperone and nemonapride show high affinities for D2-class receptors25. In D3R, the putative EBP was closed by the side chain of Phe1063.28 (Fig. 5c). If the Phe1063.28 is flipped, D3R can form an EBP similar to that of D2Rspi with the conserved residues around this region. The EBP in D2Rspi was observed at a position similar to that of the EBP in D4Rnem that binds the phenyl ring of nemonapride21 (Fig. 5d), although the contact residues were not conserved between D2R and D4R (Supplementary Table 4).
Discussion
D2Rspi was observed to differ substantially from the other inactive state structures of D2R, D2Rris, and D2Rhal, especially in ECL2, forming the entrance part of the ligand-binding pocket. This suggests that ECL2 is highly dynamic in the inactive state of D2R. The residues on ECL2 in D2R have been mapped by the substituted-cysteine accessibility method46. In that study, the sulfhydryl groups of I183C45.51 and I184C45.52 reacted with charged sulfhydryl-specific reagents, indicating that these residues are water-accessible. The binding of N-methylspiperone to I183C45.51 and I184C45.52 was inhibited by the reaction with sulfhydryl-specific reagents, indicating that Ile18345.51 and Ile18445.52 were directed toward the ligand-binding pocket. Ile18345.51 is less likely to contact the ligand because N-methylspiperone binding to I183C45.51 was inhibited only by a bulkier sulfhydryl-specific reagent, MTSET. I184C45.52 reduced the affinities for nemonapride and N-methylspiperone, suggesting that Ile18445.52 contacts nemonapride and N-methylspiperone. These results are consistent with the conformation of ECL2 observed for D2Rspi but not with that of D2Rris or D2Rhal.
The difference of the ECL2 conformation can be caused by the bound ligand, although D2Rris and D2Rhal show very similar ECL2 conformation. Indeed, ECL2 in 5-HT2AR moves slightly to bind a different ligand26. An MD simulation study also suggested these dynamics of the ECL2 of D2R47 and reported that the helical conformation of ECL2 observed in D2Rris tended to unwind toward an extended conformation, similar to that of D3Reti, regardless of the bound ligand, including spiperone, risperidone, or eticlopride47. The unwinding involves a drastic rearrangement of the side chain of Ile18345.51, dissociating from a hydrophobic pocket. The study also suggested that the ECL2 conformation observed in D2Rris represents a higher energy state than the extended conformation. Considering the structural similarity between D2Rspi and D3Reti, the conformation of ECL2 in D2Rspi likely corresponds to a lower energy state conformation.
Currently, there is a need for novel, safer antipsychotics that bind selectively to 5-HT2AR and D2R. In this study, we revealed that the ligand-binding pocket of D2R forms more than two different conformations in the inactive state. Moreover, we showed that the EBP in D2Rspi and the putative EBP in 5-HT2ARris could be used as the binding site for selective atypical antipsychotics. D2Rhal was used for the structure-based discovery of selective ligand23. The use of multiple different conformations in the structure-based design instead of a single conformation clearly increased the possibility of finding high-affinity compounds. Together with D2Rris, D2Rhal, and 5-HT2ARris, the structure of D2Rspi can be utilized for a rational, structure-based design of new antipsychotics with low side effects.
Methods
Protein engineering for structure determination
The coding sequence of human D2R (UniProt ID P14416) was synthesized by TAKARA Bio. D2R was stabilized by removing the N-terminal 34 residues, by introducing two mutations (S121K3.39 and L123W3.41)30,48 and by replacing ICL3 (Lys2215.70 to Leu3636.25) with the loop-modified cytochrome b562IIG mutant (D2R-mbIIG S121K3.39/L123W3.41)29. mbIIG contains four mutations (M7W, R98I, H102I, and R106G), and residues 41–65 have been replaced with the Gly-Ser-Gly-Ser-Gly linker to increase thermostability and reduce conformational variation. All the constructs were prepared by high-throughput fluorescent-based screening in Saccharomyces cerevisiae. A 30 ng of SmaI-linearized plasmid pDDGFP2 and 3 µl of the PCR reaction mixtures were co-transformed into S. cerevisiae strain FGY21749. Transformants harboring the plasmid encoding the receptor were selected on an agar plate without uracil [0.192% (w/v) yeast synthetic dropout media without uracil (Sigma), 0.67% (w/v) yeast nitrogen base without amino acids (BD), 2% (w/v) agar and 2% (w/v) glucose]. The S. cerevisiae transformant was cultured in 5 ml of a medium [0.192% (w/v) yeast synthetic dropout media without uracil, 0.671% (w/v) yeast nitrogen base without amino acids and 2% (w/v) glucose] at 30 °C for 24 h. The generated plasmid encoding the receptor was isolated from S. cerevisiae with the Miniprep Kit (Qiagen) by disrupting cells with 0.5 mm glass beads50. The construct was subcloned into the pFastBac1 vector (Invitrogen), with a C-terminus tobacco etch virus (TEV) protease cleavage site, green fluorescent protein (GFP), and an octa-histidine tag.
