
Abstract
Asymmetric hydrogenation of α,β-unsaturated acids catalyzed by noble metals has been well established, whereas, the asymmetric hydrogenation with earth-abundant-metal was rarely reported. Here, we describe a cobalt-catalyzed asymmetric hydrogenation of α,β-unsaturated carboxylic acids. By using chiral cobalt catalyst bearing electron-donating diphosphine ligand, high activity (up to 1860 TON) and excellent enantioselectivity (up to >99% ee) are observed. Furthermore, the cobalt-catalyzed asymmetric hydrogenation is successfully applied to a broad spectrum of α,β-unsaturated carboxylic acids, such as various α-aryl and α-alkyl cinnamic acid derivatives, α-oxy-functionalized α,β-unsaturated acids, α-substituted acrylic acids and heterocyclic α,β-unsaturated acids (30 examples). The synthetic utility of the protocol is highlighted by the synthesis of key intermediates for chiral drugs (6 cases). Preliminary mechanistic studies reveal that the carboxy group may be involved in the control of the reactivity and enantioselectivity through an interaction with the metal centre.
Similar content being viewed by others
Introduction
Chiral carboxylic acids are prevalent structural units in varieties of pharmaceutical molecules, agrochemicals, flavors, and fragrances. Some examples are exhibited in Fig. 1. Chiral carboxylic acids such as Ibuprofen, Naproxen1,2, and (R)-Tiagabine3 are well-known drugs. The key intermediates for a large number of drugs or bioactive compounds, such as Artemisinin4,5, Rupintrivir6,7, (S)-Equol8, and Sacubitril9,10,11,12 are chiral carboxylic acids. In addition, chiral carboxylic acids are versatile intermediates for organic synthesis, as they are utilized in the construction of various C–C bonds via decarboxylative coupling reaction13,14,15,16, and in the activation of C–H bond as a directing group17,18. Thus, the development of efficient methods for the preparation of chiral carboxylic acids is highly demanded in both academic research and industrial production.

Ibuprofen and Naproxen (nonsteroidal anti-inflammatory drugs); (R)-Tiagabine (γ-aminobutyric acid reuptake inhibitor); Artemisinin (antimalarial drug); Rupintrivir (rhinovirus protease inhibitor); (S)-Equol (soy isoflavonoid metabolite); Sacubitril (antihypertensive drug used in combination with Valsartan).
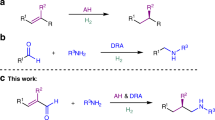
Transition-metal-catalyzed enantioselective hydrogenation of α,β-unsaturated acids is one of the most atom-economic and efficient approaches to access chiral carboxylic acids19. Various noble metal-based catalysts, such as Ru catalysts with chiral diphosphine ligands20,21,22,23,24, Rh catalysts with chiral phosphorus or nitrogen-containing ligands25,26,27,28,29,30,31, and Ir catalysts with chiral P,O-32 and P,N-ligands33,34,35,36,37,38,39,40 have been developed for the hydrogenation of different unsaturated carboxylic acids in high enantioselectivities (Fig. 2a). As modification of ligands or catalysts is necessary to achieve high enantioselectivity for different substrate types, the development of catalytic system with wide applicability is still highly desirable. On the other hand, a major concern regarding the noble metal-based hydrogenation catalysts is the sustainability. Ru, Rh, and Ir are extremely scarce elements, occurring at very low abundances in the earth’s crust (5 × 10−5–10−4 ppm)41. Owing to the low abundance and high cost of the noble metals, the development of cheap earth-abundant metal substitutes to the noble metal catalysts is of high significance42,43,44,45,46,47,48,49,50,51,52,53,54.

a Previous work: noble metal-based catalysts. b This work: cobalt-catalyzed asymmetric hydrogenation of α,β-unsaturated carboxylic acids.
Catalysts based on earth-abundant and environmentally benign cobalt are highly attractive for asymmetric alkene hydrogenation55,56,57. Early studies have showed the potential application of cobalt catalysts in the asymmetric alkene hydrogenation58,59,60,61 However, these systems suffer from limited enantioselectivities, and in many cases H2 could not be used as the stoichiometric reductant. Appreciable progress has been made in recent years, employing cobalt-based complexes for asymmetric hydrogenation of alkenes62,63,64,65,66,67,68,69,70,71,72. Chirik and coworkers report the highly enantioselective hydrogenation of styrene derivatives, cyclic alkenes, and enamides with cobalt complexes bearing C1-symmetric PNN-type pincer ligand or chiral diphosphine ligands65,71,72. Stereoselective olefin hydrogenations are also independently described by Lu and Huang group employing chiral IPO–Co and PPO–Co complexes62,66,67,69. Though important progresses have been made, the development of base metal catalyst for challenging substrates is still underexplored56.
