
Abstract
A general synthesis of carbon nitride (CN) films with extended optical absorption, excellent charge separation under illumination, and outstanding performance as a photoanode in water-splitting photoelectrochemical cells is reported. To this end, we introduced a universal method to rapidly grow CN monomers directly from a hot saturated solution on various substrates. Upon calcination, a highly uniform carbon nitride layer with tuned structural and photophysical properties and in intimate contact with the substrate is obtained. Detailed photoelectrochemical and structural studies reveal good photoresponse up to 600 nm, excellent hole extraction efficiency (up to 62%) and strong adhesion of the CN layer to the substrate. The best CN photoanode demonstrates a benchmark-setting photocurrent density of 353 µA cm−2 (51% faradaic efficiency for oxygen), and external quantum yield value above 12% at 450 nm at 1.23 V versus RHE in an alkaline solution, as well as low onset potential and good stability.
Similar content being viewed by others


Introduction
Carbon nitride (CN) materials have emerged as promising cheap and benign semiconductors for photoelectrochemical (PEC) cells during the last decade, owing to their stability under harsh conditions and suitable energy band edges for water-splitting and other chemical transformations1,2,3,4,5,6,7,8,9. However, the PEC performance of CN materials is still low compared to the state-of-art semiconductors10,11,12,13,14 and to their possible theoretical value. The CNs’ intrinsic moderate light-harvesting properties and poor charge separation properties alongside a limited variety of growth methods and monomers, which can be utilized to grow a continuous CN layer with an intimate contact to the substrate4,15,16,17,18 are obstacles to their successful PEC implementation. To date, several methods have been used to synthesize CN films, such as the doctor-blade technique, which require supramolecular assemblies as the precursor19,20, thermal vapor condensation21,22,23, solvothermal methods24,25, micro-contact printing26 and liquid-based methods27,28,29. However, despite the great progress achieved with the above-mentioned growth and deposition methods, most of them only permit the use of a restricted range of monomers as the starting precursor and some require more complicated conditions that are not suitable for all substrates and have scalability difficulties30,31,32. The small number of growth methods and usable monomers hinders the quest for CN materials with enhanced optical and electronic properties, which are mandatory characteristics for further CN-PEC improvement. Therefore, new deposition methods that allow the use of a wider range of monomers to cast multiple CN layers with diverse chemical, morphological, and photophysical properties are still highly needed to achieve the required substantial progress towards practical CN-based PECs. Moreover, to this day, most of the CN materials have only exhibited strong light response up to ~420 nm in PEC, because of their wide bandgap, which is only suitable for harvesting a small portion of the solar spectrum33,34,35,36. Therefore, increasing the photoresponse of CN layers in the visible region is crucial for further PEC activity enhancement.
Herein we demonstrate a simple and general method to grow porous CN films with extended optical absorption, improved charge separation properties under illumination, and intimate contact with the substrate via the direct and fast growth of monomers from saturated solutions, followed by calcination at high temperature. In addition, to improve the charge separation, connection to the substrate, and overall PEC properties, we introduce melamine vapor during the layer’s growth37. The best CN film, based on thiourea as a precursor, CNTM, exhibits a photoanodic photocurrent of 353 µA cm−2 at 1.23 V versus (vs.) reversible hydrogen electrode (RHE) in 0.1 M KOH aqueous solution under one-sun illumination, a low onset potential of 0.32 V, and a high incident photon-to-current conversion efficiency (IPCE) up to 600 nm. Notably, gas evolution rates of 1.88 µmol h−1 cm−2 for H2 and 0.91 µmol h−1 cm−2 for O2 are detected.
Results
Preparation and characterization of CNT films
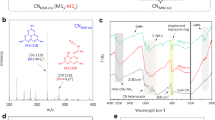
The new synthetic path is illustrated in Fig. 1a. To deposit thiourea directly on fluorine-doped tin oxide (FTO)-coated glass, a clean FTO glass is immersed into a hot (70 °C) saturated thiourea aqueous solution (ca. 0.9 g mL–1) for 1 s and subsequently taken out, yielding a uniform film layer of thiourea on FTO after the remaining liquid is left to dry. The thickness of the thiourea film can be tuned by sequential dip-dry cycles. Top-view scanning electron microscopy (SEM) of the thiourea film reveals a cohesive and oriented network structure (Fig. 1b), which is strikingly different from that of the pristine thiourea powder (Supplementary Fig. 1) and indicative of a good adhesion after recrystallization. Cross-sectional SEM (Fig. 1c) shows a uniform thickness and close contact to FTO. X-ray diffraction (XRD) patterns of the thiourea film and the pristine thiourea powder show similar crystal structures, with differences only in the relative intensity of some of the peaks, a result of variations in the orientation of the crystals in the samples (Fig. 1d).

a The proposed synthesis process of CNT film on FTO (the corresponding physical photos are shown within the dotted line). b Top-view SEM image of the thiourea film on FTO. c Cross-sectional SEM image of a 3-layer thiourea film on FTO (inset: a magnified SEM image of the squared area). d XRD patterns of clean FTO, thiourea powder, and thiourea film on FTO.
