
Abstract
Electroreduction uses renewable energy to upgrade carbon dioxide to value-added chemicals and fuels. Renewable methane synthesized using such a route stands to be readily deployed using existing infrastructure for the distribution and utilization of natural gas. Here we design a suite of ligand-stabilized metal oxide clusters and find that these modulate carbon dioxide reduction pathways on a copper catalyst, enabling thereby a record activity for methane electroproduction. Density functional theory calculations show adsorbed hydrogen donation from clusters to copper active sites for the *CO hydrogenation pathway towards *CHO. We promote this effect via control over cluster size and composition and demonstrate the effect on metal oxides including cobalt(II), molybdenum(VI), tungsten(VI), nickel(II) and palladium(II) oxides. We report a carbon dioxide-to-methane faradaic efficiency of 60% at a partial current density to methane of 135 milliampere per square centimetre. We showcase operation over 18 h that retains a faradaic efficiency exceeding 55%.
Similar content being viewed by others

Introduction
The electrochemical reduction of carbon dioxide (CO2) enables the storage of intermittent renewable energy in the form of chemical bonds1,2. CO2 emissions then become a valuable feedstock in the production of chemical fuels, enabling closing of the carbon cycle3,4,5. Methane (CH4) especially benefits from an existing widely-deployed infrastructure for its storage, distribution and utilization6,7,8.
This motivates the need for practical electrolyzers which convert CO2 to CH4 at high rates and energy efficiency9,10. Along the way to this goal, further progress is required in the electrocatalysts which facilitate the conversion chemistry11,12,13,14,15,16,17,18,19,20. By judicious adjustment of CO2 partial pressure, copper (Cu) catalysts have attained a 48% faradaic efficiency (FE) to CH4 (ref. 14). Fivefold twinned Cu nanowires have achieved a CH4 FE of 55% due to a high-density of edge sites available on twin boundaries15.
Until now, such systems have operated with good FEs of >50% only at modest current densities of <50 milliampere per square centimetre (mA cm−2), below the level needed according to technoeconomic analyses21. Electrolysis in systems where CO2 is fed in the gas phase overcomes mass transport limitations and as a result is operational at impressive current densities (>100 mA cm−2); however, this produces high local pH conditions, promoting the formation of C2+ products instead of CH4 (refs. 22,23,24).
With the goal of seeking the rational design of more efficient CH4 electrocatalysts, we began by examining the factors that influence the CO2 methanation pathway. We reasoned that, in flow cell systems, a high local pH results in a lowered driving force for pH-dependent water reduction, and reduces the availability of adsorbed hydrogen (*H) to Cu. Specifically, this militates against the protonation (transfer of *H) of *CO to generate *CHO25,26.
To remedy this, we sought to enhance the kinetics of the proton-transfer step: we would strive to increase H2O reduction to provide the necessary *H, but without significantly altering the binding energy of intermediates interacting with Cu active sites.
In this work, we design and synthesize a suite of ligand-stabilized cobalt oxide (CoO) nanoclusters. We show that judicious selection of CoO nanocluster size enables control over surface *H coverage for tuneable modulation of CO2 methanation. Optimization of CoO nanocluster size (2.5 nm) enables the cooperative system to achieve a methane FE of 60% at an operating current density of 225 mA cm−2 and continuous operation for over 18 h in a flow cell system, without significant loss in performance.
Results
Density functional theory calculations
We began by exploring CO2 methanation pathways using computational investigations. Recent studies27,28,29 of CO2 reduction pathways on Cu surface show that the *CO intermediate undergoes either a hydrogenation step (Eq. (1)) toward CH4, or a dimerization step (Eq. (2)) toward C2+ products.
Steering the CO2 reduction pathway toward CH4 requires enhancing the local *H availability to promote hydrogenation of the *CO intermediate over the dimerization (C–C coupling) step.
Coupling the Cu surface with *H generating motifs would provide a means to tune *H availability and thereby direct the reaction toward CH4. These motifs reduce H2O to produce *H, which will hydrogenate *CO on neighbouring Cu atoms to *CHO. We also compared the *CO hydrogenate energies on pure Cu via the H2O-assisting Heyrovsky mechanism vs. the *H transfer mechanism30,31 (0.63 vs. 0.43 eV, Supplementary Fig. 1): the *H transfer mechanism is more likely from a thermodynamic analysis.
We therefore introduced into density functional theory (DFT) models a series of motifs (metal and metal oxide) with varying *H binding energy on Cu as the catalyst for CO2 reduction reaction (CO2RR) and calculated the energetics of *CO hydrogenation by the *H transfer mechanism (see ‘Methods’). The *CO hydrogenation energy for the pure Cu surface is 0.43 eV (Fig. 1a); however, Cu favours the dimerization step toward C2+ products when the local *H availability is insufficient27,28,29. We considered a variety of modified Cu surfaces and found a volcano-like relationship between the *CO hydrogenation energies and the *H binding energies (Fig. 1a). Metal (as distinct from metal oxide) motifs possess strong *H binding energy, preventing them from donating *H for *CO hydrogenation. Some high-valence metal oxides (e.g. molybdenum and tungsten trioxides), exhibit a weaker *H binding energy but fail to stabilize *H adsorption near Cu.