As a continuation of our interest in transition metal-catalyzed asymmetric hydrogenation reactions, herein, we report a highly enantioselective cobalt-catalyzed hydrogenation of α,β-unsaturated carboxylic acids for the synthesis of chiral carboxylic acids. High yields and enantioselectivities are generally achieved for a wide range of substrates (up to 99% yield and up to >99% ee). Besides, the synthetic value of the methodology is demonstrated by its applications in the synthesis of important drugs (Fig. 2b). After the first submission of our paper, the asymmetric hydrogenation of α,β-unsaturated carboxylic acids is reported by Chirik and coworkers73 with a cobalt catalytic system, and high yields and enantioselectivities are achieved for a wide range of acrylic acid derivatives.
Results
Condition optimization
In the initial study, asymmetric hydrogenation of (E)-2,3-diphenylacrylic acid 1a was investigated by using 5 mol% of Co(acac)2 and 5 mol% of chiral ligand. The reaction was conducted under 60 atm H2 pressure at 50 °C in MeOH over 24 h. Several chiral diphosphine ligands were evaluated and we found that cobalt catalysts ligated by highly electron-rich and sterically demanding diphosphines were catalytically active (Fig. 3). In particular, the Ph-BPE was found to be the best one, which afforded the desired product 2a in 70% yield and 94% ee. Me-DuPhos and iPr-DuPhos were catalytically active, but only lower yields and moderate ee values were obtained. Other strongly electron-donating bis(alkylphosphine)s like Binaphine and Duanphos were also tested, but they afforded unsatisfactory yields and ee values. P-chiral diphosphine QuinoxP* and BenzP* gave no desired product. The less electron-donating bis(arylphosphine)s such as BINAP and Segphos were completely inactive in cobalt-catalyzed hydrogenation (Supplementary Table 2).

The yields were determined by 1H NMR and the enantioselectivities were determined by HPLC analysis in all cases.
Next, various solvents and additives were tested (Table 1). It became apparent that alcoholic solvents are beneficial for the catalytic asymmetric hydrogenation, with iPrOH giving the highest activity and enantioselectivity (entries 1–2). The run in more acidic fluorinated solvent, trifluoroethanol (TFE) afforded moderate ee value (entry 3). Furthermore, reactions performed in dimethoxyethane (DME) and toluene led to very low conversions (entries 4–5). When tetrahydrofuran (THF) and 1,4-dioxane were used as solvents, the reactions were totally inhibited (entries 6–7). The investigation of the effect of additives in the reaction showed that the addition of one-electron reductant is benefit for obtaining full conversions with low catalyst loading (entries 8–12). To further improve the enantiocontrol, temperature and hydrogenation pressure effects were surveyed. 97% ee of 2a could be achieved at room temperature under 40 atm H2 pressure with full conversion of 1a (entry 13). The asymmetric hydrogenation also proceeded smoothly with full conversion and high ee when reducing the catalyst loading to 0.1 mol% (entry 14). Moreover, in the presence of 0.05 mol% of catalyst, 1a reacted with H2 efficiently, giving 2a in 93% yield on 2 mmol scale without any decrease of the ee value (TON up to 1860, entry 15). The absolute configuration of the product 2a was established by comparison of its optical rotation with previous report (Supplementary Tables 3–5).
Substrate scope
To delineate the scope of the Co-catalyzed asymmetric hydrogenation, the catalytic system was applied to the reactions of different types of α,β-unsaturated acids. α-Aryl and α-alkyl cinnamic acid derivatives 1 were hydrogenated with 1 mol% catalyst loading under the optimized condition (Fig. 4). Most of the reactions proceeded smoothly under 40 atm H2 pressure at ambient temperature, providing the desired products in high isolated yields and excellent enantioselectivities (95–99% ee). The method works efficiently for α-methyl cinnamic acid derivatives bearing both electron-donating (1c–1e) and -withdrawing groups (1f–1i). Substituents at the para and meta positions of the phenyl ring are tolerated under the reaction conditions, furnishing the hydrogenation products in high yields with excellent enantioselectivity (1j–1l). Heteroaromatic α,β-unsaturated acid 1m was also efficiently hydrogenated to give the desired product 2m in 90% isolated yield with 97% ee. Increasing the steric demand in the α-substituted position by using an iPr group led to the formation of 2n with 86% ee.