The CN films were prepared by the calcination of the thiourea films at 500 °C for 2 h under nitrogen atmosphere (the proposed self-polymerization of thiourea into CN was given by Zhang et al.38); this resulted in uniform yellow-colored carbon nitride layers (CNT, T stands for thiourea) on FTO (Fig. 1a). A top-view SEM image of a CNT electrode (Fig. 2a) presents a uniform and nanostructured architecture, which is dissimilar to the corresponding CNT powder (Supplementary Fig. 2). High-magnification SEM (Fig. 2b) discloses a nanosheets structure with many pores, owing to the release of condensation products during calcination. The thickness of CNT films (Fig. 2c) can be easily tuned from 13 to 53 μm (Fig. 2d) by adjusting the starting thickness of the thiourea layer (Supplementary Fig. 3).

a Top-view SEM image (inset: digital image of CNT/FTO electrode) and b magnified top-view SEM image of CNT film. c Cross-sectional SEM images of the different thicknesses CNT films on FTO. d Relationship between the final thickness of CNT films and the corresponding thiourea layers. Each data point represents an average of three different CNT electrodes; error bars represent standard deviation.
All CNT electrodes present chemical structural properties that are similar to those of CNT powder (Supplementary Fig. 4), which indicates a successful CN layer synthesis. XRD patterns (Supplementary Fig. 4a) of all CNT electrodes exhibit a strong and sharp peak at 27.4°, which is attributed to the (002) interlayer distance of graphitic CN. Another small reflection ca. 13° is attributed to the in-plane heptazine repeating units, i.e., (100)33. The Fourier-transform infrared (FTIR) spectra exhibit pronounced absorption peaks in the 1200–1600 cm–1 range (Supplementary Fig. 4b), which are assigned to the characteristic stretch modes of aromatic CN heterocycles. The peak at 810 cm–1 corresponds to the breathing mode of the heptazine units. The stretching vibration between 3000 and 3600 cm–1 indicates the existence of unreacted NH and/or NH2 groups33,39. The UV–Vis spectra of all the CNT electrodes (Supplementary Fig. 4c) disclose a direct bandgap in the 2.15–2.38 eV range, with a long absorption tail that extends beyond 700 nm. Altering the layer thickness from 13 to 53 μm leads to a slight redshift of the optical edges, together with an increased optical density. In accordance with the absorption behavior, the photoluminescence (PL) spectra (Supplementary Fig. 4d) of all CNT electrodes indicate a decrease of the PL intensity with increasing electrode thickness probably due to the contribution of defect states (bulk recombination) and possibly also increased reabsorption of emitted photons.
The chemical composition and oxidation state of the elements in the synthesized electrode were investigated by X-ray photoelectron spectroscopy (XPS), see Supplementary Fig. 5. The high-resolution spectrum of C 1s exhibits two peaks located at 284.6 and 287.9 eV, which belong to sp2 C–C bonds and N–C=N coordination, respectively. In the N 1s spectrum, three main peaks at 398.4, 399.9, and 401.0 eV were detected and assigned to sp2 C–N=C bonds, the tertiary nitrogen N–(C)3 groups, and the amino groups (C–NH–C and C–NH2), respectively, while a peak at 403.9 eV was assigned to charging effects33,40. In the O 1s spectrum, a peak at 532.1 eV, due to adsorbed water, was observed41. The high-resolution spectrum of S 2p shows the absence of sulfur in the final CN.
PEC performance of the CNT electrode
PEC measurements in a three-electrode configuration under one-sun illumination reveal an optimal photoresponse for a CNT layer thickness of 39 µm (Fig. 3a). The photocurrent density reaches 266 µA cm–2 at 1.23 V vs. RHE at this thickness. These results imply that there is a sufficient electron diffusion length within the CNT electrode. However, the diffusion of electrons is not yet optimal as can be deduced from a front-side illumination measurement (Supplementary Fig. 6). Under front-side illumination, not all the photogenerated electrons reach the FTO glass due to the large distance that the electrons should travel. A further increase in the film’s thickness results in a decline in photocurrent, which probably stems from reduced light penetration within the electrode and increased electron–hole recombination within the active layer before they react41. In addition to the thickness, the influence of the calcination temperature during the preparation of the CNT electrode on its morphology and PEC characteristics were also explored (Supplementary Figs. 7 and 8, respectively). As shown in Supplementary Fig. 8a, the CNT electrode calcined at 500 °C exhibits the best PEC performance.

a Photocurrent–thickness relationship of CNT electrodes. b Photocurrent density curves (chronoamperometry) of CNT electrodes at 1.23 V vs. RHE in 0.1 M KOH (blue, cyan, and magenta lines) and in 0.1 M KOH containing 10% (v/v) TEOA (red line). c IPCE plots of CNT electrode as a function of incident wavelength and UV–Vis spectrum of CNT electrode. d LSV curves of CNT electrode in 0.1 M KOH with and without light. All the light conditions are calibrated one-sun illumination. Each IPCE (%) data point represents an average of three different CNT electrodes; error bars represent standard deviation.