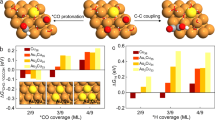
a Relationship between hydrogenation energy of *CO and surface *H binding energy for pure metal and metal oxide clusters on Cu surface. Red square for CoO, blue square for other oxides and grey circle for metals. The yellow dashed line is Cu baseline. b Schematic diagram of CO2 to CH4 on CoO cluster modified Cu surface. Tangerine for copper, violet for cobalt, red for oxygen, grey for carbon and white for hydrogen. c Hydrogenation energy of *CO for different sizes of CoO cluster. We introduced one cluster on the top of a 4-layer (5 × 2) Cu(111) surface.
The addition of CoO, which has an intermediate *H binding energy, results in the lowest reaction energy for hydrogenation of *CO (ΔE = 0.78 eV) among these motifs. This finding suggests that addition of CoO clusters could enhance the local *H availability and enable facile *CO hydrogenation formed on neighbouring Cu atoms and increase the FE toward CH4 (Fig. 1b).
We further investigated the influence of the CoO-cluster sizes on CH4 selectivity (Fig. 1c). The results show that a larger CoO-cluster size increases *CO hydrogenation activity; however, too large CoO clusters also enhance hydrogen evolution reaction (HER) activity which competes with CO2RR: indeed, calculations point to the need to optimize the cluster diameter.
Synthesis and characterization of CoO clusters
We sought experimentally to create such a system, beginning by synthesizing CoO clusters of four different sizes, 1.5, 2.1, 2.5 and 2.8 nm (see ‘Methods’). Prior studies have suggested that ligands may protect the oxidized clusters from reduction to the metallic phase during the synthesis32,33,34. The CoO clusters dispersed in solution are light purple; however, if the ligand is absent in the synthesis, this results in a black solution (Supplementary Fig. 2).
Seeking to characterize the dispersion and size distribution of the four CoO clusters, we performed scanning transmission electron microscopy (STEM, Fig. 2a–c) and high-resolution TEM (HRTEM, Fig. 2d). The homogeneous bright features in Fig. 2a–c and dark dots in Fig. 2d are the CoO clusters with different sizes of 1.5 ± 0.2, 2.1 ± 0.3, 2.5 ± 0.3 and 2.8 ± 0.3 nm, respectively (Fig. 2e–h).

a–d Transmission electron micrograph images and e–h corresponding size distribution of CoO clusters. The white light dots in (a–c) and dark black dots in (d) are the CoO clusters. Insets in (a–d) are the enlarged images showing the individual CoO cluster in red square. The scale bars in (a, b) and (c, d) are 5 and 10 nm, respectively.
Integrated CoO/Cu system
Using Nafion solution as a binding agent (see ‘Methods’), we spray-coated the CoO clusters onto a Cu sputtered polytetrafluoroethylene (Cu/PTFE) catalyst14,22 for CO2RR. To assess the impact of the clusters on the hydrogen evolution reaction (HER) activity of Cu, we performed linear sweep voltammetry in a flow cell system with Ar gas using 1 M KHCO3 electrolyte. This was carried out with CoO clusters of mean size 2.5 nm integrated with the Cu/PTFE catalyst (denoted CoO-2.5 nm/Cu/PTFE). As controls, bare Cu/PTFE and Cu/PTFE integrated with metallic Co nanoparticles (denoted Co metal/Cu/PTFE) were tested as well (Supplementary Fig. 3). We observed HER activity to follow the sequence Co metal/Cu/PTFE > CoO-2.5 nm/Cu/PTFE > Cu/PTFE. This enhancement in HER indicates that the ligand does not completely block all the *H adsorption sites present on the CoO clusters.
We then used the same flow cell system with CO2 gas and 1 M KHCO3 to assess the CO2RR performance (Supplementary Table 1). At an applied potential of −1.1 V versus reversible hydrogen electrode (V vs. RHE), CoO-2.5 nm/Cu/PTFE, Co metal/Cu/PTFE, and bare Cu/PTFE attained CH4 FEs of 60%, 0.6%, and 5%, respectively (Fig. 3a). The CoO-2.5 nm/Cu/PTFE catalyst achieved a CH4 partial current density of 135 mA cm−2; however, the bare Cu/PTFE and Co metal/Cu/PTFE controls reach only 12.5 and 1.35 mA cm−2, respectively.

a Product distribution of CoO/Cu/PTFE, Co metal/Cu/PTFE and bare Cu/PTFE at the −1.1 V vs. RHE. Red circle for methane, blue circle for hydrogen and grey circle for C2 products. b CO2-to-CH4 performance of CoO/Cu/PTFE in comparison with recent reports13,14,15,16,17,18,19,20. Blue square for the results in literature. c Stability test of CO2 methanation during 18.5 h of electrolysis under the current density of 225 mA cm−2. Red square for methane FE and grey line for potential curve. d CH4 FEs of ligand-stabilized CoO clusters with different sizes and loading contents on Cu/PTFE under an operating current density of 225 mA cm−2 in a flow cell using 1 M KHCO3 electrolyte.
