
Abstract
Organic synthesis methodology enables the synthesis of complex molecules and materials used in all fields of science and technology and represents a vast body of accumulated knowledge optimally suited for deep learning. While most organic reactions involve distinct functional groups and can readily be learned by deep learning models and chemists alike, regio- and stereoselective transformations are more challenging because their outcome also depends on functional group surroundings. Here, we challenge the Molecular Transformer model to predict reactions on carbohydrates where regio- and stereoselectivity are notoriously difficult to predict. We show that transfer learning of the general patent reaction model with a small set of carbohydrate reactions produces a specialized model returning predictions for carbohydrate reactions with remarkable accuracy. We validate these predictions experimentally with the synthesis of a lipid-linked oligosaccharide involving regioselective protections and stereoselective glycosylations. The transfer learning approach should be applicable to any reaction class of interest.
Similar content being viewed by others

Introduction
Organic synthesis is a complex problem-solving task in which the vast knowledge accumulated in the field of organic chemistry is used to create new molecules, starting from simple commercially available building blocks1. Because of its complexity, organic synthesis is believed to be one of the main bottlenecks in pharmaceutical research and development2, and having accurate models to predict reaction outcome could boost chemists’ productivity by reducing the number of experiments to perform.
Machine learning has long been present in the chemical domain, tackling challenges than range, for example for quantitative structure–activity relationship predictions3, virtual screening4 and quantum chemistry5,6. Enabled by algorithmic advances in deep learning7,8,9,10 and the availability of large reaction data sets11,12, reaction prediction methods have emerged in recent years13,14,15,16,17,18,19,20,21,22. Those reaction prediction methods can be divided into two categories23, bond change prediction methods using graph neural networks14,16,17,18,22 and product SMILES generation using sequence-2-sequence models15,19.
Reaction prediction tasks are typically evaluated on the USPTO_MIT benchmark14, which does not contain molecules with defined stereocenters. Currently, the best prediction algorithm in terms of performance is the Molecular Transformer10,19. The architecture is based on the ground-breaking work by Vaswani et al.10, which revolutionised the field of neural machine translation, where sentences in one language are translated into another language. In contrast, for reaction prediction, the model learns to translate the precursors’ Simplified molecular-input line-entry system (SMILES)24 representation into the product SMILES.
The Molecular Transformer can be accessed for free through the IBM RXN for Chemistry platform25. Compared to other methods, such as graph neural networks-based ones, the advantages of the Molecular Transformer approaches are that they do not require mapping between the product and reactant atoms in the training26 and inputs can contain stereochemistry. In fact, sequence-2-sequence approaches, like the Molecular Transformer10,19, are currently the only large-scale reaction prediction approaches capable of handling stereochemistry. Stereochemistry is systematically avoided in graph-based methods, as the connection table and adjacency matrix of two stereoisomers is identical. Although stereoselectivity can theoretically be predicted by the Molecular Transformers19, it is one of their most significant weaknesses because of the lack of clean training data. To date, their performance on predicting specific stereochemical reactions has not been investigated.
In this work, we investigate the adaptation of the Molecular Transformer to correctly predict regio- and stereoselective reactions. As study case we focus on carbohydrates, a class of molecules for which the stereochemistry and the high degree of functionalization are key reactivity factors. Carbohydrate chemistry is essential for accessing complex glycans that are used as tool compounds to investigate fundamental biological processes such as protein glycosylation27,28,29, as well as for the preparation of synthetic vaccines30,31,32. Predicting the outcome of carbohydrate transformations, such as regioselective protection/deprotection of multiple hydroxyl groups or the stereospecificity of glycosylation reactions, is a very difficult task even for experienced carbohydrate chemists33,34, implying that this field of research might particularly benefit from computer-assisted reaction prediction tools.