To determine the reproducibility of the CNT electrode PEC performance, photocurrent experiments were performed on three CNT electrodes, which were prepared in three separate synthetic batches. The photocurrent curves (Fig. 3b) of the three electrodes coincide very well (within ±10 µA cm–2), demonstrating the high repeatability of the new synthetic path of CNT electrodes. The calculated incident photon-to-current conversion efficiency (IPCE) of CNT electrodes at several illumination wavelengths (Fig. 3c) shows a direct correlation with light absorption. A high IPCE value of 13% is achieved at 400 nm, and a photoresponse can be detected up to 600 nm, which was seldom reported for unmodified CN electrodes. Linear sweep voltammetry (LSV) curves (Fig. 3d) of CNT electrode in the dark and under one-sun illumination demonstrate a typical PEC behavior, with an onset potential at 0.38 V vs. RHE, and an increase in photocurrent at higher bias potentials.
To estimate the hole extraction efficiency, 10% (v/v) triethanolamine (TEOA), an efficient hole scavenger, was added to the system. The facile hole extraction leads to an increase in the photocurrent to 470 µA cm−2 (Fig. 3b). In good agreement with this result, the IPCE value at 400 nm is improved to 19% (Fig. 3c). It is assumed that most holes are successfully extracted from the layer to oxidize TEOA, essentially removing the kinetic barrier of the oxidation half-reaction, allowing a maximal photocurrent to be obtained. Based on this assumption, the calculated charge transfer efficiency of CNT electrode at 1.23 V vs. RHE in 0.1 M KOH is 57 % (Supplementary Fig. 9), which is relatively high in the absence of any co-catalyst17,31,42. We note, that as we could not obtain a photocurrent plateau in the presence of TEOA, the actual values are probably lower.
Growing CNT films on different substrates
To show the generality of this new method, CNT film was grown on FTO with different sizes and shapes, and on other substrates, such as carbon paper, TiO2-coated electrode, and a glass slide. Figure 4a and Supplementary Fig. 10 show that regardless of the size and shape of the FTO substrate, both thiourea and the resulting CN films cover the entire FTO area well. On the other substrates, uniform and continuous CNT films were also obtained (Fig. 4b–d). The corresponding SEM characterization demonstrates that the morphologies of CNT films on different substrates are similar to those on FTO (Supplementary Figs. 11–13), suggesting a negligible effect of the substrates on the resulting CNT film morphology.

Digital images of thiourea film (left) and CNT film (right) on different substrates: a Large-sized FTO, b carbon paper, c TiO2-coated electrode, and d glass slide. e Top-view SEM image (inset: digital image of CNU electrode) and f cross-sectional SEM image of CNU film on FTO. g Photocurrent density curves (chronoamperometry) of CNU electrode at 1.23 V vs. RHE. h Top-view SEM image (inset: digital image of CND electrode) and i cross-sectional SEM image of CND film on FTO. j Photocurrent density curves (chronoamperometry) of CND electrode at 1.23 V vs. RHE.
Synthesis of CN films using different precursors
The generality of the developed CN film fabrication method was further demonstrated by using different monomers. Taking urea (ca. 2.4 g mL–1 at 70 °C in water) as an example, we showed that a uniform and ordered urea film on FTO can be obtained by using this method (Supplementary Fig. 14). The method was then extended to monomers with lower solubility than thiourea and urea, such as dicyandiamide (DCDA, ca. 0.3 g mL–1 at 70 °C). Supplementary Figure 15 shows that only a partial formation of a DCDA film is obtained on FTO, due to its low solubility in water. With the addition of a preliminary seeding layer step, the formed DCDA is evenly spread on the entire FTO (Supplementary Fig. 16). After calcination, the obtained CNU (CN synthesized from urea) electrode presents a loose coral-like morphology (Fig. 4e) while CND (CN synthesized from DCDA) looks like blades of grass (Fig. 4h). Both show a highly uniform microstructure, dissimilar to the corresponding precursor and CN powders (Supplementary Figs. 14c, 16e, and 17). Figure 4f, i demonstrate that the resulting CNU and CND films are well attached to the FTO and present good PEC performance (Fig. 4g, j, and Supplementary Fig. 18). High photocurrents of 368 and 133 µA cm−2 were obtained from the CNU electrode (Fig. 4g), and 277 and 145 µA cm−2 were achieved with the CND electrode (Fig. 4j), with or without a hole scavenger, respectively. Similarly, the two monomers can form porous and continuous CN films on other different substrates (Supplementary Figs. 19–21). In addition, two other monomers—ammonium thiocyanate (NH4SCN, abbreviated as NS, ca. 4.6 g mL–1 at 70 °C in water) and guanidine carbonate (abbreviated as GC, ca. 0.7 g mL–1 at 70 °C in water)—also allow uniform CN films formation by this method, leading to high PEC performance (Supplementary Figs. 22 and 23).
Synthesis of CNT films using other methods
For comparison, three other methods were used to synthesize CN photoelectrodes from thiourea. Direct calcination of thiourea powder spread on pristine FTO led to the formation of an uneven and cracked CN layer (Supplementary Fig. 24a). Drop-casting a slurry, followed by calcination at 300 °C for 1 h to increase adhesion gave a uniform CN film (Supplementary Fig. 24b). However, the layer peeled off upon contact with the electrolyte solution. A doctor-blade method, which is based on using thiourea as the precursor turned out to be infeasible owing to improper paste formation (Supplementary Fig. 24c).
