
Abstract
The emergence of resistance to azithromycin complicates treatment of Neisseria gonorrhoeae, the etiologic agent of gonorrhea. Substantial azithromycin resistance remains unexplained after accounting for known resistance mutations. Bacterial genome-wide association studies (GWAS) can identify novel resistance genes but must control for genetic confounders while maintaining power. Here, we show that compared to single-locus GWAS, conducting GWAS conditioned on known resistance mutations reduces the number of false positives and identifies a G70D mutation in the RplD 50S ribosomal protein L4 as significantly associated with increased azithromycin resistance (p-value = 1.08 × 10−11). We experimentally confirm our GWAS results and demonstrate that RplD G70D and other macrolide binding site mutations are prevalent (present in 5.42% of 4850 isolates) and widespread (identified in 21/65 countries across two decades). Overall, our findings demonstrate the utility of conditional associations for improving the performance of microbial GWAS and advance our understanding of the genetic basis of macrolide resistance.
Similar content being viewed by others


Introduction
Increasing antibiotic resistance in Neisseria gonorrhoeae, the causative agent of the sexually transmitted disease gonorrhea, threatens effective control of this prevalent pathogen1,2,3. Current empiric antibiotic therapy in the US comprises a combination of the cephalosporin ceftriaxone and the macrolide azithromycin, but increasing prevalence of azithromycin resistance has led some countries, such as the UK, to instead recommend ceftriaxone monotherapy4. Rapid genotypic diagnostics for antimicrobial susceptibility have been proposed as a platform to tailor therapy and to extend the clinically useful lifespan of anti-gonococcal antibiotics5,6. These rapid diagnostics rest on robust genotype-to-phenotype predictions. For some antibiotics, such as ciprofloxacin, resistance is predictable by target site mutations in a single gene, gyrA3,5. However, recent efforts to predict azithromycin minimum inhibitory concentrations (MICs) using regression-based or machine-learning approaches have indicated that a substantial fraction of phenotypic resistance is unexplained, particularly among strains with lower-level resistance3,7,8. An improved understanding of the genetic mechanisms and evolutionary pathways to macrolide resistance will therefore be critical for informing the development of diagnostics.
Macrolides function by binding to the 50S ribosome and inhibiting protein synthesis9. Increased resistance can occur in N. gonorrhoeae through target site modification, primarily via 23S rRNA mutations C2611T10 and A2059G11, and through efflux pump upregulation. The main efflux pump associated with antibiotic resistance in the gonococcus is the Mtr efflux pump, comprising a tripartite complex encoded by the mtrCDE operon under the regulation of the MtrR repressor and the MtrA activator1,12,13,14,15,16,17. Active site or frameshift mutations in the coding sequence of mtrR and promoter mutations in the mtrR promoter upregulate mtrCDE and result in increased macrolide resistance1,18. Mosaic sequences originating from recombination with homologs from commensal Neisseria donors can also result in structural changes to mtrD and increased expression of mtrCDE, which synergistically act to confer resistance19,20.
Here, we use genome-wide association on a global meta-analysis dataset to identify additional genetic variants that confer increased azithromycin resistance in N. gonorrhoeae. We find that conventional single-locus bacterial GWAS approaches lead to confounded results and reduced power, but conducting GWAS conditional on known resistance mutations in 23S rRNA reduces linkage-mediated confounding and increases power to recover mutations associated with lower-level resistance. We experimentally validate one such mutation in the 50S ribosomal protein RplD and identify other rare RplD variants associated with resistance, highlighting the ability of conditional bacterial GWAS to discover causal genes for polygenic microbial phenotypes.
Results
We previously conducted a linear mixed model GWAS using a global meta-analysis collection of 4852 N. gonorrhoeae isolates, collected across 15 studies and spanning 65 countries and 38 years7. After conducting GWAS on the 4505 isolates with associated azithromycin MICs, we identified highly significant unitigs (i.e., genetic variants generated from de novo assemblies) mapping to the 23S rRNA, associated with increased resistance, and to the efflux pump gene mtrC, associated with increased susceptibility and cervical infections7. These results highlighted the potential for GWAS to identify novel modifiers of resistance in N. gonorrhoeae. However, the characterized mutations did not fully explain azithromycin heritability and thus pointed towards unknown genetic variants.