First, we investigate transfer learning with a specialized subset of reactions as a means to adapt the Molecular Transformer to achieve high performance on carbohydrate reactions. Transfer learning, where a model is trained on a task with abundant data and either simultaneously trained or subsequently fine-tuned on another task with less data available35, has recently led to significant advancements in Natural Language Processing36,37,38,39. For instance, it has been used to improve translation performance in low-resource languages36. More recently, unsupervised pretraining transfer learning strategies have successfully been applied to sequence-2-sequence models37,40. In the chemical domain, transfer learning has enabled the development of accurate neural network potential for quantum mechanical calculations41 and shows great potential to solve other challenges42. For transfer learning we use a set of 20k carbohydrate reactions from the literature, comprising protection/deprotection and glycosylation sequences. We explore multitask learning, as well as sequential transfer learning, and show that the adapted model, called the Carbohydrate Transformer, performs significantly better than the general model on carbohydrate transformations and a model trained on carbohydrate reactions only.
Second, we perform a detailed experimental assessment of the deep learning reaction prediction model and test the Carbohydrate Transformer on unpublished reactions. Our assessment consists of a 14-step total synthesis of a modified substrate of a eukaryotic oligosaccharil transferase (OST). We also challenge our Carbohydrate Transformer to predict the reactions from the recently published total syntheses of the trisaccharide of Pseudomonas aeruginosa and Staphylococcus aureus43 as a further assessment on more complex carbohydrate reactions. Those reactions would be considered challenging to predict, even for carbohydrate experts.
Overall, we observe a consistent top-1 prediction accuracy above 70%, which roughly means a 30% increase compared to the original Molecular Transformer baseline. We find that the confidence score is a good predictor of prediction reliability and that many wrong predictions have chemical reasons such as the lack of reagent stoichiometry in the training data. The approach we used to learn carbohydrate reactions could be applied to any reaction class. Hence, it is expected to have a significant impact on the field of organic synthesis, as models like the Molecular Transformer19 can easily be specialized for the reaction subspaces that individual chemists are most interest in.
Results
Data availability scenarios
Besides the additional complexity, the main challenges for learning to predict stereochemical reactions is the data. In the largest open-source reaction data set by Lowe11,12, which fueled the recent advancements in machine learning for chemical reaction prediction, stereochemistry, and specifically reactions involving carbohydrates are underrepresented and of poor quality. Hence, those reactions are problematic to learn.
In this work, we explore two real world scenarios, where there exist a large data set of generic chemical reactions and a small data set of complex and specific reactions. In our case, we use a data set derived from the US patent reactions by Lowe12 as the large data set containing 1.1M reactions. We call this data set USPTO. For the specific reaction, we chose carbohydrates reactions, but the methods described could be applied to any reaction class of interest. We manually extracted reactions from the Reaxys44 database, selected from papers of 26 authors in the field of carbohydrate chemistry. The small data set of 25k reactions will be referred to as CARBO for the remainder of the publication. We split the USPTO and the CARBO data set into train, validation and test sets. The reaction data was canonicalised using RDKit45. A more detailed description of the data is found in Supplementary Note 1.
If the access to the large and small sets is given, the two data sets can be used simultaneously for training. We call this first scenario multitask. However, depending on the situation, direct access to the data of the generic data set may not be possible. For example, a company A may have proprietary reaction data precluded from external sharings. Company A could still train a model using their own data and share their model without revealing the exact data points. The trained model extracts some general chemical reactivity knowledge and could be shared without exposing company proprietary information. This pretrained model could then serve as a starting point to further train the model on another source of reactions. We call this scenario fine-tuning.
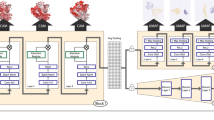
A visualisation of the model and the two scenarios can be found in Fig. 1.

Sequence-2-sequence prediction of carbohydrate reactions and the two transfer learning scenarios, namely, multitask and sequential training.