Protein expression and purification
The stabilized D2R was expressed in Sf9 cells using a Bac-to-Bac baculovirus expression system (Invitrogen). The Sf9 cells were infected at 1.5 × 106 cells/ml, at a multiplicity of infection (MOI) of 0.05, and were harvested 84 h later. Cell pellets were resuspended with hypotonic buffer (10 mM HEPES, pH 7.5, 20 mM KCl, and 10 mM MgCl2) and were repeatedly washed and centrifuged in high osmotic buffer (10 mM HEPES, pH 7.5, 1 M NaCl, 20 mM KCl, and 10 mM MgCl2) containing EDTA-free complete protease inhibitor cocktail (Roche) to purify the cell membranes. The purified membranes were solubilized for 2 h at 4 °C in solubilization buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 1% (w/v) n-dodecyl-ß-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma-Aldrich), and 20% (v/v) glycerol) supplemented with 2 mg/ml iodoacetamide (Wako Pure Chemical Industries, Ltd), 200 µM spiperone (Sigma-Aldrich), and the protease inhibitor cocktail. Insoluble materials were removed by centrifugation, and the supernatants were incubated with TALON metal affinity resin (Clontech) for 10 h at 4 °C. The resin was washed with 10 column volumes (CV) of wash buffer I (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 20 mM imidazole, 10 mM MgCl2, 8 mM ATP, and 100 µM ligand) and 10 CV of wash buffer II (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 20 mM imidazole, and 100 µM ligand). The protein was eluted in 4 CV of elution buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 200 mM imidazole, and 100 µM ligand) and concentrated to 2.5 ml with a 100-kDa molecular weight cutoff Amicon Ultra-15 concentrator (Millipore). The imidazole was removed using a PD-10 column (GE Healthcare). The desalted protein was loaded onto Ni-NTA Superflow resin (Qiagen) and incubated for 10 h. The resin was washed with 10 CV of Ni wash buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.03% (w/v) DDM, 0.006% (w/v) CHS, 20 mM imidazole, and 100 µM ligand) and eluted with 3 CV of Ni elution buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.03% (w/v) DDM, 0.006% (w/v) CHS, 400 mM imidazole, and 100 µM ligand). The imidazole was removed using a PD-10 column, and the sample was then incubated with His-tagged TEV protease (expressed and purified in-house) for 10 h. TEV protease, cleaved His-tagged GFP and uncleaved protein were removed by passing the suspension through Ni Sepharose High Performance resin (GE Healthcare).
Antibody generation
All the animal experiments conformed to the guidelines of the Guide for the Care and Use of Laboratory Animals of Japan and were approved by the Kyoto University Animal Experimentation Committee (approval no. Medkyo16043). As the antigen, we used a stabilized D2R (D2R-mbIIG S121K3.39/L123W3.41). Purified antigen was reconstituted into liposomes containing chicken egg yolk phosphatidylcholine (Avanti) and monophosphoryl lipid A (Sigma-Aldrich). MRL/lpr mice were immunized three times at two-week intervals with 0.1 mg of the proteoliposome D2R antigen. Single cells were harvested from mice spleens and were fused with NS-1 myeloma cells. To select antibodies that recognized the 3D structure of human D2R, we performed a multi-step screening method, using D2R-i3d, which lacks residues of the N-terminal and ICL3, at each step, which included liposome-ELISA, denatured ELISA, and fluorescence size-exclusion chromatography. The collected clones were evaluated using a Biacore T100 protein interaction analysis system (GE Healthcare) and were subsequently isolated by limiting dilution to establish monoclonal hybridoma cell lines. The resulting immunoglobulin-G (IgG3089) was purified with HiTrap Protein G HP (GE Healthcare) followed by proteolytic cleavage with papain (Nacalai Tesque). The Fab fragment (Fab3089) was then purified by size-exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare) and affinity chromatography with a Protein A Sepharose 4 Fast-Flow column (GE Healthcare). The sequence of Fab3089 was determined via standard 5′-RACE using total RNA isolated from hybridoma cells.