aConditions: 1 (0.1 mmol), Co(acac)2 (1 mol%), (S,S)-Ph-BPE (1 mol%) and Zn (10 mol%) in iPrOH (0.6 mL) under 40 atm H2 pressure at room temperature for 24 h. Yield of isolated products, unless noted otherwise. The ee values were determined by HPLC analysis. bUnder 60 atm H2 pressure, 50 °C. cUnder 80 atm H2 pressure, 50 °C. dWith 5 mol% of Cat. eWith 5 mol% of CoCl2 and (S,S)-Ph-BPE in MeOH (0.4 mL), 72 h. f3 (0.1 mmol), CoCl2 (5 mol%), (S,S)-Ph-BPE (5 mol%), and Zn (50 mol%) in HFIP (0.4 mL) under 60 atm H2 pressure at 50 °C for 48 h. gIn 0.6 mL solvent (MeOH/HFIP = 2/1), 72 h. hIn 0.4 mL MeOH.
Our preliminary results show that the [Co]/BPE is also active for hydrogenation of N-heterocyclic acids to produce chiral heterocyclic acids, which are present in various pharmaceuticals74. The asymmetric hydrogenation of N-Boc-1,2,5,6-tetrahydropyridine-3-carboxylic acid 1o gave the desired product 2o with an impressive 93% ee. α-Oxy-functionalized α,β-unsaturated acids were also subjected to our [Co]/BPE system due to the corresponding hydrogenation products are important building blocks for asymmetric synthesis in both pharmaceutical and agrochemical industries75. To our delight, the catalyst system showed high efficiency in the asymmetric hydrogenation of α-alkoxy- and α-aryloxy-substituted α,β-unsaturated acids. A broad range of α-aryloxy and α-alkoxy substituted α,β-unsaturated acids 1p–1u were hydrogenated smoothly to give the desired chiral α-oxy-functionalized acids in good to excellent yields and enantioselectivities (80–95% yield, 90–99% ee).
Finally, we turned our attention to the asymmetric hydrogenation of α-substituted acrylic acids (Fig. 4). It is revealed that solvents have critical roles in [Co]/BPE catalyzed hydrogenation of α-substituted acrylic acids, affecting both catalytic activity and selectivity. Full conversion and 98% ee were obtained in hexafluoroisopropanol (HFIP, Supplementary Table 6). A wide range of α-aryl acrylic acids (3a–3h) were subjected to the cobalt-catalyzed hydrogenation, affording the corresponding chiral carboxylic acids in good to excellent yields and enantioselectivities (89–99% yields, 90–99% ee). Furthermore, 2-alkyl acrylic acid 3i was also smoothly hydrogenated with 80% ee.
Synthetic applications
To demonstrate the synthetic utility of cobalt-catalyzed enantioselective hydrogenation of α,β-unsaturated acids, its synthetic application in series of chiral natural products and drugs was studied. The critical synthon to (S)-Equol, (S)-2v, could be easily accessed under standard conditions with high enantioselectivity (94% ee, Fig. 5a). Similarly, chiral carboxylic acid 2w, a key intermediate in the synthesis of rhinovirus protease inhibitor Rupintrivir, was obtained in 95% isolated yield and >99% ee (Fig. 5b). Following optimization of the reaction conditions, Sacubitril intermediate 2x was obtained in 97% isolated yield and 17/1 dr (Fig. 5c, Supplementary Table 7).

a Synthesis of (S)-Equol intermediate via cobalt-catalyzed hydrogenation. b Synthesis of Rupinnavir intermediate. c Synthesis of Sacubitril intermediate. d Asymmetric synthesis of Naproxen. e Asymmetric synthesis of Ibuprofen. f Gram-scale asymmetric hydrogenation of artemisinic acid 5.
The asymmetric hydrogenation of α-substituted acrylic acids is of highly practical value because optically pure α-substituted propionic acids, such as ibuprofen and naproxen, are well-known non-steroid anti-inflammatory drugs, which can be readily prepared through asymmetric hydrogenation of the corresponding α-aryl acrylic acids. Indeed, Naproxen (4j) and Ibuprofen (4k) were prepared with high yields and enantioselectivities by using cobalt-based catalytic system (Fig. 5d, e). Based on the excellent performance of Co/BPE in the above reaction, we carried out the gram-scale asymmetric hydrogenation of AA (artemisinic acid) 5 in the presence of 0.5 mol% of chiral cobalt catalyst. Desired product (R)-DHAA (dihydroartemisinic acid) 6 was obtained in excellent isolated yield and dr value (1.16 g, 97/3 dr, Fig. 5f). It is noteworthy that the trisubstituted olefin in 5 is unreactive under the Co-catalyzed hydrogenation conditions.