From the product distributions, it is clear that the increased CH4 FE comes at the expense of C2+ products such as ethylene and ethanol on CoO-2.5 nm/Cu/PTFE. The ratio of CH4:C2+ increases from 0.08 on bare Cu/PTFE to 7.5 on CoO-2.5 nm/Cu/PTFE (Fig. 3a). For Co metal/Cu/PTFE, the current is channelled almost entirely toward HER (~100% FE): this corresponds to the situation where *H binds strongly to the motif and is unable to promote the hydrogenation of *CO on neighbouring Cu atoms.
We noted that previous work23 has shown that modifying *H binding energy with hydroxide motifs allowing steering of post C–C coupling pathways toward ethanol instead of ethylene on Cu/PTFE; however, these experiments were performed under higher pH conditions (1 M KOH) and at lower applied potentials (−0.6 V vs. RHE) compared to this work. Herein, the neutral electrolyte and high work potential (−1.1 V vs. RHE) favour C1 products10,27, and *H binding energy has a greater effect on the branching points between methane formation and C–C coupling in CO2RR.
To exclude the influence of the poly(methacrylic acid) ligand on CO2RR activity, we spray-coated the ligand onto bare Cu/PTFE without CoO clusters. We found negligible differences in product selectivity for these samples vs. bare Cu/PTFE (Supplementary Table 1), allowing us to conclude that the ligand does not have a direct influence on CO2 reduction pathways.
We found the high CH4 partial current density of 135 mA cm−2 (Fig. 3b) and CO2-to-CH4 conversion rate of 0.17 μmol cm−2 s−1 of CoO-2.5 nm/Cu/PTFE, which improve by a factor of 2.6x relative to the best previous reports having a CH4 FE higher than 50% (Supplementary Table 2). The catalyst system reported herein achieved a half-cell energy efficiency of 27% in neutral medium (Supplementary Note 1), which is among the best previously reported catalysts with current densities above 50 mA cm−2 (Supplementary Table 2)13,14,15,16,17,18,19,20.
We then tested the stability of the CoO-2.5 nm/Cu/PTFE catalyst system. The system achieved CO2-to-CH4 generation over 18 h at a current density of 225 mA cm−2 with a CH4 FE above 55% (Fig. 3c). After 18 h, the CH4 FE decreased to below 55%. After the catalyst was washed and dried, CO2RR was resumed with the same catalyst: the CH4 FE returned to the original value of 60% (Supplementary Fig. 4), suggesting that the decrease of CH4 FE is not caused by catalyst degradation but could be due to flooding or carbonate formation on the PTFE substrate22,35.
We also investigated the CO2-to-CH4 performance as a function of CoO-cluster size. CoO-2.5 nm with a loading content of 10 ng cm−2 on Cu/PTFE catalyst (an estimated coverage of 1%, see Supplementary Note 2) gives the highest CH4 selectivity of 60% (Fig. 3d). An increase in loading content regardless of cluster size leads to an increase in H2 evolution activity which diminishes the CH4 FE (Supplementary Table 3).
When examining the selectivity of these electrocatalysts as functions of CoO-cluster size and loading content, we draw two conclusions within the size of 1–3 nm and loading content of 1–30 ng cm−2 range: (I) cluster size effects are present, and (II) there exists a maximum in CH4 selectivity with a CoO-cluster size of 2.5 nm and 10 ng cm−2 on Cu/PTFE in this work.
Characterization of CoO-cluster-localized catalysts
To investigate possible catalyst changes in structure and oxidation state following CO2RR, we carried out characterization of CoO-2.5 nm/Cu/PTFE before and after the extended electrochemical operation studies, using electron microscopy and X-ray absorption spectroscopy. Scanning electron microscopy (SEM) images of CoO-2.5 nm/Cu/PTFE catalysts demonstrate reconstruction of Cu after CO2RR (Supplementary Fig. 5). STEM images with elemental mapping reveal a uniform distribution of Cu and Co in samples before and after stability tests (Fig. 4a, b). HRTEM images show intimate contact between CoO clusters and the Cu/PTFE catalyst (Fig. 4c, d). According to the measured lattice fringes23 in the TEM images, the initial slightly oxidized Cu is reduced to its metallic state during CO2RR.