In the multitask scenario, we investigated different reaction weighting schemes between the two sets. A comparison of the top-1 accuracies on the USPTO train, USPTO test, CARBO train and CARBO test sets for models trained with different weights for the USPTO train and CARBO train sets are shown in Fig. 2a. The weights describe in what proportion reactions from the two sets are shown per training batch. For example, weight 1 on USPTO and weight 1 on CARBO means that for one USPTO reaction one CARBO reaction is shown. As can be seen in the Figure, the highest accuracy on the CARBO test set (71.2 %) is obtained with weight 9 on the USPTO set and weight 1 on the CARBO set (w9w1). As expected, training only with the CARBO train set leads to a poor CARBO test set accuracy (30.4%). As 20k reactions are not enough for the model to learn predict organic chemistry. The accuracy reached by the model trained purely on the USPTO data reaches 43.3%. It therefore performs better than the model trained purely on the CARBO reactions. In Fig. 2b, we assess the effect of the size of the CARBO train set. The accuracy continuously increases from 43.3 to 71.2% with an increasing number of reactions in the train set.

a Top-1 accuracy of models trained with different weights on the USPTO and CARBO data set (the first number corresponds to the weight on the USPTO data set and the second to the weight on the CARBO data set). b Top-1 accuracy for a model trained in the weight 9 weight 1 setting, where the number of reactions in the CARBO data set was reduced. Source data are provided as a Source Data file.
For the fine-tuning scenario, where access to the large generic data set is not given but a model, pretrained on the large data set, is available instead, the results on the CARBO and USPTO test sets are shown in Fig. 3a. After training the model on the CARBO train set, the top-1 accuracy reaches a 70.3%, similar to the model that was trained on the two data sets simultaneously. The observed behavior is the same when less CARBO reactions are available. Also for 1k CARBO reactions, the fine-tuning model matched the accuracy of the corresponding multitask model.

a CARBO random split test set performance for different training strategies. In green are the top-1 accuracies of the models that were fine-tuned on either 1k or 20k CARBO reactions shown. For comparison, we included in purple the top-1 accuracies of the models trained on the single data sets (CARBO, USPTO, and USPTO_MIT). Blue are the performances of models trained in the multitask scenario. b CARBO time split test set performance for different fine-tuning set sizes. Source data are provided as a Source Data file.
For this scenario, we analysed the effect of the train, validation, and test split in more detail. We compared the random split described above to a time split, where we included CARBO reactions first published before 2016 into the train and validation sets and the reactions published from 2016 into the test set (2831 reactions). We investigated different fine-tune set sizes (1k, 5k, 10k, 15k, and 20k). As seen in Fig. 3b, compared to the random split the top-1 accuracy with the 20k fine-tuning dropped slightly to 66% but it is still substantially larger than the accuracy that could be obtained with the generic USPTO training set only. Already with 5k CARBO reactions, an accuracy above 60% was reached. The larger the CARBO fine-tuning set, the better the performance of the fine-tuned model.
Besides the fact that the reactions in the large data set do not need to be revealed, another advantage is the short fine-tuning training time. The fine-tuning requires only 5k steps compared to 250k steps in the multitask scenario. However, if time and access to both data sets are given, it is better to train simultaneously on all data for a longer time as the performance on the large data set does not decrease, as it does in the fine-tuning scenario. If the interest is only in a specific reaction class, short adaptation times or if generic data is not available, then fine-tuning a pretrained model is better.
To further demonstrate the effectiveness of the fine-tuning approach, we performed an experiment where we pretrained a model on a data set without stereochemical information. To do so, we used the USPTO_MIT data set by Jin et al.14. As seen in Fig. 3a, although the pretrained model does not manage to predict any CARBO test set reactions, after fine-tuning for 6k steps the model reaches an accuracy of 63.3%. The accuracy was not as high as with USPTO pretraining but a significant improvement over the 0.0% correctly predicted reactions by the pretrained model. The low accuracy after pretraining was expected as none of the chiral center tokens (e.g. “[C@H]”, “[C@@H]”) were present in the training set. The fine-tuning result shows that the Molecular Transformer model is able to learn new concepts within a few thousands training steps on 20k data points.
In the next sections, we will compare the model trained only on the USPTO data, which was also used as pretrained model (USPTO model) with the model that was then fine-tuned on the 20k CARBO reactions (CARBO model).