Mechanism study
To provide insight into the possible catalyst activation mode and the mechanism of the asymmetric hydrogenation of α,β-unsaturated carboxylic acids, several control and catalytic experiments were conducted. Previous research by Chirik and coworkers revealed that additives such as Zn, Mn, or LiCH2Si(CH3)3 were necessary in the cobalt-catalyzed asymmetric hydrogenation of enamines and acted as an activator for pre-catalyst to generate catalytic active cobalt speices65,71. However, the reaction of α,β-unsaturated carboxylic acid 1a, in the absence of any additives, afford 2a in >98% conversion and 94% ee (Table 1, entry 2). The data suggested that the carboxylic acid might serve as an activator and mediate the activation process. In sharp contrast to the high reactivity and enantioselectivity observed in the case of 1b (99% yield and 96% ee, Fig. 4), no reaction occurred for the corresponding ethyl ester 1b′ under standard conditions (Fig. 6a). Moreover, no hydrogenation product was observed when AcOH or 1b was added to the reaction mixture as external carboxylic acid (Fig. 6b, c). These results indicated that the carboxy group of substrate may be involved in the control of the reactivity and enantioselectivity through interaction with the metal center19,22,23,24,33.

a Hydrogenation of ester 1b′ under standard conditions. b Hydrogenation of ester 1b′ in the presence of catalytic amount of CH3COOH. c Reaction of ester 1b′ in the presence of catalytic amount of 1b. d Deuterium-labeling experiment. e Hydrogenation of 1b in isopropanol-d8 with CH3COOD as additive.
The deuterium-labeling experiments were also conducted to elucidate the reaction mechanism. The reaction of 1b with D2 under standard condition produced 2b-d2 smoothly in >98% conversion and 96% ee (Fig. 6d). Performing the hydrogenation reaction under 40 atm of H2 in isopropanol-d8 solution with 5 eq. CH3COOD gave 2b in >98% conversion, and no deuterated products were observed (Fig. 6e). These data suggested that the H2 served as the hydrogen source and protonation of the Co-alkyl intermediate was probably not involved in the current reaction (Supplementary Figs. 1–5). Besides, EPR experiments were also conducted to monitor the process of the current reaction using 1b as model substrate. The EPR spectra changed greatly after the addition of (S,S)-Ph-BPE and substrate 1b, suggesting that the coordination of the ligand and the exchange between acac and substrate 1b probably happened (Supplementary Figs. 6–11).
Based on our experimental observations and the previous study on iridium-catalyzed enantioselective hydrogenation of α,β-unsaturated acids33, we proposed a plausible mechanism (Fig. 7). The key catalytic intermediate E could be generated through two pathways. In the absence of Zn, complex E can be generated through carboxy group mediated H2 heterolytic process. Coordination of Co(acac)2 with (S,S)-Ph-BPE generates Co(II) complex A, which then undergoes ligand exchange with more acidic substrate 1b to produce complex B, complex B and B′ are in equilibrium with each other. Heterolysis of H2 by complex B′ produces the key catalytic species E via transition state C. Intermediate E may be also produced through protonation76 of dihydride complex D65,73 with 1b when employing one-electron reductant. Chirik and coworkers73 reported an alternative mechanism which involved the migratory insertion of the dihydride complex and the subsequent reduction elimination as key step. Note that the addition of one-electron reductant is beneficial for obtaining full conversions with low catalyst loading (vide supra). The success of Zn or Mn used in this reaction is presumably due to enhanced activation of Co(II) precursor and suppressed deactivation of the active catalyst. The key intermediate E then enters the catalytic cycle. Intramolecular migratory insertion of complex E produces five-membered intermediate F. Coordination of H2 to F forms complex G, which undergoes subsequent sigma-bond metathesis to give complex H. The ligand exchange of intermediate H with unsaturated carboxylate substrate releases the hydrogenation product 2b and regenerates the cobalt hydride complex D. Mechanistic investigations indicate that the carboxy group has a pivotal role in the improvement of the reactivity and enantioselectivity via coordination with the metal center, and that is why the current reaction does not work for α,β-unsaturated esters.

a Generation of catalytic species E in the absence of Zn. b Generation of catalytic species E in the presence of Zn. c Proposed catalytic cycle.
Discussion
In summary, we have developed a highly efficient cobalt-catalyzed asymmetric hydrogenation of α,β-unsaturated carboxylic acids. The cobalt catalyst system exhibited both high activities (up to 1860 TON) and excellent enantioselectivities (up to >99% ee) for a broad spectrum of α,β-unsaturated acids. It also affords an efficient way to the key intermediates of many chiral natural products and drugs with high enantioselectivities. Moreover, this operationally simple and atom-economic protocol could be easily scaled-up in gram-scale using 0.5 mol% catalyst loading for the asymmetric synthesis of key intermediate of Artemisinin. Mechanistic studies suggest that the carboxy group may be involved in the control of the reactivity and enantioselectivity through interaction with the metal center. Additional mechanistic and DFT studies of the asymmetric transformations with such cobalt system are currently underway in our laboratory.
