
Abstract
Single-atom catalysts (SACs) have sparked broad interest recently while the low metal loading poses a big challenge for further applications. Herein, a dual protection strategy has been developed to give high-content SACs by nanocasting SiO2 into porphyrinic metal–organic frameworks (MOFs). The pyrolysis of SiO2@MOF composite affords single-atom Fe implanted N-doped porous carbon (FeSA–N–C) with high Fe loading (3.46 wt%). The spatial isolation of Fe atoms centered in porphyrin linkers of MOF sets the first protective barrier to inhibit the Fe agglomeration during pyrolysis. The SiO2 in MOF provides additional protection by creating thermally stable FeN4/SiO2 interfaces. Thanks to the high-density FeSA sites, FeSA–N–C demonstrates excellent oxygen reduction performance in both alkaline and acidic medias. Meanwhile, FeSA–N–C also exhibits encouraging performance in proton exchange membrane fuel cell, demonstrating great potential for practical application. More far-reaching, this work grants a general synthetic methodology toward high-content SACs (such as FeSA, CoSA, NiSA).
Similar content being viewed by others


Introduction
Single-atom catalysts (SACs), with the maximal utilization of metal atoms, open up a new frontier and attract increasing attention in catalysis1,2,3,4,5,6,7,8,9,10. Integrated with plenty of merits, including highly dispersed sites, high activity, excellent selectivity, and good reusability, SACs have been regarded as an ideal platform to bridge the gap between homogenous and heterogeneous catalysts1,2,3,4. Nevertheless, isolated metal atoms in SACs tend to agglomerate due to the high surface energy. Though significant progress has been achieved to ensure the atomic dispersion of metal atoms, metal loadings of SACs are basically low (<1 wt%). The construction methodology of stable SACs, especially in high metal loadings, is highly desired yet remains a grand challenge11,12,13. In addition, to boost the catalytic performance of SACs, their physical features, including porous structure and surface area, which dominate the accessibility to active sites, should also be optimized14,15,16.
Metal–organic frameworks (MOFs)17,18,19,20,21,22,23,24,25, featuring well-defined and tailored structures, present particular advantages in the precise fabrication of catalysts, especially SACs26,27,28,29,30,31,32,33. The present synthetic strategy for MOF-based SACs is to augment the distance between adjacent metal atoms based on the mixed metal/ligand and pore confinement, which effectively inhibits the agglomeration of metal atoms under pyrolysis27,28,29,30. Unfortunately, these strategies cause the decrease of metal loadings. As a result, even bearing the structural advantages in MOF-based SACs, their metal loadings are unsatisfying, such as FeSA (usually, <2 wt%)26,27,28,29,30,31,32,33. In addition, most of reported MOF-derived SACs possess micropores (<2 nm), which is unfavorable to mass transfer in catalytic process27,30. To address these issues, alternative synthetic strategies for MOF-derived SACs are imperative to improving the metal loadings and pore structures.
To realize SACs with high metal loadings, the main obstacle to overcome is their easy-to-migrate feature due to their high surface energy, especially under pyrolysis. It was found that inorganic silica (SiO2) protection method can stabilize metal nanoparticles/clusters by decreasing surface energy of metal atoms1,34,35,36,37,38, which might be effective toward the stabilization of highly loaded SACs. Making full use of the porosity of MOFs, SiO2 can be easily nanocasted into the pore space of MOFs to interact with the isolated metal atoms on MOF skeleton, which would significantly lower their surface energy. In consequence, the introduction of SiO2 into MOFs, when integrated with the merits of MOFs, should be a very promising route to improve metal loadings in SACs.