Characterization and PEC performance of the CNTM electrode
As the water-splitting reaction occurs on the photoelectrode’s surface, a surface modification could greatly affect the defect states and charge transfer to the electrolyte without varying the bulk properties of the photoelectrode15,43,44. To further improve the PEC performance of the discussed CNT electrode, melamine powder was added to the bottom of the tube, and the thiourea/FTO was then placed close to the nozzle (Fig. 5a). After calcination, a slightly darker yellow CN film was obtained, named here as CNTM. The surface modification greatly improves the PEC performance of the CN; the modified CNTM electrode calcined at 550 °C shows the best performance (Fig. 5b and Supplementary Fig. 25). The photocurrent and IPCE value at 400 nm reach up to 353 µA cm−2 and 18%, respectively (Fig. 5b, c). In addition, an IPCE above 12% was measured at 450 nm and up to 2% at 600 nm, indicating an improved visible light response. Chronoamperometric curves (Supplementary Fig. 26) of five different CNTM electrodes match very well (within ±20 µA cm–2), indicating the good repeatability of the new synthetic path of CNTM electrodes. Upon addition of 10% (v/v) TEOA hole scavenger to the system, the photocurrent increases to 565 µA cm−2 (Supplementary Fig. 27). This means an improved charge transfer efficiency of the CNTM electrode in 0.1 M KOH, up to 62% (Supplementary Fig. 28) thanks to the surface modification. The improved charge separation is also evident in the lower onset potential of the CNTM electrode (0.32 V vs. RHE) than CNT electrode (0.38 V), indicating that a non-zero photoresponse is generated at almost 1 V below the potential of water oxidation (Fig. 5d). We note that these PEC values are among the best-performing for CN materials in the absence of a hole scavenger (Supplementary Table 1). Based on the good performance of CNTM photoanodes, the gas evolution during the reaction was detected by gas chromatography (Supplementary Fig. 29a), which showed a nearly 2:1 molar ratio of H2 and O2 generation rate, about 1.88 and 0.91 µmol h−1 cm−2, respectively (Fig. 5e), with calculated faradaic efficiency (FE) of 53 ± 1% for H2 and 51 ± 1.5% for O2. After a 1-h PEC reaction, about 50% of the initial photocurrent is preserved (Supplementary Fig. 29b). Post-characterization of the CNTM electrode (Supplementary Fig. 30) shows that the crystallinity of the CNTM electrode becomes slightly weaker and a small peak appears at 288.7 eV in the C 1s XPS spectrum, which is assigned to the C–O bond45,46, suggesting partial oxidation of the CNTM electrode occurred during the 1-h reaction. This oxidation is further observed in the O 1s spectrum (Supplementary Fig. 30d), where a signal from N–C–O species appears at 531.2 eV47. Furthermore, we characterized the CN-layer after 12 h stability measurement to confirm the structural changes. Therefore, the decrease in photocurrent should be attributed to the partial oxidation of the CN photoanode and the weakening of crystallinity during the reaction in 0.1 M KOH.

a Schematic synthetic process of CNTM film on FTO. b Photocurrent density (chronoamperometry) upon 1-sun cycling illumination of CNTM electrode at 1.23 V vs. RHE in 0.1 M KOH. c IPCE plots of CNTM as a function of incident wavelength in 0.1 M KOH and the corresponding UV–Vis spectrum of the CNTM electrode. d LSV curves of CNTM electrode in 0.1 M KOH. e Time-production plot of the H2 and O2 generated from the PEC system with CNTM at 1.23 V vs. RHE in 0.1 M KOH upon 1-sun illumination. f Top-view (inset: cross-sectional) SEM images of CNTM film on FTO. (The light yellow powder labeled as CNM in frame a refers to carbon nitride obtained by calcination of melamine powder at 550 °C for 2 h at the bottom of the tube). Each IPCE (%) data point represents an average of three different CNTM electrodes; error bars represent standard deviation.
Generally, water-spitting under neutral conditions is appealing. Thus, we have also measured the performance of CNTM photoanodes in 0.1 M Na2SO4 (pH = 7.02). Supplementary Fig. 31a shows that it also exhibited an outstanding PEC performance with a photocurrent of 305 µA cm−2 at 1.23 V vs. RHE under one-sun illumination. Meanwhile, the CNTM electrode is more stable under such conditions and higher rates of gas evolution (H2 and O2) were obtained relative to alkaline conditions (0.1 M KOH). Supplementary Fig. 31b shows there is almost no loss of photocurrent during the first 30 min. After an hour, the remaining photocurrent is about 72% of the initial photocurrent. The detected H2 and O2 were 2.26 and 1.08 µmol h−1 cm−2 (Supplementary Fig. 31c–d), respectively, corresponding to FE of 55% for H2 and 52% for O2.