Methods
General procedure for asymmetric hydrogenation
In an argon-filled glovebox, Co(acac)2 (0.010 M in iPrOH, 0.10 mL, 0.001 mmol) and (S,S)-Ph-BPE (0.010 M in THF, 0.10 mL, 0.001 mmol) were stirred in a vial at room temperature for 10 min. Then zinc dust (0.65 mg, 0.01 mmol) and iPrOH (0.50 mL) were added and the mixture was stirred for 15 min. After that, substrate (0.1 mmol) was added to the reaction mixture. The vial was subsequently transferred into an autoclave and purged by three cycles of pressurization/venting with H2. The reaction was then stirred under H2 (40 atm) at room temperature for 24 h. The hydrogen gas was released slowly and carefully. The resulting solution was concentrated in vacuum and the residue was purified by chromatography on silica gel. The ee values were determined by HPLC with a chiral column.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files.
References
Morihara, T., Chu, T., Ubeda, O., Beech, W. & Cole, G. M. Selective inhibition of Aβ42 production by NSAID R-enantiomers. J. Neurochem. 83, 1009–1012 (2002).
Shen, T. Y. Perspectives in nonsteroidal anti-inflammatory agents. Angew. Chem. Int. Ed. 11, 460–472 (1972).
Bose, P., Kumar, P., Mittal, S. & Kumar, Y. Preparation of tiagabine. WO2006013550A2 (2006).
Amara, Z. et al. Applying green chemistry to the photochemical route to artemisinin. Nat. Chem. 7, 489–495 (2015).
Ro, D.-K. et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943 (2006).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Dragovich, P. S. et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 42, 1213–1224 (1999).
Chang, Y.-C. & Nair, M. G. Metabolism of daidzein and genistein by intestinal bacteria. J. Nat. Prod. 58, 1892–1896 (1995).
Seferovic, J. P. et al. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: a post-hoc analysis from the PARADIGM-HF trial. Lancet Diabet. Endocrinol. 5, 333–340 (2017).
Zile, M. R. et al. Prognostic implications of changes in N-terminal pro-B-type natriuretic peptide in patients with heart failure. J. Am. Coll. Cardiol. 68, 2425–2436 (2016).
Jhund, P. S. et al. Efficacy and safety of LCZ696 (sacubitril-valsartan) according to age: insights from PARADIGM-HF. Eur. Heart J. 36, 2576–2584 (2015).
Halama, A. et al. A method for the preparation, isolation and purification of pharmaceutically applicable forms of AHU-377. WO2016074651A1 (2016).
Park, K. & Lee, S. Transition metal-catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids. RSC Adv. 3, 14165–14182 (2013).
Rodríguez, N. & Goossen, L. J. Decarboxylative coupling reactions: a modern strategy for C–C-bond formation. Chem. Soc. Rev. 40, 5030–5048 (2011).
Baudoin, O. New approaches for decarboxylative biaryl coupling. Angew. Chem. Int. Ed. 46, 1373–1375 (2007).
Gooßen, L. J., Deng, G. & Levy, L. M. Synthesis of biaryls via catalytic decarboxylative coupling. Science 313, 662–664 (2006).
Li, S., Wang, H., Weng, Y. & Li, G. Carboxy group as a remote and selective chelating group for C-H activation of arenes. Angew. Chem. Int. Ed. 58, 18502–18507 (2019).
Zhang, Y., Zhao, H., Zhang, M. & Su, W. Carboxylic acids as traceless directing groups for the rhodium(III)-catalyzed decarboxylative C-H arylation of thiophenes. Angew. Chem. Int. Ed. 54, 3817–3821 (2015).
Zhu, S.-F. & Zhou, Q.-L. Iridium-catalyzed asymmetric hydrogenation of unsaturated carboxylic acids. Acc. Chem. Res. 50, 988–1001 (2017).
Abdel-Magied, A. F. et al. Synthesis and characterization of chiral phosphirane derivatives of [(μ-H)4Ru4(CO)12] and their application in the hydrogenation of an α,β-unsaturated carboxylic acid. J. Org. Chem. 849-850, 71–79 (2017).
Li, J. et al. Asymmetric hydrogenation of α-substituted acrylic acids catalyzed by a ruthenocenyl phosphino-oxazoline–ruthenium complex. Org. Lett. 18, 2122–2125 (2016).
Cheng, X., Zhang, Q., Xie, J.-H., Wang, L.-X. & Zhou, Q.-L. Highly rigid diphosphane ligands with a large dihedral angle based on a chiral spirobifluorene backbone. Angew. Chem. Int. Ed. 44, 1118–1121 (2005).
Pai, C.-C. et al. Highly effective chiral dipyridylphosphine ligands: synthesis, structural determination, and applications in the Ru-catalyzed asymmetric hydrogenation reactions. J. Am. Chem. Soc. 122, 11513–11514 (2000).
Fan, Q.-h, Ren, C.-y, Yeung, C.-h, Hu, W.-h & Chan, A. S. C. Highly effective soluble polymer-supported catalysts for asymmetric hydrogenation. J. Am. Chem. Soc. 121, 7407–7408 (1999).