In this work, we creatively put forward a nanocasting strategy to introduce SiO2 into the mesopores of a porphyrinic MOF, PCN-222(Fe), featuring single Fe(III) site in each porphyrin linker39,40,41. Thanks to the 1D mesochannel with a diameter of ~3.2 nm, SiO2 can be sufficiently filled into PCN-222(Fe), forming thermal stable FeN4/SiO2 interfaces. Upon high-temperature pyrolysis and SiO2 removal, the single-atom Fe catalyst, denoted FeSA–N–C, with a Fe loading as high as 3.46 wt%, is obtained (Fig. 1). During the pyrolysis, the spatial isolation of Fe atoms anchored by N atoms in porphyrin linkers is the first protective barrier to inhibit the Fe agglomeration. The silica in MOF channels serves as oxide substrate to interact with Fe atoms that can increase migration energy barrier of Fe atoms and prevent their aggregation. Meanwhile, upon removal of silica, the porosity and surface area of the resultant N-doped porous carbon can be improved, benefiting the exposure of active sites and mass transfer. As a result, the optimized FeSA–N–C exhibits excellent oxygen reduction reaction (ORR) performance, surpassing the state-of-the-art Pt/C, and almost all reported non-noble-metal catalysts, in both alkaline and the more challenging acidic solutions. Significantly, the FeSA–N–C reaches a current density of 292 mA cm−2 at 0.8 V and the highest power density of 0.68 W cm−2, comparable to that of the best non-noble metal catalysts, in H2–O2 proton exchange membrane fuel cell (PEMFC).

Illustration showing the nanocasting-assisted fabrication of FeSA–N–C from PCN-222(Fe).
Results
Synthesis and characterization of FeSA–N–C
The PCN-222(Fe) was prepared by employing trifluoroacetic acid (TFA), instead of the traditionally used benzoic acid, as a modulator (Supplementary Fig. 1)39. The scanning electron microscopy (SEM) image of PCN-222(Fe) presents the uniform spindle morphology with a diameter ~250 nm (Fig. 2a). The low boiling point of TFA makes it easy to be removed by direct degassing without additional pre-activation process (necessary for benzoic acid modulator) to deliver available pore space in PCN-222(Fe) (Supplementary Table 1). The N2 sorption with a typical type-IV isotherms present a high surface area up to 2040 m2 g−1 and the pore size distribution suggests the mesochannels centered at 3.2 nm, in good agreement with the transmission electron microscopy (TEM) observation (Fig. 2b, Supplementary Fig. 2). This greatly facilitates the subsequent introduction of tetraethylorthosilicate (TEOS) into PCN-222(Fe) mesopores for SiO2 nanocasting, after degassing the MOF at 150 °C. The facile TFA removal and mesoporosity guarantee the sufficient percolation of TEOS through the entire inner space of PCN-222(Fe). Upon HCl vapor treatment, TEOS in PCN-222(Fe) can be hydrolyzed and condensed to silica, affording SiO2@PCN-222(Fe) composite with well-retained MOF crystallinity, thanks to the ultrahigh acidic stability of the MOF (Supplementary Fig. 1)39.

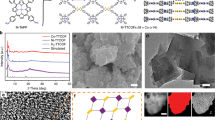
a Scanning electron microscopy (SEM) and b transmission electron microscopy (TEM) images of PCN-222(Fe). c SEM and d TEM images of FeSA–N–C. e Enlarged TEM image showing the mesoporous structure of FeSA–N–C. f Aberration-corrected HAADF-STEM image of FeSA–N–C.
The infrared peak at 1090 cm−1 assignable to Si-O-Si clearly confirms the SiO2 formation (Supplementary Fig. 3)37. The reduced surface area (1190 m2 g–1) and mesopore size (2.9 nm) in the composite, in reference to the parent MOF (BET of 2040 m2 g−1; pore size of 3.2 nm), further indicate the successful infiltration of SiO2 in PCN-222(Fe). The elemental mapping images of SiO2@PCN-222(Fe) further illustrate the homogeneous dispersion of Si in PCN-222(Fe) (Supplementary Fig. 4). After the pyrolysis of SiO2@PCN-222(Fe) at 800 °C, the composite of metal (oxides) stabilized by porous carbon is produced. Upon the removal of the oxide by acid etching, FeSA–N–C with retained spindle morphology is finally obtained and no particles are observed in TEM images (Fig. 2c–e, Supplementary Fig. 5a). The elemental mapping images clearly demonstrate the homogenous dispersion of Fe and N elements on FeSA–N–C (Supplementary Fig. 6). Aberration-corrected high-angle annular darkfield scanning transmission electron microscope (HAADF-STEM) observation shows the isolated and high-density bright spots, implying the formation of single metal atoms (Fig. 2f, Supplementary Fig. 5b). The accurate contents of Fe (3.46 wt%) and N (4.87 wt%) have been quantified by inductively coupled plasma atomic emission spectrometry (ICP) and elemental analysis, demonstrating the presence of single Fe atom and suggesting the higher Fe loading than almost all reported single-atom Fe-incorporated carbon-based materials (Supplementary Table 2). Moreover, the quantitative analysis of X-ray photoelectron spectroscopy (XPS) and energy-dispersive spectroscopy further confirm the high loading of Fe, in accordance with the ICP results (Supplementary Table 3). In addition, the content of Zr is extremely low, illustrating Fe is the dominated metal species in FeSA–N–C (Supplementary Table 3). As illustrated above, the perfect combination of PCN-222(Fe) and SiO2 can exert their respective advantages for the creation of the resultant FeSA–N–C, which are visually summarized (Supplementary Fig. 7).