a, b Dark-field scanning transmission electron micrograph images and corresponding energy-dispersive X-ray spectroscopy mapping for Cu (blue dot) and Co (red dot). c, d High-resolution transmission electron micrograph images of the CoO-2.5 nm/Cu/PTFE catalysts before and after stability test. The scale bars in (a, b) and (c, d) are 200 and 5 nm, respectively. e Co 2p X-ray photoelectron spectroscopy spectra. f The k2-weighted Fourier-transform spectra from extended X-ray absorption fine structure and g the normalized X-ray absorption near-edge structure spectra at the Co K-edge of the CoO-2.5 nm/Cu/PTFE catalysts before and after stability test. Red and blue lines for before and after stability test, respectively. h Operando Co K-edge of X-ray absorption fine structure of CoO-2.5 nm/Cu/PTFE catalyst under an operating current density of 225 mA cm−2 in a flow cell using 1 M KHCO3 electrolyte. i Fourier-transform infrared spectra of the CoO-2.5 nm/Cu/PTFE catalysts before and after stability test, compared with the bare and ligand localized Cu/PTFE. Red and blue lines for CoO-2.5 nm/Cu/PTFE catalyst before and after stability test, respectively; light grey and dark grey lines for Cu/PTFE and ligand/Cu/PTFE controls, respectively. Red and black arrows for –CO– and –COO– in the poly(methacrylic acid) ligand, respectively. a.u. presents arbitrary unit.
The lack of Co related peaks in X-ray diffraction patterns can be attributed to the small size and low loading of CoO clusters (Supplementary Fig. 6). Cu 2p high-resolution X-ray photoelectron spectroscopy (XPS) spectra reveal the existence of metallic and oxidized Cu species in the initial catalyst and mostly metallic Cu after CO2RR (Supplementary Fig. 7)23.
The Co 2p spectra show Co(II) in the CoO-2.5 nm/Cu/PTFE catalysts before and after stability testing (Fig. 4e). On the other hand, Co 2p spectra in Co metal/Cu/PTFE after the test shows metallic character, which is as expected (Supplementary Fig. 8)36. The O 1s spectra indicate the presence of metal oxides (Supplementary Fig. 9). Fourier-transformed spectra of the Co K-edge extended X-ray adsorption fine structure (EXAFS) of the CoO-2.5 nm/Cu/PTFE catalysts before and after the stability test are in Fig. 4f. The single notable peak, a feature that is at 1.0–2.0 Å, is ascribed to Co–O binding37. Figure 4g shows the normalized X-ray absorption near-edge structure (XANES) spectra. The white-line position reflects the Co(II) oxidation state of Co–O bond37,38, consistent with the view that the clusters include oxidized CoO.
To investigate whether ligand-stabilized CoO clusters remain as an oxide during CO2RR, we carried out operando X-ray absorption spectroscopy (XAS) to determine the valence states of Co during CO2 electroreduction by focusing on the Co K-edge spectra. From these studies, we see that the CoO clusters remain in their oxidized form during electrocatalysis over 3 h at an operating current density of 225 mA cm−2 (Fig. 4h). It is in these conditions that the catalyst achieves its best CO2-to-CH4 FE of 60%. We did not observe a change in the white-line position of Co species throughout electrolysis. Fitting results of the Co–O bond XAS spectra reveal a negligible change of coordination number (~4.5) and bond length (~2.0 Å) for initial vs. operando catalysts (Supplementary Table 4). This leads us to the view that the ligand may indeed protect the CoO clusters, allowing them to remain oxidized under the reducing conditions applied during CO2RR, at least over the timescales of operation (~3 h) studied herein.
We also performed operando Raman spectroscopy for CoO-2.5 nm/Cu/PTFE samples at a current density of 225 mA cm−2 over a period of 90 min (Supplementary Fig. 10a). The spectra exhibit peaks in the range of 500–600 cm−1, which is assigned to Co–O39. Upon application of a reducing potential, these peaks do not diminish in intensity even after 90 min, providing supporting evidence that CoO species are stabilized, over the timescale studied here, against reduction by the ligand. As a control, the operando Raman spectroscopy was also carried out on the bare Cu/PTFE sample under similar reaction conditions (Supplementary Fig. 10b). We observe only the presence of small peaks at 530 cm−1 related to Cu2O40, which disappears completely within 10 min of reduction. The Raman spectra for the CoO/Cu/PTFE and bare Cu/PTFE electrode before electrocatalysis (Supplementary Fig. 10c) show peaks assigned to Cu oxide40. To exclude the possibility of the Raman signals from CoO on the electronically uncontacted parts in PTFE, we performed operando Raman for two more controls: (I) commercial CoO (purchased from Sigma-Aldrich) on Cu/PTFE and (II) ligand-stabilized CoO clusters on Cu/GDL substrate (i.e., sputtered Cu on conductive carbon paper with gas diffusion layer). The Raman results of the first control (Supplementary Fig. 11) show that the Co–O bonds are completely vanished after 10 min of CO2 reduction reaction; while the second control (Supplementary Fig. 12) presents similar Raman peaks in the range of 500–600 cm−1 compared to those in the Cu/PTFE as substrate, suggesting that CoO remains over the course of the CO2 reduction studies herein.
The presence of the ligand in catalysts before and after CO2RR is indicated by Fourier-transform infrared spectroscopy analysis, showing that poly(methacrylic acid) ligand is retained during the CO2RR (Fig. 4i). In addition, the peak at 1550 cm−1 is attributed to the carboxylate groups in the ligand associated with metal ions (here is Co) in a bridging mode41.