Chen, C. et al. Enzyme-inspired chiral secondary-phosphine-oxide ligand with dual noncovalent interactions for asymmetric hydrogenation. Angew. Chem., Int. Ed. 56, 6808–6812 (2017).
Yan, Q., Kong, D., Zhao, W., Zi, G. & Hou, G. Enantioselective hydrogenation of β,β-disubstituted unsaturated carboxylic acids under base-free conditions. J. Org. Chem. 81, 2070–2077 (2016).
Li, Y., Dong, K., Wang, Z. & Ding, K. Rhodium(I)-catalyzed enantioselective hydrogenation of substituted acrylic acids with sterically similar β,β-diaryls. Angew. Chem. Int. Ed. 52, 6748–6752 (2013).
Chen, W., Spindler, F., Pugin, B. & Nettekoven, U. ChenPhos: highly modular P-stereogenic C1-symmetric diphosphine ligands for the efficient asymmetric hydrogenation of α-substituted cinnamic acids. Angew. Chem. Int. Ed. 52, 8652–8656 (2013).
Chen, W. et al. Stereoselective synthesis of ferrocene-based C2-symmetric diphosphine ligands: application to the highly enantioselective hydrogenation of α-substituted cinnamic acids. Angew. Chem. Int. Ed. 46, 4141–4144 (2007).
Hoen, R. et al. Achiral ligands dramatically enhance rate and enantioselectivity in the Rh/phosphoramidite-catalyzed hydrogenation of α,β-disubstituted unsaturated acids. Angew. Chem. Int. Ed. 44, 4209–4212 (2005).
Berens, U., Burk, M. J., Gerlach, A. & Hems, W. Chiral 1,1′-diphosphetanylferrocenes: new ligands for asymmetric catalytic hydrogenation of itaconate derivatives. Angew. Chem. Int. Ed. 39, 1981–1984 (2000).
Yang, S., Che, W., Wu, H.-L., Zhu, S.-F. & Zhou, Q.-L. Neutral iridium catalysts with chiral phosphine-carboxy ligands for asymmetric hydrogenation of unsaturated carboxylic acids. Chem. Sci. 8, 1977–1980 (2017).
Li, M.-L. et al. Mechanism studies of Ir-catalyzed asymmetric hydrogenation of unsaturated carboxylic acids. J. Am. Chem. Soc. 139, 541–547 (2017).
Song, S., Zhu, S.-F., Yu, Y.-B. & Zhou, Q.-L. Carboxy-directed asymmetric hydrogenation of 1,1-diarylethenes and 1,1-dialkylethenes. Angew. Chem. Int. Ed. 52, 1556–1559 (2013).
Song, S., Zhu, S.-F., Pu, L.-Y. & Zhou, Q.-L. Iridium-catalyzed enantioselective hydrogenation of unsaturated heterocyclic acids. Angew. Chem. Int. Ed. 52, 6072–6075 (2013).
Zhu, S.-F., Yu, Y.-B., Li, S., Wang, L.-X. & Zhou, Q.-L. Enantioselective hydrogenation of α-substituted acrylic acids catalyzed by iridium complexes with chiral spiro aminophosphine ligands. Angew. Chem. Int. Ed. 51, 8872–8875 (2012).
Yang, S. et al. Enantioselective iridium-catalyzed hydrogenation of α-arylcinnamic acids and synthesis of (S)-equol. Tetrahedron 68, 5172–5178 (2012).
Li, S. et al. Enantioselective hydrogenation of α-aryloxy and α-alkoxy α,β-unsaturated carboxylic acids catalyzed by chiral spiro iridium/phosphino-oxazoline complexes. J. Am. Chem. Soc. 132, 1172–1179 (2010).
Li, S., Zhu, S.-F., Zhang, C.-M., Song, S. & Zhou, Q.-L. Iridium-catalyzed enantioselective hydrogenation of α,β-unsaturated carboxylic acids. J. Am. Chem. Soc. 130, 8584–8585 (2008).
Roseblade, S. J. & Pfaltz, A. Iridium-catalyzed asymmetric hydrogenation of olefins. Acc. Chem. Res. 40, 1402–1411 (2007).
Alig, L., Fritz, M. & Schneider, S. First-row transition metal (de)hydrogenation catalysis based on functional pincer ligands. Chem. Rev. 119, 2681–2751 (2019).
Wang, Y. et al. Unmasking the ligand effect in manganese-catalyzed hydrogenation: mechanistic insight and catalytic application. J. Am. Chem. Soc. 141, 17337–17349 (2019).
Passera, A. & Mezzetti, A. The manganese(I)-catalyzed asymmetric transfer hydrogenation of ketones: disclosing the macrocylic privilege. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie.201912605, (2019).