Conditional GWAS identifies a resistance mutation in RplD
To identify these variants, we re-analyzed the GWAS results focusing on the remaining unitigs, which had lower effect sizes and p-values closer to the Bonferroni-corrected p-value threshold, calculated using the number of unique patterns, of 3.38 × 10−7. Numerous variants were significantly associated with increased MICs, many of which mapped to genes (e.g., hprA, WHO_F.1254, and ydfG) that had not previously been implicated in macrolide resistance in Neisseria (Supplementary Data 1). While these signals could represent novel causal resistance genes, we hypothesized that at least some of these variants could have been spuriously driven to association via genetic linkage with the highly penetrant (A2059G: β, or effect size on the log2-transformed MIC scale, = 7.14, 95% CI [6.44, 7.84]; C2611T: β = 3.67, 95% CI [3.46, 3.88]) and population-stratified 23S rRNA resistance mutations (Supplementary Fig. 1). Supporting this hypothesis, r2—a measure of linkage ranging from 0 to 1—between significant variants and 23S rRNA resistance mutations showed a bimodal distribution with a peak at 0.84 and at 0.04 (Supplementary Fig. 2). The three significant variants that mapped to hprA, WHO_F.1254, and ydfG had elevated r2 values of 0.16, 0.82, and 0.80 respectively; all three variants demonstrated clear phylogenetic overlap with 23S rRNA mutations (Supplementary Fig. 1). Additionally, we did not observe unitigs associated with previously experimentally validated resistance mutations in the mtrR promoter14 or the mtrCDE mosaic alleles19,20, suggesting decreased power to detect known causal variants with lower effect sizes.
To control for the confounding effect of the 23S rRNA mutations, we conducted a conditional GWAS by incorporating additional covariates in our linear mixed model encoding the number of copies (ranging from 0 to 4) of the resistance-conferring 23S rRNA substitutions C2611T and A2059G. We also conditioned on isolate dataset of origin to address potential spurious hits arising from study-specific sequencing methodologies. After conditioning, the previously significant genes linked to 23S rRNA (r2 > 0.80) decreased below the significance threshold, indicating that they were indeed driven to significance by genetic linkage (Fig. 1 and Supplementary Data 2). The most significant variants after the previously reported mtrC indel7 mapped to the mtrR promoter (β, or effect size, = –0.86, 95% CI [−1.05, −0.68]; p-value = 5.44 × 10−20), encoding the complement of the mtrR promoter 1 bp deletion21, and to mtrC (β = 1.23, 95% CI [0.93, 1.53]; p-value = 9.03 × 10−16), in linkage with mosaic mtr alleles19,20. The increased significance of these known efflux pump resistance mutations suggested improved power to recover causal genes with lower effects. Conditioning on dataset did not substantially affect these results but helped to remove other spurious variants arising due to study-specific biases22 (Supplementary Fig. 3 and Supplementary Data 3).

Each variant is plotted using negative log10-transformed p-values, calculated using likelihood-ratio tests, for the association with azithromycin MICs in unconditional and conditional GWASes. Genetic linkage measured by r2 to 23S rRNA mutations A2059G and C2611T is colored for significant variants as indicated on the right, ranging from 0 (blue) to 0.5 (white) to 1 (red). Variants associated with previously experimentally verified resistance mechanisms in the mtrR and mtrCDE promoters and coding regions are denoted using diamonds. Bonferroni thresholds, calculated using the number of unique patterns, for both GWASes are depicted using a gray dashed line at 3.38 × 10−7. Plot axes are limited to highlight variants associated with lower-level resistance; as a result, the highly significant 23S rRNA substitutions and mtrC indel mutations7 are not shown.
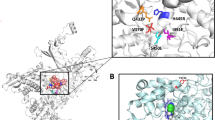
A glycine to glutamic acid substitution at site 70 of the 50S ribosomal protein L4 (RplD) was significantly associated with increased azithromycin MICs after conducting the conditional GWAS (β = 0.95, 95% CI [0.68, 1.23]; p-value = 1.08 × 10−11) (Fig. 1 and Supplementary Data 2). Structural analysis of the Thermus thermophilus 50S ribosome complexed with azithromycin suggests that this amino acid is an important residue in macrolide binding (Supplementary Fig. 4), and RplD substitutions at this binding site modulate macrolide resistance in other bacteria23,24. This substitution has previously been observed rarely in gonococcus and the association with azithromycin resistance versus susceptibility was non-significant3,25,26; as a result, the role of RplD mutations in conferring macrolide resistance was unclear. To assess the contribution of RplD mutations to continuous azithromycin MIC levels, we modeled MICs using a linear regression framework with known genetic resistance determinants as predictors (Supplementary Data 4 and 5)7,27. Compared to this baseline model, inclusion of the RplD G70D mutation decreased the number of strains with unexplained MIC variation (defined as absolute model error greater than one MIC dilution) from 1514 to 1463, improved adjusted R2 from 0.691 to 0.704, and significantly improved model fit (p-value < 10−10; χ2 test statistic = 288.51; Likelihood-ratio χ2 test for nested models). These results indicate that RplD G70D is a strong candidate for addressing a portion of the unexplained azithromycin resistance in N. gonorrhoeae.