Crystallization
The D2R–Fab3089 complex was prepared by mixing the purified D2R-mbIIG S121K3.39/L123W3.41 and Fab3089 at a molar ratio of 1:1.2 for 1 h on ice. The mixture was injected onto a Superdex 200 10/300 GL column (GE Healthcare), and the fractions containing the complex were concentrated to approximately 30 mg/ml with a 50-kDa molecular weight cutoff Amicon Ultra-15 concentrator (Millipore). The D2R–Fab3089 in complex with spiperone was reconstituted in LCP by mixing approximately 30 mg/ml protein solution with monoolein and 10% w/w cholesterol at a volume ratio of 2:3 (protein:lipid) using two 100-μl Hamilton syringes and a syringe coupler. One syringe and a coupler were then removed, and a cleaning wire was inserted into the protein-laden LCP in the other syringe51,52. Approximately 10 μl of the protein-laden LCP was extruded from the syringe with the wire and was soaked in a 0.6-ml tube filled with precipitant solution (0.1 M Tris-HCl, pH 8.0, 0.1 M CH3COOLi, 28–32% PEG400, 5% dimethyl sulfoxide, 0.01 M ATP, and 1 mM spiperone) and incubated at 20 °C. Microcrystals appeared after 2 days, growing to a maximum size of 20 × 2 × 2 µm3 within a week.
Data collection using an X-ray free-electron laser
The data were collected at beamline BL3 of the SPring-8 Angstrom Compact Free-Electron Laser (SACLA)53 (Hyogo, Japan) by the serial femtosecond crystallography technique using 1.5 × 1.5 µm2 microbeams focused by Kirkpatrick–Baez mirrors54 with a short-working-distance octal multiport CCD detector with eight sensor modules55. The data were collected at 7 keV with a pulse duration of approximately 10 fs and a repetition rate of 30 Hz. To inject the microcrystals, LCP was loaded into a sample cartridge through a needle connected to a syringe.
After centrifugation at 2000g for 10 s, the cartridge was mounted in a high-viscosity micro-extrusion injector with a nozzle diameter of 100 µm51,52,56. The injector was set in a chamber filled with helium gas in the Diverse Application Platform for Hard X-ray Diffraction in SACLA (DAPHNIS) set-up57 and was maintained at a constant 20 °C. A total volume of 60 μl of LCP was injected at a flow rate of 420 nl/min. Data collection was guided by a real-time data processing pipeline58 based on Cheetah59. Data processing and indexing were performed using CrystFEL 0.8.060,61 and XGANDALF (https://onlinelibrary.wiley.com/iucr/doi/10.1107/S2053273319010593), respectively. The total number of collected, hit, and indexed images were 351,326, 11,373, and 9,464, respectively. The dataset was merged by process_hkl in the CrystFEL suite, without scaling. A per-image resolution cutoff was applied by using the –push-res = 1.2 option to account for variations in the crystal quality.
Structure determination and refinement
The structure was determined by molecular replacement with Phaser62 software using the structures of the transmembrane region of D3R (PDB ID: 3PBL), cytochrome b562RIL (PDB ID: 1M6T), and the Fab fragment (PDB ID: 1NGZ) as the search models. Refinements were performed using phenix.refine63 in reciprocal space against experimental structure factors, followed by manual examination and rebuilding of the refined coordinates in Coot45. Fourteen TLS groups were used that were chosen by the phenix.find_tls_groups tool63. Spiperone was modeled using 2Fo-Fc map, Fo-Fc map, and polder map64 (Supplementary Fig. 4d). Statistics for the data collection and refinement are shown in Table 1. The Ramachandran statistics analyzed using MolProbity65 were as follows: 98.0% in the favored region, 2.0% allowed, with no outliers. Figures were prepared using Cuemol (http://www.cuemol.org/) and PyMOL (https://www.pymol.org/).