Hu, Y. et al. Cobalt-catalyzed asymmetric hydrogenation of C=N bonds enabled by assisted coordination and nonbonding interactions. Angew. Chem. Int. Ed. 58, 15767–15771 (2019).
Duan, Y.-N. et al. Homogeneous hydrogenation with a cobalt/tetraphosphine catalyst: a superior hydride donor for polar double bonds and N-heteroarenes. J. Am. Chem. Soc. 141, 20424–20433 (2019).
Jing, Y., Chakraborty, S., Brennessel, W. W. & Jones, W. D. Additive-free cobalt-catalyzed hydrogenation of esters to alcohols. ACS Catal. 7, 3735–3740 (2017).
Garbe, M. et al. Manganese(I)-catalyzed enantioselective hydrogenation of ketones using a defined chiral PNP pincer ligand. Angew. Chem. Int. Ed. 56, 11237–11241 (2017).
Zhang, D. et al. Enantioselective hydrogenation of ketones catalyzed by chiral cobalt complexes containing PNNP ligand. Asian J. Org. Chem. 5, 1323–1326 (2016).
Xu, R. et al. Iron-catalyzed homogeneous hydrogenation of alkenes under mild conditions by a stepwise, bifunctional mechanism. ACS Catal. 6, 2127–2135 (2016).
Gorgas, N., Stoeger, B., Veiros, L. F. & Kirchner, K. Highly efficient and selective hydrogenation of aldehydes: a well-defined Fe(II) catalyst exhibits noble-metal activity. ACS Catal. 6, 2664–2672 (2016).
Roesler, S., Obenauf, J. & Kempe, R. A highly active and easily accessible cobalt catalyst for selective hydrogenation of C=O bonds. J. Am. Chem. Soc. 137, 7998–8001 (2015).
Lagaditis, P. O. et al. Iron(II) complexes containing unsymmetrical P-N-P’ pincer ligands for the catalytic asymmetric hydrogenation of ketones and imines. J. Am. Chem. Soc. 136, 1367–1380 (2014).
Gaertner, D., Welther, A., Rad, B. R., Wolf, R. & von Wangelin, A. J. Heteroatom-free arene-cobalt and arene-iron catalysts for hydrogenations. Angew. Chem. Int. Ed. 53, 3722–3726 (2014).
Zhang, G., Scott, B. L. & Hanson, S. K. Mild and homogeneous cobalt-catalyzed hydrogenation of C=C, C=O, and C=N bonds. Angew. Chem. Int. Ed. 51, 12102–12106 (2012).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953 (2019).
Chirik, P. J. Iron- and cobalt-catalyzed alkene hydrogenation: catalysis with both redox-active and strong field ligands. Acc. Chem. Res. 48, 1687–1695 (2015).
Bullock, R. M. Catalysis Without Precious Metals (Wiley-Blackwell, 2010).
Nindakova, L. O., Shainyan, B. A. & Shmidt, F. K. Enantioselective hydrogenation over chiral cobalt complexes with (+)-(1S,2S,5R)-neomenthyldiphenylphosphine and (-)-(R,R)-2,2-dimethyl-4,5-bis(diphenylphosphinomethyl)-1,3-dioxolane. Russ. J. Org. Chem. 40, 973–975 (2004).
Corma, A., Iglesias, M., del Pino, C. & Sánchez, F. Optically active complexes of transition metals (RhI, RuII, CoII and NiII) with 2-aminocarbonylpyrrolidine ligands. Selective catalysts for hydrogenation of prochiral olefins. J. Organomet. Chem. 431, 233–246 (1992).
Leutenegger, U., Madin, A. & Pfaltz, A. Enantioselective reduction of α,β-unsaturated carboxylates with NaBH4 and catalytic amounts of chiral cobalt semicorrin complexes. Angew. Chem. Int. Ed. 28, 60–61 (1989).
Yoshiaki, O., Seiji, T., Yukikazu, N. & Juji, Y. Asymmetric reactions. X. Asymmetric hydrogenation catalyzed by bis(dimethylglyoximato)cobalt(II)–chiral cocatalyst (amino alcohol) system. Bull. Chem. Soc. Jpn. 54, 2124–2135 (1981).
Zuo, Z. et al. Cobalt-catalyzed asymmetric hydrogenation of vinylsilanes with a phosphine–pyridine–oxazoline ligand: synthesis of optically active organosilanes and silacycles. Organometallics 38, 3906–3911 (2019).
Zhong, H., Friedfeld, M. R. & Chirik, P. J. Syntheses and catalytic hydrogenation performance of cationic bis(phosphine) cobalt(I) diene and arene compounds. Angew. Chem. Int. Ed. 58, 9194–9198 (2019).