Genomic epidemiology of RplD macrolide binding site mutations
We next assessed the population-wide prevalence and diversity of RplD-azithromycin binding site mutations. The RplD G70D mutation was present in 231 out of 4850 isolates (4.76%) with multiple introductions observed across varied genetic backgrounds (Fig. 2). An additional 34 isolates contained mutations at amino acids 68 (G68D, G68C), 69 (T69I), and 70 (G70S, G70A, G70R, G70duplication) (Fig. 3a). These other putative RplD binding site mutations were associated with significantly higher azithromycin MICs compared to both RplD G70D and RplD wild-type strains, indicating multiple avenues for disruption of macrolide binding (Fig. 3b). Grouping all RplD binding site mutations together resulted in increased effect size (β = 1.02) and p-value (9.25 × 10−18) in the conditional GWAS linear mixed model compared to the association with just RplD G70D (β = 0.95, p-value = 1.08 × 10−11). Strains with RplD binding site mutations were identified from 21 countries from 1993 to 2015 with prevalence reaching over 10% in some datasets (New York City 2011–201528 and Japan 1996–201529; Supplementary Table 1), in line with sustained transmission of RplD G70D strains (Fig. 2). Our results suggest that macrolide binding to the 50S ribosome can be disrupted via multiple mutations and that these mutations are widespread contributors to azithromycin resistance in some populations.

A midpoint rooted recombination-corrected maximum likelihood phylogeny of 4852 genomes based on 68697 SNPs non-recombinant from Ma and Mortimer et al.7 was annotated with the presence of RplD macrolide binding site mutations (orange for G70D and dark orange for other binding site mutations). Branch length represents total number of substitutions after removal of predicted recombination.

Mean (a) and distribution (b) of azithromycin MICs for RplD macrolide binding site variants. Previously reported mutations are cited with the first reporting publications. Violin and swarm plots and statistical analyses were limited to isolates with MICs < 8 to exclude isolates with 23S rRNA mutations. Rare RplD mutations (denoted as “Other (n = 31)”) were grouped for visualization and statistical analysis, and thus were assumed to all have the same effect. Quartiles within violin plots are depicted using dotted lines. Statistical significance between RplD variants and RplD wildtype MIC distributions was assessed by two-sided Mann–Whitney U Test: *p < 0.05, **p < 0.01, and ***p < 0.001. Exact p-values from left to right were 3.09 × 10−7 (WT vs. G70D), 3.91 × 10−10 (WT vs. Other), 5.74 × 10−5 (G70D vs. Other).
Experimental validation and growth dynamics of RplD G70D strains
To experimentally verify that RplD G70D contributes to macrolide resistance, we constructed an isogenic derivative of the laboratory strain 28Bl with the G70D substitution (using two biological replicates: C5 and E9) and tested for MIC differences across a panel of macrolides. Azithromycin and erythromycin MICs increased by three-fold, and clarithromycin MICs increased by six-fold on average in the G70D strains compared to the wild-type strain (Table 1). We also compared the experimental results with our modeling analysis: the estimate from our linear model for the azithromycin MIC of a strain that contains the RplD G70D mutation and no other resistance mutations was 0.363, which agrees well with the experimental results. Macrolide resistance has been associated with a fitness cost in other species30, prompting us to measure the in vitro growth dynamics of the RplD G70D strain. Time-course growth curves of the wild-type strain 28Bl and isogenic G70D strain E9 were similar (Supplementary Fig. 5) with overlapping estimates of doubling times: 28Bl doubling time = 1.756 h, 95% CI [1.663, 1.861] versus 28Bl RplDG70D (E9) doubling time = 1.787 h, 95% CI [1.671, 1.920] (Supplementary Table 2). These results confirm the role of RplD G70D in mediating macrolide resistance and indicate a lack of severe associated in vitro fitness costs.
Discussion
Azithromycin resistance in N. gonorrhoeae is a polygenic trait involving contributions from mutations in different 50S ribosomal components, up- and down-regulation of efflux pump activity, and additional unknown factors (Supplementary Table 3). Genome-wide association methods offer one approach for uncovering the genotypic basis of unexplained resistance in clinical isolates, but novel causal genes associated with lower effects have been difficult to identify with traditional microbial GWAS approaches24. Our results indicate that extending the GWAS linear mixed model to incorporate known causal genetic variants could address some of these challenges, particularly when known genes exhibit strong penetrance and population stratification, obfuscating signals with lower effects. After conducting conditional GWAS on azithromycin MICs, we observed a reduction in spurious results attributable to genetic linkage with known high-level resistance mutations in the 23S rRNA, and an increase in power to recover secondary resistance mutations in the MtrCDE efflux pump. We also identified a resistance-associated mutation in the macrolide binding site of 50S ribosomal protein RplD as significant only after conditioning. These results are in line with studies of multi-locus methods in the human GWAS field showing increased power31,32,33 and complementary methods using whole-genome elastic nets for microbial genome data34,35.
The situations under which conditional GWAS improves the power to detect new causal genes will need to be further characterized in other bacterial species and through simulations35. Here, we observed both increased magnitude of effect (rplD β increased from 0.52 to 0.95) and increased model precision (rplD standard error decreased from 0.223 to 0.140) after conditioning, both of which could improve power. The success of this conditional analysis using a relatively small sample size compared to human GWAS studies may also be attributable to the degree of homoplasy, as RplD mutations have been acquired multiple times across the phylogeny (Fig. 2). The extent of genetic linkage between true positives and the dominant resistance gene is likely key: if the degree of linkage is high (e.g., because a few bacterial lineages repeatedly acquire different mechanisms of resistance to the same drug), then a conditional GWAS could lead to a loss of power to detect the causal genes.