Experimental assessment
Although the accuracy of the transformer has been widely assessed19, an experimental validation is still missing. Here, we decided to validate both the transformer and the augmented precision of the CARBO model on a recently realized synthetic sequence from our own laboratory, absent from the training data. This sequence is a 14-step synthesis of lipid-linked oligosaccharide (LLO) 15 to be used as a substrate to study OST46,47 (Fig. 4). The sequence contains typical carbohydrate chemistry: protecting group manipulations (steps: b, h, i, l n, p), functional group manipulations (step c, d), regioselective protections (step e), a β-selective glycosylation (step g) and an α-selective phosphorylation (step m). The latter regio- and stereoselective transformations are of particular interest because their selectivity is generally difficult to control and to predict, even for experienced synthetic chemists.

Reaction conditions: a BnOH, Yb(OTf)3, DCE, 90 °C, 2h, 78%. b MeONa, MeOH, sonication, 30 min. c PPh3, I2, imidazole, THF, 1h, reflux, 88% over two steps. d Pd/C, NH4OH, H2, THF/H2O, 30 min, 77%. e BzCl, pyr, −35°, 70%. f BF3Et2O, 4 MS, DCM, 26 h, 73%. g Zn, Ac2O, AcOH, DCE 50°, 3 h, 96%. h MeONa, MeOH/DMF, 4 days. i Ac2O, 4-(Dimethylamino)pyridine, pyr, 76% over three steps. l H2, THF/H2O, 10 bar, 16 h. m LiHMDS, tetrabenzylpyrophosphate, 53%. n H2, THF/MeOH, 1 h. o farnesylnerol, CDI, DMF, then 11, 5 days, 18%. p MeOH, NH4OH, 16 h, qte. An asterisk represents “*” reaction present in the training set.
We used both the general USPTO model and the fine-tuned CARBO model to predict 13 of the 14 steps in the sequence (step b was removed since it appeared in the training set). The USPTO only made four correct predictions (31%), which were either standard protecting group manipulations (step a, g, n) or functional group exchanges (step c). The CARBO model also correctly predicted these four simple reactions, but additionally, made another six correct predictions, including the regioselective benzoylation (5–6, step e) and the β-selective phosphorylation (11–12, step m), corresponding to a 77% success rate and a 46% improvement over the USPTO model, in line with the overall statistics presented above.
In detail, the CARBO model only made three mistakes. The first one concerns the reduction of the primary iodide 4 to a methyl group in 5 by hydrogenation, which is mistakenly predicted to also reduce the benzyl glycoside. The USPTO model makes the same mistake. Both models have not learned that carrying out the reaction in the presence of ammonia reduces the catalyst activity and avoids debenzylation, as no such reaction was present in the training sets. The second mistake concerns a similar reduction of the benzyl glycoside in 10 (step l), which is predicted to yield the β-lactol while the product 11 is in fact formed as an anomeric mixture. Again, the USPTO model makes the same mistake. Both models ignore that the initially formed β-lactol equilibrates spontaneously to the anomeric mixture via ring opening. Finally, the CARBO model predicts a shortened prenyl chain in the phosphate coupling reaction forming the protected LLO 14 (step o), which does not make chemical sense. In this case it should be noted that the CARBO training set does not contain a single LLO molecule, and that the USPTO model performs worse since it returns an invalid SMILES for this reaction.
We obtained similar prediction performances from both models when analyzing a recently published total syntheses of the trisaccharide repeating unit of Pseudomonas aeruginosa and Staphylococcus aureus43. Those synthetic sequences comprises four difficult regio- and stereoselective glycosylation steps and five regioselective protection steps that are of particular interest. Out of the 38 reactions that are absent from the training set in this sequence (Supplementary Figs. 2–7), the USPTO model predicts only 15 reactions (39%) correctly, and none of the difficult steps mentioned above. The CARBO model performs much better and correctly predicts 26 of the 38 reactions, corresponding to a 68% overall accuracy and a 29% gain over the USPTO model. In particular, the CARBO model correctly predicts the regioselectivity of the dimethyltin chloride mediated benzoylation of L-Rhamnopyranoside 16 (step no. 10), the difficult regio- and stereoselective glycosylation at position 3 of the terminal fucosyl in disaccharide 18 (step no. 24) as well as the regioselective protection of the same disaccharide at position 3 (step no. 29), all of which are nonobvious even for synthetic chemists (Fig. 5). Interestingly, the CARBO model predicts a double substitution of bis-triflate 19 instead of the correct single substitution at position 2, which the USPTO model correctly predicts. In this case it should be noted that the outcome of the reaction is dictated by stoichiometry (only one equivalent of the azide nucleophile), an information which is absent from the training data. In contrast to the USPTO training set, that contains only single azide substitutions, the CARBO training set contains single, as well as double substitutions. An analysis of the stereo centres in both data sets can be sound in Supplementary Table 1 and Supplementary Fig. 1.