Despite the introduction of melamine, the chemical structure, composition, as well as the morphology of the synthesized CNTM film remains similar to CNT with slight surface modification of the nanosheets (Fig. 5f and Supplementary Figs. 32–34). In addition, a small redshift of the direct optical bandgap from 2.33 to 2.26 eV is observed (Fig. 5c and Supplementary Fig. 35a). Combined with the results of Mott-Schottky measurements (Supplementary Fig. 35b, c), the conduction band and valence band positions of CNTM electrode are –1.21 and 1.05 eV vs. Ag/AgCl, respectively (see scheme in Supplementary Fig. 35d), which thermodynamically allow both water reduction and oxidation. Moreover, electrochemical active surface area (ECSA) measurements (Supplementary Fig. 36) demonstrate that the ECSA of the CNTM electrode is higher than that of the CNT electrode, which is in line with the calculated Brunauer–Emmet–Teller (BET) specific surface area from N2 sorption experiments (9.2 m2 g–1 for CNTM vs. 6.1 m2 g–1 for CNT, Supplementary Fig. 37). The ECSA and BET analysis suggest that the CNTM layer has more accessible sites for PEC reactions. In addition, electrochemical impedance spectroscopy (EIS) measurements (Supplementary Figs. 8 and 38) reveal that the introduction of melamine decreases the charge transfer resistance of holes from the CN layer to the solution.
Adhesion test
On the other hand, the addition of melamine during the synthesis increases the pressure inside the tube, which results in better contact between the CN and the FTO substrate (Fig. 5f, inset). We note that melamine can be replaced with another monomer (e.g., NH4SCN, who’s photoresponse is shown in Supplementary Fig. 39) that produces significant vapor pressure, leading to enhanced PEC performance. The latter accentuates the importance of the induced pressure during the film synthesis over the final performance. To confirm the strong adhesion, the prepared CNTM electrode was subjected to ultrasonication and adhesive tape testing. After 5 min of ultrasonication, only slight turbidity is detected in the liquid (Supplementary Fig. 40), which mainly resulted from CN powder at the surface of the photoactive CN film. Additional ultrasonication for a total of 30 min did not increase the turbidity and the CN film remained unchanged. The tape adhesion experiment was then carried out on the same CNTM electrode (after sonication, Supplementary Fig. 41). The CNTM film remained intact even after an adhesive tape was pasted onto the film under pressure for 2 h. A control experiment with CNT electrode (Supplementary Fig. 42a) shows that the CNT solution is more turbid than the CNTM one after 5 min of ultrasonication. It was clearly observed that with increasing ultrasonication time, the edges of the CNT film are slowly falling off. This shows that the adhesion between CNTM film and FTO is stronger than that of the CNT film. The better contact between the FTO and CNTM layer leads to lower resistance, which in turn enables the higher photocurrent and better stability. However, we note that most of the CNT film remains intact after 30 min of ultrasonication and a subsequent tape-test (Supplementary Fig. 42b), indicating that it is still robust.
Discussion
The reported simple, scalable, and versatile method allows the deposition of uniform carbon nitride layers with enhanced optical absorption and charge separation under illumination on various substrates. This deposition method relies on the fast growth of CN monomers directly on a substrate, followed by calcination at high temperature. Furthermore, we demonstrate that the introduction of melamine vapor during the calcination step significantly enhances the PEC activity of the CN electrodes thanks to surface modification. Detailed PEC studies unveil excellent hole extraction efficiency, up to 62%, together with good photoresponse over the visible range up to 600 nm (IPCE > 12% at 450 nm). The best CN film, based on thiourea as the starting monomer, exhibits a high photocurrent density of 353 µA cm−2, with FE of 53 ± 1% for H2 and 51 ± 1.5% for O2, and an IPCE value of 18% at 400 nm at 1.23 V vs. RHE in alkaline (0.1 M KOH) aqueous solution without sacrificial agents, providing a benchmark for CN photoanode materials.
Methods
Chemicals
All the chemical reagents were used without further purification. Thiourea (99%) and guanidine carbonate (GC) (99%) from Acros Organics. Urea (99%), dicyandiamide (DCDA) (99%), and melamine (99%) from Sigma-Aldrich. NH4SCN (NS) (98%) from Alfa Aesar. KOH (85%) and Na2SO4 (99%) from Loba-Chemie. Methanol (≥99.8%), ethanol (≥99%), and acetone (≥99%) from Bio-Lab Ltd. Triethanolamine (>99%) from Carl Roth. Fluorine-doped tin oxide (FTO)-coated glass (12–14 Ω sq−1) was purchased from Xop Glass company, Spain. Before using, the FTOs were thoroughly washed with an aqueous detergent (Alconox) solution, acetone, and ethanol in sequence and dried at 60 °C. Deionized water (18.2 MΩ cm resistivity at 25 °C, purified using a Millipore Direct-Q3 system) was used as the solvent for all the reported experiments.
Characterization
A PANalytical’s Empyrean diffractometer was employed to conduct X-ray diffraction (XRD) measurements. Fourier transformed infrared (FTIR) spectra were collected on a Thermo Scientific Nicolet iN 10Mx infrared microscope. UV–Vis spectra were recorded on a Cary 100 spectrophotometer equipped with a diffuse reflectance accessory (DRA). An Edinburgh FI/FSTCSPC 920 fluorimeter was used to collect photoluminescence (PL) spectra. The morphologies of the samples were characterized using a scanning electron microscope (SEM, JEOL JSM-7400F equipped with a FEG source) operated at 3.5 kV using a secondary electrons detector. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Thermo Fisher Scientific ESCALAB 250 using monochromated Kα X-rays (1486.6 eV), and all the binding energies obtained in XPS spectra were calibrated using the C 1s peak at 284.6 eV. N2 (99.999%) adsorption-desorption measurements were performed using a Quantachrome autosorb IQ2 at 77 K. The specific surface area was calculated using the Brunauer–Emmet–Teller (BET) model. Pore‐size distribution was calculated using non‐localized density functional theory (NLDFT) from the nitrogen sorption measurements. The amount of generated gases in the cell of the PEC set up were analyzed using a gas chromatograph (Agilent 7820 GC system) equipped with a thermal conductivity detector (TCD).