Powder X-ray diffraction (PXRD) pattern of FeSA–N–C gives two broad peaks in the ranges of 20–30° and 40–45°, corresponding to (002) and (101) planes of graphitized carbon, and no diffraction of Fe-based species is identifiable, in accordance with the TEM observation results (Fig. 2c–e, Supplementary Fig. 8). Raman scattering spectrum for FeSA–N–C gives low intensity ratio (ID/IG = 0.95) of D band (~1345 cm−1) and G band (~1590 cm−1), illustrating the high graphitization degree (Supplementary Fig. 9). N2 sorption measurement for FeSA–N–C manifests its high BET surface area up to 1615 m2 g−1 (Supplementary Fig. 10). The mesoporous pore size distribution extends up to 10 nm (Supplementary Fig. 10), which can be also seen in enlarged TEM image (Fig. 2e). The large surface area and hierarchical pore of FeSA–N–C would make the single Fe atoms readily accessible and guarantee high-flux mass transfer, which are essential to boost the catalysis42. The existing states of Fe and N elements have been examined by XPS. The N 1 s XPS spectrum of FeSA–N–C is fitted into five configurations, including pyridinic N (398.5 eV), Fe–N (399.2 eV), pyrrolic N (400.2 eV), graphitic N (401.1 eV), and oxidized N (402.9 eV), respectively (Supplementary Fig. 11a)27,28,29,34,35,36,37,38,43. The Fe 2 p3/2 binding energy in FeSA–N–C centers at 710.5 eV (close to Fe3+), suggesting the positively charged Fe atoms (Supplementary Fig. 11b). No Fe0 belonging to Fe particle can be identified from XPS spectrum, in consistent with the absence of Fe particles from PXRD and TEM results (Fig. 2d–f, Supplementary Fig. 8). In addition, no obvious Si residual can be detected from the XPS result of FeSA–N–C (Supplementary Fig. 12). It is noteworthy that the direct pyrolysis of PCN-222(Fe) without SiO2 leads to the formation of Fe nanoparticles in the resultant catalyst (denoted FeNP–N–C), manifesting the important role of SiO2 in inhibiting the agglomeration of Fe atoms under pyrolysis (Supplementary Fig. 13). The absence of Fe peaks in the XRD pattern of FeNP–N–C should be due to the small amount of Fe NPs (Supplementary Fig. 8).
X-ray absorption spectroscopy studies
To gain more information of the electronic structure and coordination environment of single Fe atoms in FeSA–N–C, X-ray absorption near-edge structure (XANES) and Fourier transform-extended X-ray absorption fine structure (FT-EXAFS) spectra have been examined. The XANES spectra of Fe in SiO2/FeSA–N–C and FeSA–N–C show almost the same absorption edge located at between Fe foil and Fe2O3 (Fig. 3a), illustrating the positive valence state of Fe close to +3. The Fourier transformed EXAFS spectra of both SiO2/FeSA–N–C and FeSA–N–C present a dominated peak at ~1.4 Å respecting to the Fe–N scattering path, and no Fe–Fe bond is detected (Fig. 3b). Furthermore, the curve fitting for EXAFS data of FeSA–N–C further verifies the coordination structure around Fe centers. The best fitting result for the first shell indicates that Fe atoms are fourfold coordinated by N atoms in average (Fig. 3c, Supplementary Fig. 14, Supplementary Table 4).