a, b Reactions correctly predicted. c wrongly predicted reaction (red structure) due to missing reagent stoichiometry in the model: only one equivalent of NaN3 was used resulting in single substitution, while the model predicts double substitution.
Every predicted reaction is associated with a confidence score19, which is calculated from the product of the probabilities of the predicted product tokens. Interestingly, the confidence score correlates with the correctness of the prediction (Fig. 6). For both models most of the correct predictions have a score higher than 0.8.

Predictions (ordered by confidence score) for the experimental assessment. Source data are provided as a Source Data file.
To have a closer look at the capabilities of the model to self-estimate its own uncertainty, we analyzed every reaction in detail. In some cases, we observe epimerization or rearrangements that have little chemical significance and are associated with low score values. This even occurs in more trivial transformations, such as amine acetylation of the trisaccharide in reaction 27 (scheme S3). Although the model is not able to predict the correct product, its low score seems to indicate that the model senses its own mistake. The second class are arguably wrong predictions that have high confidence for chemical reasons. Such an example is the previously discussed reaction 12 (Scheme 2, entry c) whose outcome is influenced by stoichiometry that together with other reaction conditions, is excluded from the training data, making these reactions extremely difficult to predict.
Similar to previous work19, one of the limitations of current SMILES-2-SMILES models is that environmental reaction conditions like temperature and pressure are not taken into account. Those conditions are often missing in the data sets, and even if present, it would not be straightforward to codify temperature profiles applied during chemical reactions. Another limitation is the data coverage and quality. As pointed out above, most of the wrong predictions can be explained with the data that the models have seen during training.
The availability of large high-quality open-source reaction data set containing information detailed on amounts, stoichiometry, and reaction conditions could substantially improve reaction prediction models.
Discussion
In this work, we demonstrated that transfer learning can be successfully applied to a generally trained transformer model using as few as 20k data points to derive a specific model that predicts reactions from a specific class with significantly improved performance. Transfer learning of the general molecular transformer model, trained on the USPTO data set to a specific set of reactions, to obtain a high-performance specialized model as demonstrated here should be generally applicable towards any subclass of specific reactions of interest.
Here we used transfer learning to improve predictions of regio- and stereoselectivity, a central aspect of synthetic chemistry that has not been systematically evaluated previously by reaction prediction models, in part due to the fact that the Molecular Transformer is currently the only model able to handle stereochemistry. As a test case we examined carbohydrates, a well-defined class of molecules for which reactions are difficult to predict even for experienced chemists, and subjected our model to experimental validation. We anticipate that the Carbohydrate Transformer will serve the practical purpose of improving the efficiency of complex carbohydrate syntheses. The model can guide chemists by predicting and scoring potential carbohydrates reactions before performing them experimentally. The fact that the confidence score correlates with prediction accuracy offers a simple metric to judge the quality of predictions. The shortcomings noted should be addressable by extending the training set with reactions that are not predicted well.
Methods
Reaction prediction model
All the experiments in this work were run with the Molecular Transformer model19, which is illustrated in Fig. 1. For details on the architecture we refer the reader to10,19. We used Pytorch48 and the OpenNMT49 framework to build, train and test our models. Hyperparameters and a detailed description of the data sets can be found in the supplementary information. The investigated task is reaction prediction, where the aim is to predict the exact structural formula, including stereochemistry, of the products that are formed from a given a set of precursors as input. In the inputs, no difference is made between reactant and reagent molecules19. Following previous work13,15,19, we use accuracy as the evaluation metric. The reported accuracies describe the percentage of correct reactions. A reaction is counted as correct only if the predicted products exactly matches the products reported in the literature after canonicalisation using RDKit45. The canonicalisation is required as multiple SMILES can represent the same molecule.