Radioligand-binding assay
The mutants were prepared using the primers listed in Supplementary Table 5. The radioligand-binding assay was performed using HEK293 cell or Sf9 cell membranes that expressed the receptor. The wild-type or a mutant D2R was transfected with a pCAGGS plasmid into HEK293 cells using a FuGENE HD transfection reagent (10 μg of plasmid, 50 μl of FuGENE HD solution per 10-cm culture dish). The membranes were prepared as described in the “Protein expression and purification” section. The protein concentration of the membrane was determined by the bicinchoninic acid (BCA) method (Thermo Fisher Scientific) with bovine serum albumin as a standard. The membranes were stored at –80 °C until use. All the experiments were performed in triplicate (with independent expressions) in a total volume of 200 µl. The membranes were dispersed with binding assay buffer (50 mM Tris-HCl, pH 7.5, and 150 mM NaCl). Then, 0.5–5 µg of the membranes were incubated for 2 h at room temperature with [3H]-spiperone (Perkin Elmer) at concentrations of 0.31–20 nM or [3H]-raclopride (Perkin Elmer) at concentrations of 0.20–100 nM. Unifilter-96 GF/B filter plates (Perkin Elmer) were pre-soaked in 0.3% polyethyleneimine (PEI, Nacalai Tesque) for 30 min to reduce non-specific binding. Non-specific binding was determined in the presence of 100 μM spiperone (Sigma) or raclopride (Tocris). For the competition-binding assay, 2 μg of membranes were incubated for 2 h at room temperature with 30 nM of [3H]-raclopride and unlabeled eticlopride at concentrations ranging from 0.01 nM to 1.0 μM. Samples were harvested with Unifilter-96 GF/B filter plates and the unbound ligand was washed three times with distilled water using a FilterMate Cell Harvester system (Perkin Elmer). After adding 20 μl of MicroScint-20 (PerkinElmer), the bound [3H]-spiperone or [3H]-raclopride was quantified with a MicroBeta2 scintillation counter (PerkinElmer). The data were analyzed by nonlinear curve fitting using GraphPad Prism 5 software. To determine Kd value of the receptors for spiperone, we used the equation accounting for ligand depletion. Binding data are reported as the mean ± SEM.
TGFα shedding assay
The antagonist activity of spiperone for the mutant D2Rs was determined by the TGFα shedding assays32. Briefly, a pCAGGS plasmid encoding the human wild-type or a mutant D2R (full-length, untagged), together with pCAGGS plasmids that encoded the chimeric Gαq/i3 subunit and alkaline phosphatase-tagged TGFα (AP-TGFα; human codon optimized), were transfected into HEK293A cells that were negative for mycoplasma contamination (MycoAlert Mycoplasma detection kit, Lonza) by using a PEI transfection reagent (PEI MAX MW 40,000; Polysciences). The chimeric Gαq/i3 subunit comprises the Gαq backbone and the Gαi3-derived 6-amino acid C-terminus, and it couples with Gi-coupled D2R, but induces a Gq-dependent TGFα shedding response32. For each 10-cm culture dish, we used 1 µg of D2R plasmid, 0.5 µg of Gαq/i3 plasmid, 2.5 µg of AP-TGFα plasmid and 25 µl of 1 mg/ml PEI solution. After culturing for one day at 37 °C in a 5% CO2 incubator, the transfected cells were harvested by trypsinization, washed once with Hank’s balanced salt solution (HBSS) containing 5 mM HEPES (pH 7.4), and resuspended in 30 ml of the HBSS-containing HEPES. The cell suspension was seeded in a 96-well culture plate (“cell plate”) at a volume of 80 µl per well and incubated for 30 min in the CO2 incubator. To determine the antagonist activity of spiperone or risperidone, cells were pretreated with 3.2-fold-titrated concentration of the antagonists (final concentrations of 32 pM–1 µM for spiperone or 320 pM–10 µM for risperidone; 10 µl per well) for 5 min and stimulated with dopamine (final concentration of 1 µM; 10 µL per well). To determine the agonist activity of dopamine, vehicle (10 µL per well) were predispensed before cell seeding and 3.2-fold-titrated concentration of dopamine (final concentrations of 1 nM–32 µM; 10 µl per well) was added to the cells. For all the experiments, the compounds were diluted in 0.01% bovine serum albumin- and HBSS-containing HEPES. After incubation with dopamine for 1 h, the cell plate was spun at 190 × g for 2 min and conditioned medium (CM; 80 μl per well) was transferred to an empty 96-well plate (“CM plate”). Alkaline phosphatase reaction solution (10 mM p-nitrophenylphosphate, 120 mM Tris-HCl, pH 9.5, 40 mM NaCl, and 10 mM MgCl2) was dispensed into the cell plates and CM plates (80 µl). The absorbance of the plates at 405 nm was measured using a microplate reader (SpectraMax 340 PC384, Molecular Devices) before and after incubation for 1 or 2 h at room temperature. Ligand-induced AP-TGFα release was obtained by calculating AP activity in conditioned media and subtracting a vehicle-treated spontaneous AP-TGFα signal32. Using Prism 8 software (GraphPad Prism), the AP-TGFα release signals were fitted with a four-parameter sigmoidal concentration–response curve, from which EC50 or IC50 and Emax values were obtained. Negative logarithmic values of EC50 (pEC50) were used to calculate the mean and SEM.