The role of RplD G70D mutations in conferring azithromycin resistance has previously been unclear, in part because of its lower effect size relative to 23S rRNA mutations. The G70D mutation was first observed in isolates from France 2013–201425 and in the US Centers for Disease Control Gonococcal Isolate Surveillance Program (CDC GISP) surveillance isolates from 2000 to 20133, and a related G68D mutation was described in the GISP collection and in European isolates from 2009 to 201426. However, these analyses reported no clear association with categorical resistance versus susceptibility. In line with this, we observed lower significance for the RplD unitig in the conditional GWAS model when the isolates were dichotomized into azithromycin susceptible versus non-susceptible (p-value = 3.38 × 10−09 versus 1.08 × 10−11 in the continuous case). Follow up studies in the US, Eastern China, and a historical Danish collection also reported strains with the G70D mutation36,37,38, but other surveillance datasets from Canada, Switzerland, and Nanjing did not10,39,40,41, indicating geography-specific circulation. As a result of this ambiguity, previous studies modeling phenotypic azithromycin resistance from genotype did not include RplD mutations27,42.
Here, we provided confirmatory evidence that the RplD G70D mutation increases macrolide MICs several-fold and that inclusion of the mutation in resistance regression models improves model fit, in line with the GWAS analyses. While RplD G70D mutations on their own are not predicted to confer resistance levels above the clinical CLSI non-susceptibility threshold of 1.0 µg/mL, there is growing appreciation of the role that sub-breakpoint increases in resistance can play in mediating treatment failure43. For example, treatment failures in Japan after a 2 g azithromycin dose were associated with MICs as low as 0.5 µg/mL44, and treatment failures in several case studies of patients treated with a 1 g azithromycin dose were associated with MICs of 0.125 to 0.25 µg/mL45. Low level azithromycin resistance may also serve as a stepping stone to higher level resistance, as suggested by an analysis of an outbreak of a high level azithromycin resistant N. gonorrhoeae lineage in the UK46.
We also observed multiple previously undescribed mutations in the RplD macrolide binding site associated with even higher MICs than the G70D mutation. The transmission of these isolates has been relatively limited, potentially due to increased fitness costs commensurate with increased resistance. In contrast, several lines of evidence suggest that the G70D mutation carries a relatively minimal fitness cost. Time-course growth experiments indicated that the RplD G70D isogenic pair of strains have similar doubling times, and phylogenetic analyses suggest multiple acquisitions of G70D in distinct genetic backgrounds, with a lineage in New York City showing evidence of sustained transmission.
As macrolide use continues to select for increased resistance in N. gonorrhoeae, both the RplD G70D and rarer binding site mutations should be targets for surveillance in future whole-genome sequencing studies. The rapid increase in the prevalence of strains with mosaic mtr alleles conferring azithromycin reduced susceptibility underscores how quickly the molecular landscape of resistance can change47,48 and highlights the value of early and proactive surveillance studies. Systematic genomic surveillance in turn allows for novel resistance mutations to be identified using conditional GWAS and other complementary approaches. With an increasingly refined understanding of the molecular basis of resistance, sequence-based diagnostics can then be developed by leveraging emerging point-of-care technologies such as Nanopore sequencing and CRISPR-based paper diagnostics49,50. The methods here can also be easily extended for other antibiotics used to treat gonococcal infections such as ceftriaxone, where resistance is of paramount concern and molecular mechanisms underlying resistance are still being uncovered51.
In summary, by reducing genetic confounders and amplifying true signals through bacterial GWAS conditional on known effects, we identified and experimentally characterized mutations in the 50S ribosome that contribute to increased macrolide resistance in N. gonorrhoeae.
Methods
Genomics and GWAS
All isolates included in this study are listed in Supplementary Data 6. We conducted whole-genome sequencing assembly, resistance allele calling, phylogenetic inference, genome-wide association, and significant unitig mapping using methods from a prior GWAS7. Briefly, reads were downloaded using fastq-dump in SRA toolkit (version 2.8.1). We then created a recombination-corrected phylogeny by running Gubbins (version 2.3.4)52 on an alignment of pseudogenomes generated from filtered SNPs from Pilon (version 1.16)53 after mapping reads in BWA-MEM (version 0.7.17-r1188)54 to the NCCP11945 reference genome (RefSeq accession: NC_011035.1). 23S rRNA mutations were called by mapping reads to a copy of the 23S rRNA locus and analyzing the frequency of variants55. Read mapping quality control was conducted in FastQC (version 0.11.7, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and BamQC in Qualimap (version 2.2.1)56, and read deduplication was conducted using Picard (version 2.8.0, https://github.com/broadinstitute/picard). We also annotated assemblies with Prokka (version1.13)57 and clustered core genes using Roary (version 3.12)58.
All phylogenies and annotation rings were visualized in iTOL (version 5.5)59. As in the prior study, azithromycin MICs prior to 2005 from the CDC GISP dataset3 were doubled to account for an MIC protocol testing change60. For analyses using susceptible versus non-susceptible categories as the outcome variable, isolates with adjusted azithromycin MICs of 1.0 μg/mL or lower were classified as susceptible.