We noted that stronger-binding organic ligands on Co(II) did not prevent Co metal formation under negative cathodic potentials in prior literature reports42; however, in these prior studies, the electrolyte was acidic phosphate buffer (pH 2.2), and the cathodic reaction was hydrogen evolution, compared to the 1 M KHCO3 solution (pH 8.9) studied in CO2 reduction herein. The local pH is higher during electrochemical CO2 reduction, particularly so at high current density (such as 225 mA cm−2 when we obtained the best CH4 FE)43. The stability of Coδ+ based catalysts, including Coδ+ complexes44 and ultra-small/thin cobalt oxides45,46, under negative potentials for electrocatalytic CO2 reduction, has previously been reported. In a broader context, there are other examples in which metal oxide surface remain during electrochemical CO2 reduction, even though the Pourbaix diagram suggests that the metallic phase is thermodynamically stable47,48. We propose that the CoO clusters are ligand-stabilized CoO to the point that, under the conditions of pH and applied potential discussed here, the oxidation state and catalytic enhancement can be maintained under the conditions of current and experimental duration reported herein.
We also prepared the molybdenum(VI), tungsten(VI), nickel(II) and palladium(II) oxides clusters as controls (Supplementary Fig. 13), with the ligand stabilization and similarly spray-coated on Cu/PTFE. The microscopy and spectroscopy analyses show that these clusters are oxidized49,50,51,52 and these catalysts exhibit similar structures to CoO/Cu/PTFE (Supplementary Figs. 14–17). Similar catalytic trends are obtained using these catalysts with their different respective ligand concentrations and cluster loading contents (Supplementary Figs. 18–20 and Tables 5–8). Of the catalysts, CoO-2.5 nm/Cu/PTFE exhibits the highest CH4 selectivity.
Discussion
We developed a cooperative system designed on the principle that local *H availability can provide enhanced CO2 methanation at a catalyst/electrolyte interface, with size effects playing a role in localized CoO clusters. The CoO-cluster size is controlled from 1.5 to 2.8 nm and the clusters are reduction-resistant under a cathodic potential, a finding we attribute to stabilization using a poly(methacrylic acid) ligand. These clusters serve to modulate the CO2 reduction pathways toward CH4 formation. On the CoO/Cu/PTFE catalyst, the reaction energetics are altered to favour CO hydrogenation. The catalysts exhibit a CH4 FE of 60% at a current density of 225 mA cm−2; these operate over 18 h while retaining initial performance to within 91%.
Methods
Catalyst preparation
For CoO clusters, 8.8 mg of cobalt(II) chloride (99.9%, Sigma-Aldrich) and 20 mg of poly(methacrylic acid) ligand32,33,34 were added into a screw-neck glass bottle containing 5 ml of methanol. After the solution becoming uniform by sonication, 0.5 ml of freshly prepared NaBH4 solution (5 mg ml−1 in methanol) was injected into the solution under vigorous stirring (2000 r.p.m.). The stirring speed of the solution was kept at the same for another 10 min. In the washing process, the precipitate was sonicated to ensure all clusters dispersing uniformly in methanol, and then separated by centrifuge. The precipitate was washed for two times and stored in methanol for further use. For different sizes of CoO clusters, the ligand concentrations are 2, 4, 8 and 16 mg ml−1 for synthesize cluster with mean size of 2.8, 2.5, 2.1 and 1.5 nm, respectively. The metal Co nanoparticles were synthesized using the similar process without adding the ligand. For different clusters, we only changed the corresponding chloride salts (tungsten(VI) chloride, 99.9%, Sigma-Aldrich; molybdenum(V) chloride, 99.99%, Sigma-Aldrich; nickel(II) chloride, 99.99%, Sigma-Aldrich; palladium(II) chloride, 99.9%, Sigma-Aldrich) as the precursor and the concentration is the same 0.8 mg ml−1 (metal-based). Cu/PTFE electrodes were prepared by sputtering a Cu layer of 200 nm in thickness onto a PTFE membrane (average pore size of 450 nm) using a Cu target (99.99%) at a rate of 1 Å s−1. Then, 10 μl of 5% Nafion was dispersed in 1 ml the cluster suspension (0.2 mg ml−1 in methanol) and the mixture was under ultrasonication for 30 min. The suspension was deposited on a Cu/PTFE electrode using spray-coating.
Materials characterization
The morphology of the electrodes was characterized using scanning electron microscopy (SEM, Hitachi S-5200) with a 5-kV beam voltage. Transmission electron microscopy (TEM) and elemental mapping images were collected using a Hitachi HF-3300, at an acceleration voltage of 300 kV, equipped with a Bruker energy-dispersive X-ray spectroscopy (EDX) detector. The acquisition time in the EDX studies was 3 min. The samples for TEM measurements are dip-coated on the carbon-coated nickel grids. Fourier-transform infrared spectroscopy (FTIR) was performed using a Nicolet 6700 FTIR spectrometer. Measurements were carried out using the attenuated total reflection (ATR) mode using the iS50-ATR-FTIR system. Spectra were obtained using 16 scans with a resolution of 4 cm−1. Powder X-ray diffraction (XRD) patterns were recorded using a Bruker D8 using Cu-Kα radiation (λ = 0.15406 nm). X-ray photoelectron spectroscopy (XPS) was conducted on a PHI 5700 ESCA System using Al Kα X-ray radiation (1486.6 eV) for excitation. Some exposure to air during cell disassembly and the transfer to XPS equipment is expected. Operando Raman spectroscopy measurements were carried out using a Renishaw inVia Raman microscope in a modified flow cell (Supplementary Fig. 10d) using a water immersion objective with a 785-nm laser.