a Fe K-edge XANES and b FT-EXAFS spectra of SiO2/FeSA–N–C, FeSA–N–C, and FeNP–N–C (represented by blue, red, and orange lines, respectively). c EXAFS fitting for FeSA–N–C (red line: fitting curve; gray cycles: experimental data). Inset: schematic model of Fe coordination environment in FeSA–N–C. The red, blue, and gray spheres represent Fe, N, and C atoms, respectively. d The relative energy along the intrinsic reaction coordinate for FeN4/SiO2 (red line) and FeN4 (black line) migrating from initial state to final state.
Insight into the formation process of FeSA–N–C
To unveil the critical role of the SiO2 nanocasting for the generation of atomically dispersed Fe in FeSA–N–C, as a control, pure PCN-222(Fe) without SiO2 was directly pyrolyzed at 800 °C in N2 atmosphere. The Fe-based particles with clear Fe–Fe bond are observed in the EXAFS spectrum of FeNP–N–C, in stark contrast to that of FeSA–N–C derived from SiO2@PCN-222(Fe) (Fig. 3b). The results above unambiguously demonstrate the crucial role of SiO2 in the prevention of Fe atom migration/growth at a high loading under pyrolysis. To further illustrate the stabilizing effect of SiO2 for Fe atoms in SiO2/FeSA–N–C, the energy changes along the intrinsic reaction coordinate were also investigated through density functional theory (DFT) calculation. By selecting the FeN4 and FeN4/SiO2 as representative models for FeSA–N–C and SiO2/FeSA–N–C, respectively, the migration energies of each model from the initial state to the final state have been studied in detail (Supplementary Figs. 15–17). The migration energy barrier of Fe atom in FeN4/SiO2 is found to be 8.35 eV, obviously larger than 6.26 eV of FeN4 without SiO2, suggesting the more thermally stable Fe atoms in SiO2/FeSA–N–C due to the stabilization effect of SiO2 for Fe atoms (Fig. 3d). Moreover, SiO2 also serves as a hard template, creating much larger surface area of FeSA–N–C (1615 m2 g−1) than those of FeNP–N–C (445 m2 g−1) and the N-doped porous carbon (simply as N–C, 612 m2 g−1) derived by the pyrolysis of PCN-222 without Fe centers (Supplementary Fig. 10). Therefore, the SiO2-assisted MOF pyrolysis strategy is able to control the dispersion of active sites and tailor the microstructure of FeSA–N–C, which would be of great significance for subsequent catalysis.
Based on the results above, the formation process of FeSA–N–C from SiO2@PCN-222(Fe) is readily understood. The periodic array of porphyrin linkers in the MOF guarantees the Fe isolation. The N atoms in porphyrin linker serve as the first safeguard to stabilize the Fe species. Moreover, the SiO2 in the MOF pores behaves as an oxide support to further offer anchoring effect, upraising migration energy barrier of Fe atoms, and preventing their migration/growth upon pyrolysis (Fig. 3d). Based on the synergistic interactions endowed by the MOF and SiO2, the thermal agglomeration of Fe atoms is suppressed, leading to the high Fe loadings in atomically dispersed form in FeSA–N–C. In addition to FeSA–N–C, CoSA–N–C, and NiSA–N–C have also been successfully fabricated from SiO2-nanocasted PCN-222(Co) and PCN-222(Ni), respectively, further manifesting the reliability and universality of this powerful strategy (Supplementary Fig. 18).