We use a linear mixed model-based GWAS to control for population structure:
Here, Y is the vector of azithromycin MICs, W is the covariate matrix and α their fixed effects, X is the genetic variant/unitig under consideration and β its fixed effect, u is a random effect parameterized with population structure matrix K and additive genetic variance \(\sigma _g^2\), and ϵ is a random effect that models the non-genetic effects parameterized with variance \(\sigma _e^2\) and identity matrix I. This model is fit individually for all variants and the p-value for β is estimated using the likelihood-ratio test. The covariates can include isolate metadata such as country of origin or dataset as well as genetic information encoding known resistance genes.
To conduct the GWAS in Pyseer (version 1.2.0)61, unitigs were generated from genomes assembled with SPAdes (version 3.12.0)62 using GATB, and a population structure matrix was generated from the Gubbins phylogeny for the linear mixed model. We conducted conditional GWAS in Pyseer (version 1.2.0)61 by including additional columns in the covariate file encoding the number of 23S rRNA mutations and including flags --covariates and --use-covariates.
We assessed genetic linkage by calculating r2, or the squared correlation coefficient between two variants defined as r2 = (pij – pipj)2/(pi (1 – pi) pj (1 – pj)), where pi is the proportion of strains with variant i, pj is the proportion of strains with variant j, and pij is the proportion of strains with both variants63,64. For a given GWAS variant, we calculated r2 between that variant and the significant unitig from the GWAS mapping to 23 S rRNA C2611T. We repeated the calculation for the same variant but with the unitig mapping to 23 S rRNA A2059G, and took the maximum r2 value from the two calculations.
Azithromycin MIC regression models
Azithromycin log-transformed MICs were modeled using a panel of resistance markers7,65 and country of origin in R (version 3.5.1) using the lm function, with and without inclusion of RplD G70D and proximal mutations:
Model 1: Log_AZI ~ Country + MtrR 39 + MtrR 45 + MtrR LOF + MtrC LOF + MtrR promoter + mtrCDE BAPS Cluster + 23S rRNA 2059 + 23S rRNA 2611
Model 2: Log_AZI ~ Country + MtrR 39 + MtrR 45 + MtrR LOF + MtrC LOF + MtrR promoter + mtrCDE BAPS Cluster + 23S rRNA 2059 + 23S rRNA 2611 + RplD G70D + RplD other 68-70 mutations
Improvement in model fit was assessed using Anova for likelihood-ratio tests for nested models in R (version 3.5.1). BAPS clusters for mtrCDE were called as previously described using FastBAPS (version 1.0.0) as a way to flexibly group resistance-conferring mosaic alleles for inclusion in the regression model as a categorical covariate7,20. Variance explained by predictors was calculated using the relaimpo R package (version 2.2.3), which assesses the change in model R2 after inclusion of a predictor. Three approaches were used to calculate this change: the “first” metric compares a model without any predictors to a model with just the predictor of interest, the “last” metric compares a model with all predictors except the one of interest to a model with all predictors, and the “lmg” method averages the change in R2 over all possible model subsets.
Diversity of RplD macrolide binding site mutations
We ran BLASTn (version 2.6.0)66 on the de novo assemblies using a query rplD sequence from FA1090 (RefSeq accession: NC_002946.2). rplD sequences were aligned using MAFFT (version 7.450)67. Binding site mutations were identified after in silico translation of nucleotide alignments in Geneious Prime (version 2019.2.1, https://www.geneious.com). Subsequent analyses identifying prevalence, geometric mean azithromycin MIC, and MIC distribution differences were conducted in Python (version 3.6.5) using the Biopython package (version 1.69)68 and R (version 3.5.1).
Experimental validation
We cultured N. gonorrhoeae on GCB agar (Difco) plates supplemented with 1% Kellogg’s supplements (GCBK) at 37 °C in a 5% CO2 incubator69. We conducted antimicrobial susceptibility testing using Etests (bioMérieux) placed onto GCB agar plates supplemented with 1% IsoVitaleX (Becton Dickinson). We selected laboratory strain 28Bl for construction of isogenic strains and measured its MIC for azithromycin, clarithromycin, and erythromycin20. rplD encoding the G70D mutation was PCR amplified from RplD G70D isolate GCGS10433 using primers rplD_FWD_DUS (5′ CATGCCGTCTGAACAAGACCCGGGTCGCG 3′) (containing a DUS tag to enhance transformation70) and rplD_REV (5′ TTCAGAAACGACAGGCGCC 3′). The resulting ~1 kb amplicon was spot transformed69 into 28Bl. We selected for transformants by plating onto GCBK plates with clarithromycin 0.4 μg/mL and erythromycin 0.4 μg/mL. We confirmed via Sanger sequencing that transformants had acquired the RplD G70D mutation and selected one transformant from each selection condition (strain C5 for clarithromycin and strain E9 for erythromycin) for further characterization. We confirmed that for all macrolides used for selection, no spontaneous resistant mutants were observed after conducting control transformations in the absence of GCGS1043 PCR product. We did not construct strains with the mutation complemented.