XAS measurements were carried out at 9-BM beamline at the advanced photon source (APS) light source (IL, USA). An in-house custom-made electrochemical cell with a three-electrode configuration was used to perform operando XAS. A platinum mesh and Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. The catalyst was prepared on the front side of PTFE and connected electrically by Cu tape while the back side was stuck to a thin Kapton tape. The sample was then mounted on the window of the electrochemical cell so that the back side (PTFE gas diffusion layer) faced the beam and the catalyst (front side) is in direct contact with the electrolyte. The ex situ XAS experiments, including XANES and EXAFS, were conducted on the front side of the samples. Data was collected in fluorescence mode on a passivated implanted planar silicon (PIPS)/Lytle detector placed at an angle of 45°. Several spectra were collected and averaged at each condition to improve the quality of the data and increase the signal to noise ratio. Data post-processing and fitting was performed using the Demeter software package.
Electrochemical measurements
We used an electrochemical flow cell containing a gas chamber, a cathodic chamber, an anodic chamber and an anion exchange membrane (separating the anodic and cathodic chambers) to study the electrochemical CO2 reduction performance. We fixed the working electrode (such as CoO/Cu/PTFE) with a geometric active surface area of 1 cm2 and place it between the gas and cathodic chambers. We made the catalysts layer side of the working electrode face to the cathodic chamber. We chose 1 M KHCO3 solution as the electrolyte, and used peristaltic pumps to circulate the electrolyte through the cathodic and anodic chambers with a rate of 10 ml min−1. We controlled the CO2 gas flow rate through the gas chamber as 50 sccm by a gas flow controller. We operated the electrochemical tests on an Autolab PGSTAT204, with a reference Ag/AgCl electrode and a counter Ni foam electrode. We calibrated the potentials to those vs. RHE after iR-compensation. We collected the gas products from the end of the gas chamber and analysed them by a gas chromatography (PerkinElmer Clarus 600) with a flame ionization detector (FID) and a thermal conductivity detector (TCD). We evaluated the hydrogen evolution activities of the electrodes in the same flow cell using argon as the only flow gas.
DFT calculations
We performed density functional theory calculations with the Vienna Ab Initio Simulation Package (VASP) code53,54. The exchange-correlation energy was described by Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)55. The ionic cores were simplified using the projector augmented wave (PAW) pseudo-potentials56. After a series of tests, we set up the cutoff energy of 450 eV. In all calculations, the Hellmann–Feynman forces criteria between atoms were set as 0.02 eV Å−1 and the electronic iterations convergence was 10−5 eV using the Normal algorithm. A 4-layer (5 × 2) Cu (111) supercell was built to simulate the most stable exposed surface of copper accompanied with a sufficient vacuum gap of 15 Å. Pure metal (Co, Ni, Pd, W and Mo) and metal oxide (CoO, NiO, PdO, WO3 and MoO3) clusters were added on the fixed positions of Cu (111) surface. Structural optimizations were then performed on all modified slab models. *CO, *CHO and *(CO+H) were considered as key intermediates to simulate the CO2-to-methane reaction on catalyst surfaces. During all adsorption calculations, the top two layers were fully relaxed while the other layers were fixed at the tested lattice position. After comparing the hydrogenation activity of Co atom and O atom (Supplementary Fig. 21), we consider Co sites in clusters as the active sites for *CO hydrogenation. The adsorption energies and hydrogenation energies were calculated according to Supplementary Note 3.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Qiao, J., Liu, Y., Hong, F. & Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014).
Kim, D., Kley, C. S., Li, Y. & Yang, P. Copper nanoparticle ensembles for selective electroreduction of CO2 to C2–C3 products. Proc. Natl Acad. Sci. USA 114, 10560–10565 (2017).
Yang, D. et al. Selective electroreduction of carbon dioxide to methanol on copper selenide nanocatalysts. Nat. Commun. 10, 677 (2019).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016).
Liu, Y., Chen, S., Quan, X. & Yu, H. Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J. Am. Chem. Soc. 137, 11631–11636 (2015).
Howarth, R. W., Ingraffea, A. & Engelder, T. Should fracking stop? Nature 477, 271–275 (2011).
Connolly, B. M. et al. Tuning porosity in macroscopic monolithic metal-organic frameworks for exceptional natural gas storage. Nat. Commun. 10, 2345 (2019).
Gan, Y. et al. Carbon footprint of global natural gas supplies to China. Nat. Commun. 11, 824 (2020).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Liu, X. et al. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 8, 15438 (2017).