Morello, G. R., Zhong, H., Chirik, P. J. & Hopmann, K. H. Cobalt-catalysed alkene hydrogenation: a metallacycle can explain the hydroxyl activating effect and the diastereoselectivity. Chem. Sci. 9, 4977–4982 (2018).
Friedfeld, M. R., Zhong, H., Ruck, R. T., Shevlin, M. & Chirik, P. J. Cobalt-catalyzed asymmetric hydrogenation of enamides enabled by single-electron reduction. Science 360, 888–893 (2018).
Guo, J., Shen, X. & Lu, Z. Regio- and enantioselective cobalt-catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Angew. Chem. Int. Ed. 56, 615–618 (2017).
Guo, J., Cheng, B., Shen, X. & Lu, Z. Cobalt-catalyzed asymmetric sequential hydroboration/hydrogenation of internal alkynes. J. Am. Chem. Soc. 139, 15316–15319 (2017).
Friedfeld, M. R., Shevlin, M., Margulieux, G. W., Campeau, L.-C. & Chirik, P. J. Cobalt-catalyzed enantioselective hydrogenation of minimally functionalized alkenes: isotopic labeling provides insight into the origin of stereoselectivity and alkene insertion preferences. J. Am. Chem. Soc. 138, 3314–3324 (2016).
Chen, J., Chen, C., Ji, C. & Lu, Z. Cobalt-catalyzed asymmetric hydrogenation of 1,1-diarylethenes. Org. Lett. 18, 1594–1597 (2016).
Hopmann, K. H. Cobalt–bis(imino)pyridine-catalyzed asymmetric hydrogenation: electronic structure, mechanism, and stereoselectivity. Organometallics 32, 6388–6399 (2013).
Friedfeld, M. R. et al. Cobalt precursors for high-throughput discovery of base metal asymmetric alkene hydrogenation catalysts. Science 342, 1076–1080 (2013).
Monfette, S., Turner, Z. R., Semproni, S. P. & Chirik, P. J. Enantiopure C1-symmetric bis(imino)pyridine cobalt complexes for asymmetric alkene hydrogenation. J. Am. Chem. Soc. 134, 4561–4564 (2012).
Zhong, H., Shevlin, M. & Chirik, P. J. Cobalt-catalyzed asymmetric hydrogenation of α,β-unsaturated carboxylic acids by homolytic H2 cleavage. J. Am. Chem. Soc. 142, 5272–5281 (2020).
Dinges, J., Lamberth, C. Bioactive Heterocyclic Compound Classes: Pharmaceuticals (Wiley-VCH Verlag GmbH & Co. KGaA, 2012).
Wade, L. G. Total synthesis of natural products: the ‘chiron’ approach. J. Chem. Ed. 62, A190 (1985).
Wiedner, E. S., Appel, A. M., DuBois, D. L. & Bullock, R. M. Thermochemical and mechanistic studies of electrocatalytic hydrogen production by cobalt complexes containing pendant amines. Inorg. Chem. 52, 14391–14403 (2013).
Acknowledgements
X. Zhang is indebted to Southern University of Science and Technology (start-up fund), Shenzhen Science and Technology Innovation Committee (Nos. KQTD20150717103157174 and JSGG20170821140353405), Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002), National Natural Science Foundation of China (Nos. 21991113), and SZDRC Discipline Construction Program for financial support. G.-Q. Chen gratefully acknowledges the Youth Fund from National Natural Science Foundation of China (No. 21901107). We thank Dr. Yanxia Zhang and Dr. Dongyang Wang from Shanghai Institute of Organic Chemistry (SIOC) and Dr. Yinhua Yang from Southern University of Science and Technology Core Research Facilities for their help in EPR experiments.
Author information
Authors and Affiliations
Contributions
X.D. and Y.X. contributed equally to this work. X.D. and Y.X. contributed to the conception and design of the experiments. X.D. and Y.X. performed the experiments and analyzed the data. J.-M.H., Y.Z., Y.-N.D. and H.W. synthesized several substrates. C.S. carried out the HPLC analysis of DHAA. G.-Q.C. and X.Z. directed the project. X.D. and G.-Q.C. co-wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Kuo-wei Huang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, X., Xiao, Y., Huang, JM. et al. Cobalt-catalyzed highly enantioselective hydrogenation of α,β-unsaturated carboxylic acids. Nat Commun 11, 3239 (2020). https://doi.org/10.1038/s41467-020-17057-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-17057-z
This article is cited by
-
Nickel-catalyzed asymmetric hydrogenation for the preparation of α-substituted propionic acids
Nature Communications (2024)
-
Cooperative triple catalysis enables regioirregular formal Mizoroki–Heck reactions
Nature Synthesis (2022)
-
Ni-catalyzed asymmetric hydrogenation of N-aryl imino esters for the efficient synthesis of chiral α-aryl glycines
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