Chemical synthesis
All reagents were purchased from commercial sources and used without further purifications unless otherwise stated. All reactions were carried out in flame-dried round-bottomed-flask under an argon atmosphere, except if specified. Room temperature (rt) refers to ambient temperature. Temperatures of 0 °C were maintained using an ice-water, −78 °C with acetone/dry ice bath and the other temperatures using a cryostat. Dry solvents were obtained by passing commercially available pre-dried, oxygen-free formulations through activated alumina columns. Hydrogenation was performed at room pressure using H2 filled balloon. Chromatographic purifications were performed with silica gel pore size 60, 230–400 mesh particle size (Sigma-Aldrich). Thin layer chromatography was performed using ALUGRAM Xtra Sil G/UV on pre-coated aluminium sheets, using UV light as a visualizing, and a basic aqueous potassium permanganate solution and ceric ammonium molybdate as developing agents. NMR spectra for 1H, 13C, DEPT, 31P, COSY, HSQC, HMBC, and NOE were recorded at rt with a Bruker AV (400 MHz 1H). Spectra were and processed using TopSpin 3.6.1 software. Chemical shifts are reported in δ (ppm) relative units to residual solvent peaks CDCl3 (7.26 ppm for 1H and 77.2 ppm for 13C) and MeOD (3.31 ppm for 1H and 49.00 ppm for 13C). Splitting patterns are assigned as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), multiplet (m), dd (doublet of doublets), and td (triplet of doublets). High-resolution mass spectra was provided by the “Service of Mass Spectrometry” at the Department of Chemistry and Biochemistry in Bern and were obtained by electron spray ionization in positive or negative mode recorded on a Thermo Scientific LTQ Orbitrap XL. For the experimental procedures, NMR spectra and physical data of compounds 2–15, see Supplementary Note 3.
Data availability
The USPTO data set derived from Lowe12 that we used for training and evaluation, our carbohydrate reactions, as well as the ones from the work of Behera et al.43 are available from (https://github.com/rxn4chemistry/OpenNMT-py/tree/carbohydrate_transformer). Source data are provided with this paper.
Code availability
The code and trained models are available from (https://github.com/rxn4chemistry/OpenNMT-py/tree/carbohydrate_transformer). The models are compatible with OpenNMT-py49,50, which was used for training and evaluation. The SMILES tokenization function for preprocessing the inputs is found on the Molecular Transformer repository19,51. The setup and hyperparameters can also be found in Supplementary Note 2.
References
Corey, E. J. The logic of chemical synthesis: multistep synthesis of complex carbogenic molecules. Angew. Chem. Int. Ed. 30, 455–465 (1991).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Lo, Y.-C., Rensi, S. E., Torng, W. & Altman, R. B. Machine learning in chemoinformatics and drug discovery. Drug Discov. today 23, 1538–1546 (2018).
Melville, J. L., Burke, E. K. & Hirst, J. D. Machine learning in virtual screening. Comb. Chem. High. Throughput Screen. 12, 332–343 (2009).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Dral, P. O. Quantum chemistry in the age of machine learning. J. Phys. Chem. Lett. 11, 2336–2347 (2020).
Sutskever, I., Vinyals, O. & Le, Q. V. Sequence to sequence learning with neural networks. In Advances in Neural Information Processing Systems, 3104–3112 (2014).
Luong, M.-T., Pham, H. & Manning, C. D. Effective approaches to attention-based neural machine translation. In Proceedings of the 2015 Conference on Empirical Methods in Natural Language Processing, 1412–1421 (2015).
Duvenaud, D. K. et al. Convolutional networks on graphs for learning molecular fingerprints. In Advances in Neural Information Processing Systems, 2224–2232 (2015).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems, 5998–6008 (2017).