Zhou, Y. et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 10, 974–980 (2018).
Zhang, S. et al. Polymer-supported CuPd nanoalloy as a synergistic catalyst for electrocatalytic reduction of carbon dioxide to methane. Proc. Natl Acad. Sci. USA 112, 15809–15814 (2015).
Reske, R., Mistry, H., Behafarid, F., Cuenya, B. R. & Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 136, 6978–6986 (2014).
Wang, X. et al. Efficient methane electrosynthesis enabled by tuning local CO2 availability. J. Am. Chem. Soc. 142, 3525–3531 (2020).
Li, Y. et al. Structure-sensitive CO2 electroreduction to hydrocarbons on ultrathin 5-fold twinned copper nanowires. Nano Lett. 17, 1312–1317 (2017).
Wang, Y. et al. Single-atomic Cu with multiple oxygen vacancies on ceria for electrocatalytic CO2 reduction to CH4. ACS Catal. 8, 7113–7119 (2018).
Wang, Y., Chen, J., Wang, G., Li, Y. & Wen, Z. Perfluorinated covalent triazine framework derived hybrids for the highly selective electroconversion of carbon dioxide into methane. Angew. Chem. Int. Ed. 57, 13120–13124 (2018).
Han, L. et al. Stable and efficient single-atom Zn catalyst for CO2 reduction to CH4. J. Am. Chem. Soc. 142, 12563–12567 (2020).
Chang, C.-J. et al. Dynamic reoxidation/reduction-driven atomic interdiffusion for highly selective CO2 reduction toward methane. J. Am. Chem. Soc. 142, 12119–12132 (2020).
Hu, Q. et al. Facile synthesis of sub‐nanometric copper clusters by double confinement enables selective reduction of carbon dioxide to methane. Angew. Chem. Int. Ed. 59, 19054–19059 (2020).
Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of CO2 electrolysis systems. Ind. Eng. Chem. Res. 57, 2165–2177 (2018).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Luo, M. et al. Hydroxide promotes carbon dioxide electroreduction to ethanol on copper via tuning of adsorbed hydrogen. Nat. Commun. 10, 5814 (2019).
Li, J. et al. Enhanced multi-carbon alcohol electroproduction from CO via modulated hydrogen adsorption. Nat. Commun. 11, 3685 (2020).
Cheng, T., Xiao, H. & Goddard, W. A. Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl Acad. Sci. USA 114, 1795–1800 (2017).
Cheng, T., Xiao, H. & Goddard, W. A. Free-energy barriers and reaction mechanisms for the electrochemical reduction of CO on the Cu(100) surface, including multiple layers of explicit solvent at pH 0. J. Phys. Chem. Lett. 6, 4767–4773 (2015).
Peterson, A. A. & Nørskov, J. K. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts. J. Phys. Chem. Lett. 3, 251–258 (2012).
Liu, X. et al. pH effects on the electrochemical reduction of CO(2) towards C2 products on stepped copper. Nat. Commun. 10, 32 (2019).
Kortlever, R., Shen, J., Schouten, K. J. P., Calle-Vallejo, F. & Koper, M. T. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 6, 4073–4082 (2015).
Nie, X., Esopi, M. R., Janik, M. J. & Asthagiri, A. Selectivity of CO2 reduction on copper electrodes: the role of the kinetics of elementary steps. Angew. Chem. Int. Ed. 52, 2459–2462 (2013).
Feng, Y. et al. Electrochemical CO2 reduction reaction on M@Cu(211) bimetallic single-atom surface alloys: mechanism, kinetics, and catalyst screening. ACS Sustain. Chem. Eng. 8, 210–222 (2020).
Li, Y. H. et al. Unidirectional suppression of hydrogen oxidation on oxidized platinum clusters. Nat. Commun. 4, 2500 (2013).
Majeed, M. I., Guo, J., Yan, W. & Tan, B. Preparation of magnetic iron oxide nanoparticles (MIONs) with improved saturation magnetization using multifunctional polymer ligand. Polymers 8, 392 (2016).
Wen, L. et al. Ultrasmall biocompatible WO3−x nanodots for multi‐modality imaging and combined therapy of cancers. Adv. Mater. 28, 5072–5079 (2016).
Nwabara, U. O., Cofell, E. R., Verma, S., Negro, E. & Kenis, P. J. A. Durable cathodes and electrolyzers for the efficient aqueous electrochemical reduction of CO2. ChemSusChem 13, 855–875 (2020).
Lukashuk, L. et al. Operando insights into CO oxidation on cobalt oxide catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 8, 8630–8641 (2018).
Li, X., Clatworthy, E. B., Bartlett, S., Masters, A. F. & Maschmeyer, T. Structural investigation of cobalt oxide clusters derived from molecular cobalt cubane, trimer, and dimer oligomers in a phosphate electrolyte. J. Phys. Chem. C. 121, 11021–11026 (2017).
Jacobs, G. et al. Fischer-Tropsch synthesis: study of the promotion of Pt on the reduction property of Co/Al2O3 catalysts by in situ EXAFS of Co K and Pt LIII edges and XPS. J. Synchrotron Rad. 11, 414–422 (2004).