The equilibrium dissociation constant (KB) was calculated for each experiment performed in parallel from the IC50 values (for spiperone and risperidone), an EC50 value (for dopamine), a Hill slope (K, for dopamine), and the tested concentration of dopamine (A; 1 µM), as follows66:
The resulting KB values were logarithmically transformed and their negative values (pKB) were used to calculate the difference between the pKB values (∆pKB) for a mutant (MT) and the wild-type (WT) receptor, derived from parallelly conducted experiments, as follows:
Mean and SEM values of the pKB and the ∆pKB values were calculated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The coding sequence of human D2R is available in UniProt with accession code P14416. The protein coordinate and atomic structure factor have been deposited in the Protein Data Bank (PDB) with accession code 7DFP. The raw diffraction images have been deposited to CXIDB (https://cxidb.org/) with accession code 110. Other data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Kebabian, J. W. Multiple classes of dopamine receptors in mammalian central nervous-system - involvement of dopamine-sensitive adenylyl cyclase. Life Sci. 23, 479–483 (1978).
Spano, P. F., Govoni, S. & Trabucchi, M. Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochemical Psychopharmacol. 19, 155–165 (1978).
Schwartz, J.-C., Giros, B., Martres, M.-P. & Sokoloff, P. The dopamine receptor family: molecular biology and pharmacology. Semin. Neurosci. 4, 99–108 (1992).
Grandy, D. K. et al. Cloning of the cDNA and gene for a human D2 dopamine receptor. Proc. Natl. Acad. Sci. USA 86, 9762–9766 (1989).
Sokoloff, P., Giros, B., Martres, M. P., Bouthenet, M. L. & Schwartz, J. C. Molecular-cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347, 146–151 (1990).
Vantol, H. H. M. et al. Cloning of the gene for a human dopamine D4-receptor with high-affinity for the antipsychotic clozapine. Nature 350, 610–614 (1991).
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M. & Caron, M. G. Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225 (1998).
Gerfen, C. R. Molecular effects of dopamine on striatal-projection pathways. Trends Neurosci. 23, S64–S70 (2000).
Snyder, S. H., Taylor, K. M., Coyle, J. T. & Meyerhoff, J. L. The role of brain dopamine in behavioral regulation and the actions of psychotropic drugs. Am. J. Psychiatry 127, 199–207 (1970).
Creese, I., Burt, D. R. & Snyder, S. H. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192, 481–483 (1976).
Howes, O. D. & Kapur, S. The dopamine hypothesis of schizophrenia: version III-the final common pathway. Schizophr. Bull. 35, 549–562 (2009).
Lang, A. E. & Lozano, A. M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 339, 1044–1053 (1998).
Lang, A. E. & Lozano, A. M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 339, 1130–1143 (1998).
Seeman, P., Chau-Wong, M., Tedesco, J. & Wong, K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc. Natl. Acad. Sci. USA 72, 4376–4380 (1975).
Meltzer, H. Y., Matsubara, S. & Lee, J. C. Classification of typical and atypical antipsychotic-drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharm. Exp. Ther. 251, 238–246 (1989).
Roth, B. L., Sheffler, D. J. & Kroeze, W. K. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Disco. 3, 353–359 (2004).
Muench, J. & Hamer, A. M. Adverse effects of antipsychotic medications. Am. Fam. Physician 81, 617–622 (2010).
de Graaf, C. et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor. J. Med. Chem. 54, 8195–8206 (2011).