Lowe, D. M. Extraction of chemical structures and reactions from the literature. Ph.D. thesis, University of Cambridge (2012).
Lowe, D. Chemical reactions from US patents (1976–2016) (2017). https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873.
Nam, J. & Kim, J. Linking the neural machine translation and the prediction of organic chemistry reactions. Preprint at https://arxiv.org/abs/1612.09529 (2016).
Jin, W., Coley, C., Barzilay, R. & Jaakkola, T. Predicting organic reaction outcomes with weisfeiler-lehman network. In Advances in Neural Information Processing Systems, 2607–2616 (2017).
Schwaller, P., Gaudin, T., Lanyi, D., Bekas, C. & Laino, T. Found in translation: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 9, 6091–6098 (2018).
Bradshaw, J., Kusner, M., Paige, B., Segler, M. & Hernández-Lobato, J. A generative model for electron paths. In International Conference on Learning Representations (2019).
Do, K., Tran, T. & Venkatesh, S. Graph transformation policy network for chemical reaction prediction. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 750–760 (2019).
Coley, C. W. et al. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 10, 370–377 (2019).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019).
Nair, V. H., Schwaller, P. & Laino, T. Data-driven chemical reaction prediction and retrosynthesis. CHIMIA 73, 997–1000 (2019).
Schwaller, P. et al. Predicting retrosynthetic pathways using a combined linguistic model and hyper-graph exploration strategy. Chem. Sci. 11, 3316–3325 (2020).
Qian, W. W. et al. Integrating deep neural networks and symbolic inference for organic reactivity prediction. Preprint at https://doi.org/10.26434/chemrxiv.11659563.v1 (2020).
Schwaller, P. & Laino, T. Data-driven learning systems for chemical reaction prediction: an analysis of recent approaches. In Machine Learning in Chemistry: Data-Driven Algorithms, Learning Systems, and Predictions, 61–79 (ACS Publications, 2019).
Weininger, D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
IBM RXN for chemistry. https://rxn.res.ibm.com. Accessed 13 Sep 2019.
Griffiths, R.-R., Schwaller, P., Lee, A. Dataset bias in the natural sciences: a case study in chemical reaction prediction and synthesis design. Preprint at https://doi.org/10.26434/chemrxiv.7366973.v1 (2018).
Ernst, B., Hart, G. W. & Sinaÿ, P. Carbohydrates in chemistry and biology (Wiley Blackwell, 2008).
Stallforth, P., Lepenies, B., Adibekian, A. & Seeberger, P. H. Carbohydrates: a frontier in medicinal chemistry. J. Med. Chem. 52, 5561–5577 (2009).
Boilevin, J. M. & Reymond, J.-L. Synthesis of lipid-linked oligosaccharides (llos) and their phosphonate analogues as probes to study protein glycosylation enzymes. Synthesis 50, 2631–2654 (2018).
Mettu, R., Chen, C.-Y. & Wu, C.-Y. Synthetic carbohydrate-based vaccines: challenges and opportunities. J. Biomed. Sci. 27, 1–22 (2020).
Broecker, F. & Seeberger, P. H. Identification and design of synthetic b cell epitopes for carbohydrate-based vaccines. In Methods in Enzymology, vol. 597, 311–334 (Elsevier, 2017).
Barel, L.-A. & Mulard, L. A. Classical and novel strategies to develop a shigella glycoconjugate vaccine: from concept to efficacy in human. Hum. Vaccines Immunother. 15, 1338–1356 (2019).
Kamat, M. N. & Demchenko, A. V. Revisiting the armed- disarmed concept rationale: S-benzoxazolyl glycosides in chemoselective oligosaccharide synthesis. Org. Lett. 7, 3215–3218 (2005).
Dhakal, B. & Crich, D. Synthesis and stereocontrolled equatorially selective glycosylation reactions of a pseudaminic acid donor: importance of the side-chain conformation and regioselective reduction of azide protecting groups. J. Am. Chem. Soc. 140, 15008–15015 (2018).
Ruder, S.Neural transfer learning for natural language processing. Ph.D. thesis, NUI Galway (2019).