Growth assays
We streaked 28Bl and 28Bl RplDG70D (E9) onto GCBK plates and grew them overnight for 16 hours at 37 °C in a 5% CO2 atmosphere. We prepared 1 L of fresh Graver Wade (GW) media71 and re-suspended overnight cultures into 1 mL of GW. After normalizing cultures to OD 0.1, we diluted cultures 1:105 and inoculated central wells of a 24-well plate with 1.5 mL GW and cells in triplicate. Edge wells were filled with 1.5 mL water. After growth for 1 hour to acclimate to media conditions, we sampled CFUs every 2 hours for a total of 12 hours. For each timepoint, we aspirated using a P1000 micropipette to dissolve clumps and then plated serial dilutions onto a GCBK plate. We counted CFUs the following day and used GraphPad Prism (version 8.2.0 for Windows, GraphPad Software) to graph the data and estimate exponential phase growth rates following removal of lag phase data points and log-transformation of CFUs/mL.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
In Supplementary Data 6, we have included accession numbers (via publicly hosted database NCBI SRA) for accessing all raw sequence data used for N. gonorrhoeae analyses. Intermediate outputs from the genomics pipeline (e.g., de novo assemblies) may also be available from the authors upon request. An interactive and downloadable version of the phylogeny and annotation rings used in Fig. 2 and Supplementary Fig. 1 is hosted at https://itol.embl.de/tree/1281032245351351597338246. Source data are available at https://github.com/gradlab/rplD-conditional-GWAS (https://doi.org/10.5281/zenodo.4042334)72.
Code availability
Code to reproduce the analyses and figures is available at https://github.com/gradlab/rplD-conditional-GWAS (https://doi.org/10.5281/zenodo.4042334)72 or from the authors upon request.
References
Unemo, M. & Shafer, W. M. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin. Microbiol. Rev. 27, 587–613 (2014).
Wi, T. et al. Antimicrobial resistance in Neisseria gonorrhoeae: global surveillance and a call for international collaborative action. PLoS Med. 14, e1002344 (2017).
Grad, Y. H. et al. Genomic epidemiology of gonococcal resistance to extended-spectrum cephalosporins, macrolides, and fluoroquinolones in the United States, 2000-2013. J. Infect. Dis. 214, 1579–1587 (2016).
Fifer, H., Saunders, J., Soni, S., Sadiq, S. T. & Fitzgerald, M. British Association for Sexual Health and HIV national guideline for the management of infection with Neisseria gonorrhoeae https://www.bashhguidelines.org/media/1208/gc-2019.pdf (2019).
Allan-Blitz, L.-T. et al. A cost analysis of gyrase A testing and targeted ciprofloxacin therapy versus recommended 2-drug therapy for Neisseria gonorrhoeae infection. Sex. Transm. Dis. 45, 87–91 (2018).
Tuite, A. R. et al. Impact of rapid susceptibility testing and antibiotic selection strategy on the emergence and spread of antibiotic resistance in gonorrhea. J. Infect. Dis. 216, 1141–1149 (2017).
Ma, K. C. et al. Adaptation to the cervical environment is associated with increased antibiotic susceptibility in Neisseria gonorrhoeae. Nat. Commun. 11, 4126 (2020).
Hicks, A. L. et al. Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole genome sequencing data. PLoS Comput. Biol. 15, e1007349 (2019).
Gaynor, M. & Mankin, A. S. Macrolide antibiotics: binding site, mechanism of action, resistance. Curr. Top. Med. Chem. 3, 949–961 (2003).
Ng, L. K., Martin, I., Liu, G. & Bryden, L. Mutation in 23S rRNA associated with macrolide resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 46, 3020–3025 (2002).
Zhang, J. & van der Veen, S. Neisseria gonorrhoeae 23S rRNA A2059G mutation is the only determinant necessary for high-level azithromycin resistance and improves in vivo biological fitness. J. Antimicrob. Chemother. 74, 407–415 (2019).
Rouquette, C., Harmon, J. B. & Shafer, W. M. Induction of the mtrCDE-encoded efflux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol. Microbiol. 33, 651–658 (1999).
Shafer, W. M. et al. in National Institute of Allergy and Infectious Diseases, NIH 55-63 (Humana Press, 2008).
Veal, W. L., Nicholas, R. A. & Shafer, W. M. Overexpression of the MtrC-MtrD-MtrE efflux pump due to an mtrR mutation is required for chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. J. Bacteriol. 184, 5619–5624 (2002).
Warner, D. M., Folster, J. P., Shafer, W. M. & Jerse, A. E. Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J. Infect. Dis. 196, 1804–1812 (2007).
Warner, D. M., Shafer, W. M. & Jerse, A. E. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 70, 462–478 (2008).
Zalucki, Y. M., Dhulipala, V. & Shafer, W. M. Dueling regulatory properties of a transcriptional activator (MtrA) and repressor (MtrR) that control efflux pump gene expression in Neisseria gonorrhoeae. MBio 3, e00446–00412 (2012).