Material preparation
Thiourea/FTO: To deposit a thiourea film onto FTO, a clean FTO plate was immersed into a hot (70 °C) saturated thiourea aqueous solution and subsequently taken out. After the surface liquid was dried, a uniform film layer of thiourea on FTO was obtained. This step can be repeated several times to get different layers of thiourea film on FTO. Finally, the thiourea/FTO samples were dried in a 60 °C oven for subsequent use. Thiourea films on other substrates were obtained similarly.
Urea/FTO, NS/FTO, and GC/FTO: The preparing procedure of urea/FTO, NS/FTO, and GC/FTO is similar to that of thiourea/FTO, i.e., using urea, NH4SCN, and guanidine carbonate instead of thiourea, respectively. Urea on other substrates was also prepared using the same method.
DCDA/FTO: First, a clean FTO was dipped into a saturated DCDA solution in methanol at room temperature and was dried under air to form DCDA seeds. Secondly, the DCDA seeds/FTO was immersed into a hot (70 °C) saturated DCDA aqueous solution and subsequently taken out. A uniform layer of DCDA on FTO was thus obtained and dried at 60 °C. DCDA on other substrates was prepared using the same method.
CNT/FTO: To obtain a carbon nitride film on FTO, thiourea/FTO substrates were encapsulated in a glass tube filled with nitrogen. The glass tube was inserted into a ceramic tube furnace with N2 atmosphere. The oven was heated at a rate of 5 °C min−1 to 500 °C, when a calcination step at 500 °C for 2 h took place, resulting in a uniform yellow CNT layer on FTO.
CNU/FTO, CND/FTO, CNNS/FTO, and CNGC/FTO: CNU/FTO, CNNS/FTO, and CNGC/FTO were obtained by calcination of the corresponding precursor films at 500 °C for 2 h under N2. CND/FTO was obtained by calcination of DCDA/FTO at 500 °C for 4 h under N2.
Carbon nitride (CNT, CNU, and CND) films on other substrates were similarly obtained by changing the substrate on which the precursor deposition occurred.
CNTM/FTO: First, 1.0 g of melamine powder was added to the bottom of the tube, then the thiourea/FTO was placed close to the nozzle. The tube was then purged with N2, closed, and placed into an N2 oven. After calcination at 550 °C for 2 h, the CNTM/FTO was finally obtained. CNTNS/FTO was similarly obtained by using NH4SCN powder instead of the melamine powder.
The dimensions of all the reported substrates (FTO, carbon paper, a glass slide) were 1.2 cm × 2.5 cm unless otherwise specified.
Photoelectrochemical analysis
A Newport 300 W Xe arc lamp equipped with air mass AM 1.5G and water filters was used as a photoexcitation light source. The intensity of incident light simulated as one-sun illumination was calibrated using a Newport 919P thermopile detector. The electrochemical analysis measurements were conducted in a conventional three-electrode cell on an Autolab potentiostat (Metrohm, PGSTAT302N) system, using a 1 cm2 Pt foil electrode as the counter electrode and an Ag/AgCl electrode as the reference electrode, respectively. 0.1 M KOH aqueous solution (pH = 13) and 0.1 M Na2SO4 aqueous solution (pH = 7.02) were used as the electrolyte and were purged with argon to remove O2 before the electrochemical measurements. All the potentials were converted to reversible hydrogen electrode (RHE) values using the Nernst equation at room temperature:
Photocurrent measurements were conducted at 1.23 V vs. RHE and all photocurrents in the text and supporting materials were measured after three minutes of equilibration time unless otherwise specified. Mott-Schottky measurements were carried out in 1 M Na2SO4 aqueous solution using the same equipment.
Incident photon-to-current conversion efficiency (IPCE) values were calculated using:
where J is the photocurrent density; λ is the wavelength of the incident monochromic light, which is controlled by different optical bandpass filters (Newport 10BBPF10 series) of 400, 420, 450, 480, 510, 540, 570, and 600 nm; I is the incident light power.
Hole extraction efficiency was calculated using:
It is assumed that the extraction rate of photogenerated holes in the system is 100% after the addition of a hole scavenger (10% v/v TEOA). JKOH is the photocurrent density obtained in 0.1 M KOH aqueous solution, while JTEOA is the photocurrent density obtained in 0.1 M KOH containing 10% (v/v) TEOA.
The faradaic efficiency (FE) was calculated using:
where m is the number of moles of gas produced; n is the number of reaction electrons; F is the Faraday constant; I is the photocurrent; t is the reaction time.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Wang, X. et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76 (2009).
Xiong, W., Huang, F. & Zhang, R. Q. Recent developments in carbon nitride based films for photoelectrochemical water splitting. Sustain. Energy Fuels 4, 485–503 (2020).
Ong, W. J., Tan, L. L., Ng, Y. H., Yong, S. T. & Chai, S. P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016).