Ravindra, A. V., Behera, B. C. & Padhan, P. Laser induced structural phase transformation of cobalt oxides nanostructures. J. Nanosci. Nanotechnol. 14, 5591–5595 (2014).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Lu, Y. & Miller, J. D. Carboxyl stretching vibrations of spontaneously adsorbed and LB-transferred calcium carboxylates as determined by FTIR internal reflection spectroscopy. J. Colloid Interface Sci. 256, 41–52 (2002).
Kaeffer, N. et al. The dark side of molecular catalysis: diimine–dioxime cobalt complexes are not the actual hydrogen evolution electrocatalyst in acidic aqueous solutions. ACS Catal. 6, 3727–3737 (2016).
Zhang, Z. et al. pH matters when reducing CO2 in an electrochemical flow cell. ACS Energy Lett. 5, 3101–3107 (2020).
Wang, M. et al. CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine. Nat. Commun. 10, 3602 (2019).
Gao, S. et al. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 529, 68–71 (2016).
Sekar, P. et al. Cobalt spinel nanocubes on N-doped graphene: a synergistic hybrid electrocatalyst for the highly selective reduction of carbon dioxide to formic acid. ACS Catal. 7, 7695–7703 (2017).
Luc, W. et al. Ag–Sn bimetallic catalyst with a core–shell structure for CO2 reduction. J. Am. Chem. Soc. 139, 1885–1893 (2017).
Deng, P. et al. Bismuth oxides with enhanced bismuth–oxygen structure for efficient electrochemical reduction of carbon dioxide to formate. ACS Catal. 10, 743–750 (2020).
Kröger, M. et al. Role of the deep-lying electronic states of MoO3 in the enhancement of hole-injection in organic thin films. Appl. Phys. Lett. 95, 123301 (2009).
Kwon, Y. T., Song, K. Y., Lee, W. I., Choi, G. J. & Do, Y. R. Photocatalytic behavior of WO3-loaded TiO2 in an oxidation reaction. J. Catal. 191, 192–199 (2000).
Grosvenor, A. P., Biesinger, M. C., Smart, R. S. C. & Mclntyre, N. S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 600, 1771–1779 (2006).
Brun, M., Berthet, A. & Bertolini, J. C. XPS, AES and auger parameter of Pd and PdO. J. Electron. Spectrosc. Relat. Phenom. 104, 55–60 (1999).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computat. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Payne, M. C., Teter, M. P., Allan, D. C., Arias, T. A. & Joannopoulos, J. D. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys. 64, 1045–1097 (1992).
Acknowledgements
This work was supported by the Natural Gas Innovation Fund, the Natural Sciences and Engineering Research Council (NSERC) of Canada, the Natural Resources Canada - Clean Growth Program, and the Ontario Research Fund—Research Excellence program. All DFT computations were performed on the Niagara supercomputer at the SciNet HPC Consortium. SciNet is funded by the Canada Foundation for Innovation, the Government of Ontario, Ontario Research Fund Research Excellence Program, and the University of Toronto. This research used synchrotron resources of the Advanced Photon Source (APS), an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners. The authors thank T. P. Wu, Y. Z. Finfrock and L. Ma for technical support at 9-BM beamline of APS. Y.L. acknowledges the financial support from the program of China Scholarships Council (201906745001).
Author information
Authors and Affiliations
Contributions
C.L. and E.H.S. jointly supervised this work. Yu.L. designed and carried out the experiments. A.X. carried out the DFT calculations. B.C. collected the STEM and TEM images and did the EDX mapping. J.A., S.F.H., F.L. and Yu.L. performed and analysed the XAS measurements. A.S.R., J.W. and A.X. performed the XPS and XRD measurements. J.E.H. performed the Raman spectroscopy measurements. L.K.S. performed the FTIR measurements. T.P. collected the SEM images. X.W., Z.W., Y.X., D.S., A.H.I. and H.J. contributed to data analysis and manuscript editing. Yu.L., Ya.L., C.L. and E.H.S. co-wrote the paper. All authors discussed the results and assisted during manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Wei An, Feng Jiao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Xu, A., Lum, Y. et al. Promoting CO2 methanation via ligand-stabilized metal oxide clusters as hydrogen-donating motifs. Nat Commun 11, 6190 (2020). https://doi.org/10.1038/s41467-020-20004-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20004-7
This article is cited by
-
Selective hydrocarbon or oxygenate production in CO2 electroreduction over metallurgical alloy catalysts
Nature Synthesis (2023)
-
Manipulating local coordination of copper single atom catalyst enables efficient CO2-to-CH4 conversion
Nature Communications (2023)
-
High-rate and selective conversion of CO2 from aqueous solutions to hydrocarbons
Nature Communications (2023)
-
Atomically dispersed Co−Cu alloy reconstructed from metal-organic framework to promote electrochemical CO2 methanation
Nano Research (2023)
-
Single-site decorated copper enables energy- and carbon-efficient CO2 methanation in acidic conditions
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