Zarantonelli, L., Borthagaray, G., Lee, E.-H. & Shafer, W. M. Decreased azithromycin susceptibility of Neisseria gonorrhoeae due to mtrRMutations. Antimicrob. Agents Chemother. 43, 2468–2472 (1999).
Rouquette-Loughlin, C. E. et al. Mechanistic basis for decreased antimicrobial susceptibility in a clinical isolate of Neisseria gonorrhoeae possessing a mosaic-like mtr efflux pump locus. MBio. https://doi.org/10.1128/mBio.02281-18 (2018).
Wadsworth, C. B., Arnold, B. J., Sater, M. R. A. & Grad, Y. H. Azithromycin resistance through interspecific acquisition of an epistasis-dependent efflux pump component and transcriptional regulator in Neisseria gonorrhoeae. MBio. https://doi.org/10.1128/mBio.01419-18 (2018).
Cousin, S. L. Jr, Whittington, W. L. H. & Roberts, M. C. Acquired macrolide resistance genes and the 1 bp deletion in the mtrR promoter in Neisseria gonorrhoeae. J. Antimicrob. Chemother. 51, 131–133 (2003).
Lees, J. A. et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat. Commun. 7, 12797 (2016).
Diner, E. J. & Hayes, C. S. Recombineering reveals a diverse collection of ribosomal proteins L4 and L22 that confer resistance to macrolide antibiotics. J. Mol. Biol. 386, 300–315 (2009).
Wheeler, N. E. et al. Contrasting approaches to genome-wide association studies impact the detection of resistance mechanisms in Staphylococcus aureus. bioRxiv. https://doi.org/10.1101/758144 (2019).
Belkacem, A. et al. Molecular epidemiology and mechanisms of resistance of azithromycin-resistant Neisseria gonorrhoeae isolated in France during 2013-14. J. Antimicrob. Chemother. 71, 2471–2478 (2016).
Jacobsson, S. et al. WGS analysis and molecular resistance mechanisms of azithromycin-resistant (MIC > 2 mg/L) Neisseria gonorrhoeae isolates in Europe from 2009 to 2014. J. Antimicrob. Chemother. 71, 3109–3116 (2016).
Demczuk, W. et al. Equations to predict antimicrobial minimum inhibitory concentrations in Neisseria gonorrhoeae using molecular antimicrobial resistance determinants. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02005-19 (2019).
Mortimer, T. D. et al. The distribution and spread of susceptible and resistant Neisseria gonorrhoeae across demographic groups in a major metropolitan center. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa1229 (2020).
Yahara, K. et al. Genomic surveillance of Neisseria gonorrhoeae to investigate the distribution and evolution of antimicrobial-resistance determinants and lineages. Microb. Genom. https://doi.org/10.1099/mgen.0.000205 (2018).
Zeitouni, S., Collin, O., Andraud, M., Ermel, G. & Kempf, I. Fitness of macrolide resistant Campylobacter coli and Campylobacter jejuni. Microb. Drug Resist. 18, 101–108 (2012).
Ma, L., Han, S., Yang, J. & Da, Y. Multi-locus test conditional on confirmed effects leads to increased power in genome-wide association studies. PLoS ONE 5, e15006 (2010).
Segura, V. et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet 44, 825–830 (2012).
Bennett, D., Morris, D. & Seoighe, C. Controlling for off-target genetic effects using polygenic scores improves the power of genome-wide association studies. bioRxiv. https://doi.org/10.1101/2020.05.21.097691 (2020).
Lees, J. A. et al. Improved prediction of bacterial genotype-phenotype associations using interpretable pangenome-spanning regressions. mBio 11, (2020).
Saber, M. & Shapiro, B. Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes. Microb. Genom. https://doi.org/10.1099/mgen.0.000337 (2020).
Zheng, Z. et al. Antimicrobial resistance and molecular characteristics among neisseria gonorrhoeae clinical isolates in a Chinese tertiary hospital. Infect. Drug Resist. 12, 3301–3309 (2019).
Thomas, J. C. et al. Evidence of recent genomic evolution in gonococcal strains with decreased susceptibility to cephalosporins or azithromycin in the United States, 2014–2016. J. Infect. Dis. https://doi.org/10.1093/infdis/jiz079 (2019).
Golparian, D. et al. Genomic evolution of Neisseria gonorrhoeae since the preantibiotic era (1928–2013): antimicrobial use/misuse selects for resistance and drives evolution. BMC Genomics 21, 116 (2020).
Demczuk, W. et al. Genomic epidemiology and molecular resistance mechanisms of azithromycin-resistant Neisseria gonorrhoeae in Canada from 1997 to 2014. J. Clin. Microbiol. 54, 1304–1313 (2016).
Endimiani, A. et al. Characterization of Neisseria gonorrhoeae isolates detected in Switzerland (1998-2012): emergence of multidrug-resistant clones less susceptible to cephalosporins. BMC Infect. Dis. 14, 106 (2014).
Wan, C. et al. Increasing resistance to azithromycin ofNeisseria gonorrhoeaein eastern Chinese cities: mechanisms and genetic diversity of resistant Nanjing isolates. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02499-17 (2018).