Roth, B. L., Irwin, J. J. & Shoichet, B. K. Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol. 13, 1143–1151 (2017).
Chien, E. Y. et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 (2010).
Wang, S. et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386 (2017).
Wang, S. et al. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555, 269 (2018).
Fan, L. et al. Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 11, 1074 (2020).
Yin, J. et al. Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 584, 125–129 (2020).
Roth, B. L., Lopez, E., Patel, S. & Kroeze, W. K. The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches? Neuroscientist 6, 252–262 (2000).
Kimura, K. T. et al. Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 26, 121–128 (2019).
Peng, Y. et al. 5-HT2C receptor structures reveal the structural basis of GPCR polypharmacology. Cell 172, 719 (2018).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Chu, R. et al. Redesign of a four-helix bundle protein by phage display coupled with proteolysis and structural characterization by NMR and X-ray crystallography. J. Mol. Biol. 323, 253–262 (2002).
Yasuda, S. et al. Hot-spot residues to be mutated common in G protein-coupled receptors of class A: identification of thermostabilizing mutations followed by determination of three-dimensional structures for two example receptors. J. Phys. Chem. B 121, 6341–6350 (2017).
Toyoda, Y. et al. Ligand binding to human prostaglandin E receptor EP4 at the lipid-bilayer interface. Nat. Chem. Biol. 15, 18–26 (2019).
Inoue, A. et al. TGF alpha shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021 (2012).
Michino, M., Free, R. B., Doyle, T. B., Sibley, D. R. & Shi, L. Structural basis for Na+-sensitivity in dopamine D2 and D3 receptors. Chem. Commun. 51, 8618–8621 (2015).
Wheatley, M. et al. Lifting the lid on GPCRs: the role of extracellular loops. Br. J. Pharm. 165, 1688–1703 (2012).
Cho, W., Taylor, L. P., Mansour, A. & Akil, H. Hydrophobic residues of the D2 dopamine receptor are important for binding and signal transduction. J. Neurochem. 65, 2105–2115 (1995).
Mansour, A. et al. Site-directed mutagenesis of the human dopamine D2 receptor. Eur. J. Pharmacol.-Mol. Pharmacol. 227, 205–214 (1992).
Cox, B. A., Henningsen, R. A., Spanoyannis, A., Neve, R. L. & Neve, K. A. Contributions of conserved serine residues to the interactions of ligands with dopamine D2 receptors. J. Neurochem. 59, 627–635 (1992).
Woodward, R., Coley, C., Daniell, S., Naylor, L. H. & Strange, P. G. Investigation of the role of conserved serine residues in the long form of the rat D2 dopamine receptor using site-directed mutagenesis. J. Neurochem. 66, 394–402 (1996).
Michino, M. et al. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharm. Rev. 67, 198–213 (2015).
Shimamura, T. et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65–70 (2011).
Wacker, D. et al. Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 (2013).
Tang, L., Todd, R. D., Heller, A. & O’Malley, K. L. Pharmacological and functional characterization of D2, D3 and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J. Pharm. Exp. Ther. 268, 495–502 (1994).
Ioerger, T. R., Du, C. G. & Linthicum, D. S. Conservation of cys-cys trp structural triads and their geometry in the protein domains of immunoglobulin superfamily members. Mol. Immunol. 36, 373–386 (1999).
Bhattacharyya, R., Pal, D. & Chakrabarti, P. Disulfide bonds, their stereospecific environment and conservation in protein structures. Protein Eng. Des. Sel. 17, 795–808 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Shi, L. & Javitch, J. A. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc. Natl. Acad. Sci. USA 101, 440–445 (2004).
Lane, J.R. et al. Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. Elife 9, e52189 (2020).
Roth, C. B., Hanson, M. A. & Stevens, R. C. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J. Mol. Biol. 376, 1305–1319 (2008).
Shiroishi, M. et al. Platform for the rapid construction and evaluation of GPCRs for crystallography in Saccharomyces cerevisiae. Micro. Cell Fact. 11, 78 (2012).
Shiroishi, M. et al. Production of the stable human histamine H1 receptor in Pichia pastoris for structural determination. Methods 55, 281–286 (2011).
Nango, E. et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 354, 1552–1557 (2016).
Suno, R. et al. Crystal structures of human Orexin 2 receptor bound to the subtype-selective antagonist EMPA. Structure 26, 7–19 (2018). e15.