Dong, G., Zhao, L., Wu, X., Zhu, M. & Wang, F. Photocatalysis removing of NO based on modified carbon nitride: The effect of celestite mineral particles. Appl. Catal. B 245, 459–468 (2019).
Xiao, K. et al. Artificial light-driven ion pump for photoelectric energy conversion. Nat. Commun. 10, 74 (2019).
Zhang, G. et al. Ionothermal synthesis of Triazine–Heptazine-based copolymers with apparent quantum yields of 60% at 420 nm for solar hydrogen production from “Sea Water”. Angew. Chem. Int. Ed. 57, 9372–9376 (2018).
Volokh, M., Peng, G., Barrio, J. & Shalom, M. Carbon nitride materials for water splitting photoelectrochemical cells. Angew. Chem. Int. Ed. 58, 6138–6151 (2019).
Zhao, T. et al. Ultrafast condensation of carbon nitride on electrodes with exceptional boosted photocurrent and electrochemiluminescence. Angew. Chem. Int. Ed. 59, 1139–1143 (2020).
Li, X. et al. Recent advances in 3D g-C3N4 composite photocatalysts for photocatalytic water splitting, degradation of pollutants and CO2 reduction. J. Alloy. Compd 802, 196–209 (2019).
Huang, M., Bian, J., Xiong, W., Huang, C. & Zhang, R. Low-dimensional Mo: BiVO4 photoanodes for enhanced photoelectrochemical activity. J. Mater. Chem. A 6, 3602–3609 (2018).
Kim, T. W. & Choi, K. S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science 343, 990–994 (2014).
Huang, C., Bian, J. & Zhang, R. Q. Role of Cl ion desorption in photocurrent enhancement of the annealed rutile single-crystalline TiO2 nanorod arrays. J. Phys. Chem. C 121, 18892–18899 (2017).
Chang, K. et al. Targeted synthesis of 2H-and 1T-phase MoS2 monolayers for catalytic hydrogen evolution. Adv. Mater. 28, 10033–10041 (2016).
May, M. M., Lewerenz, H. J., Lackner, D., Dimroth, F. & Hannappel, T. Efficient direct solar-to-hydrogen conversion by in situ interface transformation of a tandem structure. Nat. Commun. 6, 8286 (2015).
Fang, Y., Li, X. & Wang, X. Synthesis of polymeric carbon nitride films with adhesive interfaces for solar water splitting devices. ACS Catal. 8, 8774–8780 (2018).
Ruan, Q. et al. A nanojunction polymer photoelectrode for efficient charge transport and separation. Angew. Chem. Int. Ed. 56, 8221–8225 (2017).
Peng, G., Albero, J., Garcia, H. & Shalom, M. A water-splitting carbon nitride photoelectrochemical cell with efficient charge separation and remarkably low onset potential. Angew. Chem. Int. Ed. 130, 16033–16037 (2018).
Fang, Y., Xu, Y., Li, X., Ma, Y. & Wang, X. Coating polymeric carbon nitride photoanodes on conductive Y: ZnO nanorod arrays for overall water splitting. Angew. Chem. Int. Ed. 57, 9749–9753 (2018).
Peng, G., Xing, L., Barrio, J., Volokh, M. & Shalom, M. A general synthesis of porous carbon nitride films with tunable surface area and photophysical properties. Angew. Chem. Int. Ed. 57, 1186–1192 (2018).
Lou, S. et al. Comparison study of the photoelectrochemical activity of carbon nitride with different photoelectrode configurations. ACS Appl. Mater. Interfaces 8, 22287–22294 (2016).
Bian, J. et al. Thermal vapor condensation of uniform graphitic carbon nitride films with remarkable photocurrent density for photoelectrochemical applications. Nano Energy 15, 353–361 (2015).
Bian, J. et al. Efficiency enhancement of carbon nitride photoelectrochemical cells via tailored monomers design. Adv. Energy Mater. 6, 1600263 (2016).
Huang, M. et al. Collaborative enhancement of photon harvesting and charge carrier dynamics in carbon nitride photoelectrode. Appl. Catal. B 237, 783–790 (2018).
Fan, X. et al. Layered double hydroxides decorated graphic carbon nitride film as efficient photoanodes for photoelectrochemical water splitting. Catal. Today 335, 423–428 (2019).
Xie, X., Fan, X., Huang, X., Wang, T. & He, J. In situ growth of graphitic carbon nitride films on transparent conducting substrates via a solvothermal route for photoelectrochemical performance. RSC Adv. 6, 9916–9922 (2016).
Liu, J. et al. Microcontact-printing‐assisted access of graphitic carbon nitride films with favorable textures toward photoelectrochemical application. Adv. Mater. 27, 712–718 (2015).
Xu, J. et al. Liquid-based growth of polymeric carbon nitride layers and their use in a mesostructured polymer solar cell with Voc exceeding 1 V. J. Am. Chem. Soc. 136, 13486–13489 (2014).
Zhang, W. et al. One-pot synthesis of nickel-modified carbon nitride layers toward efficient photoelectrochemical cells. ACS Appl. Mater. Interfaces 9, 32667–32677 (2017).
Zhang, J., Zhang, M., Lin, L. & Wang, X. Sol processing of conjugated carbon nitride powders for thin‐film fabrication. Angew. Chem. Int. Ed. 54, 6297–6301 (2015).