Zoph, B., Yuret, D., May, J. & Knight, K. Transfer learning for low-resource neural machine translation. In Proceedings of the Conference on Empirical Methods in Natural Language Processing, 1568–1575 (2016).
Ramachandran, P., Liu, P. & Le, Q. Unsupervised pretraining for sequence to sequence learning. In Proceedings of the Conference on Empirical Methods in Natural Language Processing, 383–391 (2017).
Howard, J. & Ruder, S. Universal language model fine-tuning for text classification. In Proceedings of the Annual Meeting of the Association for Computational Linguistics, 328–339 (2018).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: pre-training of deep bidirectional transformers for language understanding. In Proceedings of the Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, 4171–4186 (2019).
Song, K., Tan, X., Qin, T., Lu, J. & Liu, T.-Y. Mass: masked sequence to sequence pre-training for language generation. In International Conference on Machine Learning, 5926–5936 (2019).
Smith, J. S. et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 10, 1–8 (2019).
Öztürk, H., Özgür, A., Schwaller, P., Laino, T. & Ozkirimli, E. Exploring chemical space using natural language processing methodologies for drug discovery. Drug Discov. Today 25, 689–705 (2020).
Behera, A., Rai, D. & Kulkarni, S. S. Total syntheses of conjugation-ready trisaccharide repeating units of Pseudomonas aeruginosa o11 and Staphylococcus aureus type 5 capsular polysaccharide for vaccine development. J. Am. Chem. Soc. 142, 456–467 (2019).
Reaxys database. https://www.reaxys.com. Accessed 29 Oct 2019.
Landrum, G. et al. RDKit: Open-Source Cheminformatics Software, Release 2019_03_4. https://doi.org/10.5281/zenodo.3366468. Accessed 29 Oct 2019.
Ramírez, A. S. et al. Characterization of the single-subunit oligosaccharyltransferase stt3a from trypanosoma brucei using synthetic peptides and lipid-linked oligosaccharide analogs. Glycobiology 27, 525–535 (2017).
Bloch, J. S. et al. Structure and mechanism of the er-based glucosyltransferase alg6. Nature 579, 443–447 (2020).
Paszke, A. et al. Pytorch: an imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems, 32, 8024–8035 (2019).
Klein, G., Kim, Y., Deng, Y., Senellart, J. & Rush, A. M. OpenNMT: open-source toolkit for neural machine translation. In Proceedings of ACL (2017).
OpenNMT-py. https://github.com/OpenNMT/OpenNMT-py. Accessed 29 Oct 2019.
Molecular Transformer. https://github.com/pschwllr/MolecularTransformer. Accessed 29 Aug 2019.
Acknowledgements
This research was supported by the Swiss National Science Foundation (SNF) Sinergia programme GlycoStart (CRSII5 173709). We thank the anonymous reviewers for their careful reading of our manuscript and their many insightful comments and suggestions.
Author information
Authors and Affiliations
Contributions
The project was conceived and designed by G.P., P.S., and J.L.R. and supervised by T.L. and J.L.R. G.P. performed the experiments. P.S. trained the models. All authors discussed the results and approved the manuscript. G.P. and P.S. contributed equally to this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pesciullesi, G., Schwaller, P., Laino, T. et al. Transfer learning enables the molecular transformer to predict regio- and stereoselective reactions on carbohydrates. Nat Commun 11, 4874 (2020). https://doi.org/10.1038/s41467-020-18671-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18671-7
This article is cited by
-
Transfer learning across different chemical domains: virtual screening of organic materials with deep learning models pretrained on small molecule and chemical reaction data
Journal of Cheminformatics (2024)
-
MATEO: intermolecular α-amidoalkylation theoretical enantioselectivity optimization. Online tool for selection and design of chiral catalysts and products
Journal of Cheminformatics (2024)
-
A general model for predicting enzyme functions based on enzymatic reactions
Journal of Cheminformatics (2024)
-
Augmenting large language models with chemistry tools
Nature Machine Intelligence (2024)
-
Predicting the formation of NADES using a transformer-based model
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