Tono, K. Beamline, experimental stations and photon beam diagnostics for the hard x-ray free electron laser of SACLA. New J. Phys. 15, 083035 (2013).
Yumoto, H. et al. Focusing of X-ray free-electron laser pulses with reflective optics. Nat. Photonics 7, 43–47 (2013).
Kameshima, T. et al. Development of an X-ray pixel detector with multi-port charge-coupled device for X-ray free-electron laser experiments. Rev. Sci. Instrum. 85, 033110 (2014).
Shimazu, Y. et al. High-viscosity sample-injection device for serial femtosecond crystallography at atmospheric pressure. J. Appl. Crystallogr. 52, 1280–1288 (2019).
Tono, K. et al. Diverse application platform for hard X-ray diffraction in SACLA (DAPHNIS): application to serial protein crystallography using an X-ray free-electron laser. J. Synchrotron Radiat. 22, 532–537 (2015).
Nakane, T. et al. Data processing pipeline for serial femtosecond crystallography at SACLA. J. Appl. Crystallogr. 49, 1035–1041 (2016).
Barty, A. et al. Cheetah: software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 47, 1118–1131 (2014).
White, T. A. et al. CrystFEL: a software suite for snapshot serial crystallography. J. Appl. Crystallogr. 45, 335–341 (2012).
White, T. A. et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 49, 680–689 (2016).
Mccoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Liebschner, D. et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D 73, 148–157 (2017).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Cheng, H. C. The power issue: determination of KB or Ki from IC50: a closer look at the Cheng-Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 46, 61–71 (2001).
Acknowledgements
This work was supported by grants from the Research Acceleration Program of the JST (S.I.), the X-ray Free-Electron Laser Priority Strategy Program from MEXT (T.S., S.I.), JSPS KAKENHI (Grant Nos. 24370044, 24121715, 26102725, 15H04338, 17K19349, 18H02388, and 20K21392 to T.S.; Grant No. 15K18376 to D.I.; 17K08264 to A.I.; JP19H05777 to S.I.) and the Mitsubishi Foundation (T.S.). This research was also supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) and the Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from AMED under Grant Numbers JP17am0101070 and JP20am0101079; the PRIME JP17gm5910013 (A.I.) and the LEAP JP17gm0010004 (A.I. and J.A.) from AMED. We thank Kayo Sato, Yuko Sugamura and Ayumi Inoue (Tohoku University) for optimization and technical assistance of the TGFα shedding assay. We thank the Radioisotope Research Center and Medical Research Support Center at the Kyoto University for instrumental support. X-ray crystallographic data were collected at SACLA (Proposal No. 2016B8060, 2017A8019, and 2017B8022). We acknowledge computational support from the SACLA HPC system and the Mini-K supercomputer system.
Author information
Authors and Affiliations
Contributions
D.I. designed constructs, expressed, purified, and crystallized the receptor. D.I. and T.U. generated the antibody. D.I., T.F., and C.M. prepared microcrystals. D.I., T.F., Y.Y., T.T., A.Y., and E.N. collected data at SACLA. T.N. processed the data. K.T. contributed to the beamline operation at SACLA. Y.S. and N.N. prepared mutants. A.I. and F.M.N.K. designed, performed, and analyzed the functional assay under J.A.’s supervision. H.A. and K.T.K. performed the binding assay. T.S. designed the constructs and solved and refined the structure as well as supervised the project. D.I, S.I., and T.S. wrote the manuscript. S.I. advised T.S. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature communications thanks Lei Shi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Im, D., Inoue, A., Fujiwara, T. et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat Commun 11, 6442 (2020). https://doi.org/10.1038/s41467-020-20221-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20221-0
This article is cited by
-
Neurotransmitter recognition by human vesicular monoamine transporter 2
Nature Communications (2024)
-
A bitopic agonist bound to the dopamine 3 receptor reveals a selectivity site
Nature Communications (2024)
-
Structural insights into the agonists binding and receptor selectivity of human histamine H4 receptor
Nature Communications (2023)
-
Structural basis of α1A-adrenergic receptor activation and recognition by an extracellular nanobody
Nature Communications (2023)
-
Dopamine receptor D4 (DRD4) negatively regulates UCP1- and ATP-dependent thermogenesis in 3T3-L1 adipocytes and C2C12 muscle cells
Pflügers Archiv - European Journal of Physiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