Guo, B. et al. Vertically aligned porous organic semiconductor nanorod array photoanodes for efficient charge utilization. Nano Lett. 18, 5954–5960 (2018).
Peng, G., Qin, J., Volokh, M., Liu, C. & Shalom, M. Graphene oxide in carbon nitride: from easily processed precursors to a composite material with enhanced photoelectrochemical activity and long-term stability. J. Mater. Chem. A 7, 11718–11723 (2019).
Safaei, J. et al. Graphitic carbon nitride (g-C3N4) electrodes for energy conversion and storage: a review on photoelectrochemical water splitting, solar cells and supercapacitors. J. Mater. Chem. A 6, 22346–22380 (2018).
Qin, J., Wang, S., Ren, H., Hou, Y. & Wang, X. Photocatalytic reduction of CO2 by graphitic carbon nitride polymers derived from urea and barbituric acid. Appl. Catal. B 179, 1–8 (2015).
Xiang, Q., Yu, J. & Jaroniec, M. Preparation and enhanced visible-light photocatalytic H2 production activity of graphene/C3N4 composites. J. Phys. Chem. C 115, 7355–7363 (2011).
Ye, C. et al. Enhanced driving force and charge separation efficiency of protonated g-C3N4 for photocatalytic O2 evolution. ACS Catal. 5, 6973–6979 (2015).
Zhang, G. et al. Optimizing optical absorption, exciton dissociation, and charge transfer of a polymeric carbon nitride with ultrahigh solar hydrogen production activity. Angew. Chem. Int. Ed. 56, 13445–13449 (2017).
Abisdris, L. et al. Electrophoretic deposition of supramolecular complexes for the formation of carbon nitride films. Sustain. Energy Fuels 4, 3879–3883 (2020).
Zhang, G., Zhang, J., Zhang, M. & Wang, X. Polycondensation of thiourea into carbon nitride semiconductors as visible light photocatalysts. J. Mater. Chem. 22, 8083–8091 (2012).
Ong, W. J., Tan, L. L., Chai, S. P., Yong, S. T. & Mohamed, A. R. Surface charge modification via protonation of graphitic carbon nitride (g-C3N4) for electrostatic self-assembly construction of 2D/2D reduced graphene oxide (rGO)/g-C3N4 nanostructures toward enhanced photocatalytic reduction of carbon dioxide to methane. Nano Energy 13, 757–770 (2015).
Barrio, J. et al. Unprecedented centimeter-long carbon nitride needles: synthesis, characterization and applications. Small 14, 1800633 (2018).
Chang, F. et al. Enhanced visible-light-driven photocatalytic performance of porous graphitic carbon nitride. Appl. Surf. Sci. 358, 270–277 (2015).
Peng, G., Volokh, M., Tzadikov, J., Sun, J. & Shalom, M. Carbon nitride/reduced graphene oxide film with enhanced electron diffusion length: an efficient photo‐electrochemical cell for hydrogen generation. Adv. Energy Mater. 8, 1800566 (2018).
Chang, S. M. & Liu, W. S. Surface doping is more beneficial than bulk doping to the photocatalytic activity of vanadium-doped TiO2. Appl. Catal. B 101, 333–342 (2011).
Matis, B. R. et al. Surface doping and band gap tunability in hydrogenated graphene. ACS nano 6, 17–22 (2012).
Liu, X. et al. Ozone treatment of graphitic carbon nitride with enhanced photocatalytic activity under visible light irradiation. J. Colloid Interface Sci. 505, 919–928 (2017).
Ming, L., Yue, H., Xu, L. & Chen, F. Hydrothermal synthesis of oxidized g-C3N4 and its regulation of photocatalytic activity. J. Mater. Chem. A 2, 19145–19149 (2014).
Li, J. et al. A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem. Commun. 48, 12017–12019 (2012).
Acknowledgements
This work is financially supported by the Israel Science Foundation, Grant No. 1161/17 and ISF-NSFC Grant No. 2969/19. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement No. 849068). J.Q. thanks to the National Natural Science Foundation of China (No. 21902095).
Author information
Authors and Affiliations
Contributions
J.Q. performed most of the experiments, analyzed the data, and wrote the initial draft of the manuscript. J.B., G.P., J.T., L.A., and M.V. performed some material characterization. M.V. took part in analysis and writing. M.S. supervised the study and co-wrote the paper. All the authors discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Robert Godin and other, anonymous, reviewers for their contributions to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, J., Barrio, J., Peng, G. et al. Direct growth of uniform carbon nitride layers with extended optical absorption towards efficient water-splitting photoanodes. Nat Commun 11, 4701 (2020). https://doi.org/10.1038/s41467-020-18535-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18535-0
This article is cited by
-
Metal-free photoanodes for C–H functionalization
Nature Communications (2023)
-
Integrated molten and vapor condensation of polymeric carbon nitride photoelectrode towards efficient water splitting
Science China Materials (2023)
-
Ultrasensitive broadband position-sensitive detector based on graphitic carbon nitride
Nano Research (2023)
-
Lamellar carbon nitride membrane for enhanced ion sieving and water desalination
Nature Communications (2022)
-
Laser-driven growth of structurally defined transition metal oxide nanocrystals on carbon nitride photoelectrodes in milliseconds
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