Harrison, O. B. et al. Genomic analyses of Neisseria gonorrhoeae reveal an association of the gonococcal genetic island with antimicrobial resistance. J. Infect. 73, 578–587 (2016).
Colangeli, R. et al. Bacterial factors that predict relapse after tuberculosis therapy. N. Engl. J. Med. 379, 823–833 (2018).
Yasuda, M., Ito, S., Hatazaki, K. & Deguchi, T. Remarkable increase of Neisseria gonorrhoeae with decreased susceptibility of azithromycin and increase in the failure of azithromycin therapy in male gonococcal urethritis in Sendai in 2015. J. Infect. Chemother. 22, 841–843 (2016).
Tapsall, J. W. et al. Failure of azithromycin therapy in gonorrhea and discorrelation with laboratory test parameters. Sex. Transm. Dis. 25, 505–508 (1998).
Fifer, H. et al. Sustained transmission of high-level azithromycin-resistant Neisseria gonorrhoeae in England: an observational study. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(18)30122-1 (2018).
Williamson, D. A. et al. Bridging of Neisseria gonorrhoeae lineages across sexual networks in the HIV pre-exposure prophylaxis era. Nat. Commun. 10, 3988 (2019).
Gernert, K. M. S. et al. Azithromycin susceptibility of Neisseria gonorrhoeae in the USA in 2017: a genomic analysis of surveillance data. Lancet Microbe 1(Aug), e154–e164 (2020).
Břinda, K. et al. Rapid inference of antibiotic resistance and susceptibility by genomic neighbour typing. Nature Microbiology 5, 455–464 (2020).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017).
Palace, S. G. et al. RNA polymerase mutations cause cephalosporin resistance in clinical Neisseria gonorrhoeae isolates. Elife. https://doi.org/10.7554/eLife.51407 (2020).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint 1303.3997 (2013).
Johnson, S. R., Grad, Y., Abrams, A. J., Pettus, K. & Trees, D. L. Use of whole-genome sequencing data to analyze 23S rRNA-mediated azithromycin resistance. Int J. Antimicrob. Agents 49, 252–254 (2017).
Garcia-Alcalde, F. et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Kersh, E. N. et al. Rationale for a Neisseria gonorrhoeae susceptible only interpretive breakpoint for azithromycin. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciz292 (2019).
Lees, J. A., Galardini, M., Bentley, S. D., Weiser, J. N. & Corander, J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics 34, 4310–4312 (2018).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Arnold, B. et al. Fine-scale haplotype structure reveals strong signatures of positive selection in a recombining bacterial pathogen. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msz225 (2019).
Hill, W. G. & Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl Genet 38, 226–231 (1968).
Demczuk, W. et al. Equations to predict antimicrobial MICs in Neisseria gonorrhoeae using molecular antimicrobial resistance determinants. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02005-19 (2020).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Cock, P. J. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Dillard, J. P. Genetic Manipulation of Neisseria gonorrhoeae. Curr. Protoc. Microbiol. Chapter 4 (Unit4A), 2 (2011).
Ambur, O. H., Frye, S. A. & Tønjum, T. New functional identity for the DNA uptake sequence in transformation and its presence in transcriptional terminators. J. Bacteriol. 189, 2077–2085 (2007).
Wade, J. J. & Graver, M. A. A fully defined, clear and protein-free liquid medium permitting dense growth of Neisseria gonorrhoeae from very low inocula. FEMS Microbiol. Lett. 273, 35–37 (2007).
Ma, K. C. gradlab/rplD-conditional-gwas 1.0.0 (Version 1.0.0). Zenodo, https://doi.org/10.5281/zenodo.4042334 (2020).
Acknowledgements
This work was supported by the NIH/NIAID grant 1R01AI132606-01 and the Smith Family Foundation. T.D.M. is additionally supported by the NIH/NIAID 1 F32 AI145157-01, and K.C.M. is additionally supported by the NSF GRFP grant DGE1745303. Portions of this research were conducted on the O2 high-performance computing cluster, supported by the Research Computing Group at Harvard Medical School. We additionally thank Daniel Rubin, Samantha Palace, Crista Wadsworth, and other members of the Grad Lab for helpful comments during development of the project.
Author information
Authors and Affiliations
Contributions
K.C.M., T.D.M., A.L.H., N.E.W., and L.S.B. performed and interpreted genomic analyses. K.C.M. and M.A.D. performed and interpreted experimental analyses. Y.H.G. supervised the project. K.C.M. and Y.H.G. wrote the paper with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks John Lees and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, K.C., Mortimer, T.D., Duckett, M.A. et al. Increased power from conditional bacterial genome-wide association identifies macrolide resistance mutations in Neisseria gonorrhoeae. Nat Commun 11, 5374 (2020). https://doi.org/10.1038/s41467-020-19250-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19250-6
This article is cited by
-
Genome-wide association study reveals serovar-associated genetic loci in Riemerella anatipestifer
BMC Genomics (2024)
-
Next-generation microbiology: from comparative genomics to gene function
Genome Biology (2021)
-
A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch
Genome Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