Electrocatalytic performance for ORR and fuel cells
Encouraged by the high-content single-atom Fe sites and pore structure of FeSA–N–C, its ORR performance has been investigated. To our delight, when firstly tested in 0.1 M KOH solution, FeSA–N–C exhibits the highest half-wave potential (E1/2 = 0.90 V) and kinetic current density (Jk) at 0.85 V (37.19 mA cm−2) among FeNP–N–C, N–C and the commercial Pt/C. The mass activity of FeSA–N–C is calculated to be 21.36 mA g−1 at 0.9 V, much better than FeNP–N–C and N–C counterparts (Supplementary Table 5). The ideal 4e− transfer process, as well as the excellent durability and methanol tolerance of FeSA–N–C all manifest the superior performance to Pt/C and most non-noble metal catalysts ever reported under alkaline conditions (Fig. 4a, Supplementary Figs. 19–22, Supplementary Table 6). Encouraged by the excellent ORR performance of FeSA–N–C in alkaline media, we further explore its performance under more challenging acidic conditions. When tested in 0.1 M HClO4, the linear sweep voltammetry (LSV) curve of FeSA–N–C shows remarkable ORR activity with much higher E1/2 (0.80 V) than that of FeNP–N–C (0.67 V), and N–C (0.51 V; Fig. 4b, c). Also, FeSA–N–C shows better mass activity 1.12 mA g−1 at 0.9 V than that of FeNP–N–C and N–C counterparts (Supplementary Table 7). In addition, FeSA–N–C demonstrates superior Jk at 0.80 V (6.14 mA cm−2) to that of FeNP–N–C (0.30 mA cm−2) and N–C (0.17 mA cm−2), revealing the more favorable kinetics of FeSA–N–C (Fig. 4c). The superior performance of FeSA–N–C to FeNP–N–C and N–C clearly manifests that single-atom Fe sites are the real origin of the high activity for ORR. It is worth noting that, although much endeavor has been devoted, very limited non-noble metal catalysts were reported to present excellent ORR performance in acidic media. The results highlight the particular superiority of FeSA–N–C toward ORR among all reported non-noble-metal catalysts (Supplementary Fig. 23, Supplementary Table 8). Given the superb activity of FeSA–N–C, the deeper ORR investigations have been further conducted in HClO4. To understand the electron transfer mechanism, LSV curves at different rotating rates of rotating disk electrode are recorded. The Koutechy-Levich (K-L) plots obtained from the LSV curves present good linearity, manifesting the first-order reaction kinetics for FeSA–N–C with a potential-independent electron transfer rate (Supplementary Fig. 24a)44,45. The electron transfer number calculated by K-L equation is determined to be 4, in accordance with the result of the rotating ring disk electrode test (Supplementary Fig. 24a, b). Furthermore, the LSV curves of FeSA–N–C after 20,000 cycles, and the methanol addition present its excellent durability and methanol tolerance, in stark contrast to the significant decline of Pt/C (Supplementary Figs. 25 and 26). The XPS spectrum of Fe for FeSA–N–C after stability test shows identical peak to the as-prepared catalyst (Supplementary Fig. 27). From the aberration-corrected HAADF-STEM image, Fe atoms in FeSA–N–C after stability test still maintain atomic dispersion on the porous carbon (Supplementary Fig. 28). The results above manifest that the single Fe atoms in FeSA–N–C can retain the structure after stability test. In addition, to ascertain the critical role of single Fe atoms for the excellent ORR, SCN−, with strong affinity to Fe, was employed to act as a probe to poison Fe–N sites46,47. Upon the addition of KSCN solution into 0.1 M HClO4, the deactivation of FeSA–N–C for ORR, with the half-wave potential decreased significantly by 52 mV, clearly manifests that single Fe atoms are responsible for the excellent ORR performance of FeSA–N–C (Fig. 4d). The excellent ORR activity in acid media of FeSA–N–C has been further approved by the PEMFC measurements. The FeSA–N–C produces a remarkable current density of 292 mA cm−2 at 0.8 V (or 463 mA cm−2 at 0.8 ViR-free), which is among the highest activities of platinum group metals-free cathodes reported in real PEMFCs (Fig. 4e, Supplementary Fig. 29, Supplementary Table 9). Moreover, the peak power density reaches a considerable value of 0.68 W cm−2, ~54% the power density of the Pt-cathode under the same operating conditions (Fig. 4e). The fuel cell stability test for FeSA–N–C indicates a stabilized current density ~0.3 A cm−2 after 20 h testing at 0.5 V (Supplementary Fig. 30). Furthermore, SiO2@FeSA–N–C, featuring much smaller pore size and volume than FeSA–N–C, has also been tested for fuel cell application (Supplementary Fig. 31). It can be seen that SiO2@FeSA–N–C shows much inferior performance to FeSA–N–C, clearly demonstrating the vital importance of porous structure in FeSA–N–C for the improvement of fuel cell performance (Supplementary Fig. 32).

LSV curves for FeSA–N–C (red line), FeNP–N–C (blue line), N–C (olive line) and Pt/C (black line) in a 0.1 M KOH and b 0.1 M HClO4. c Comparison of E1/2 and Jk at 0.80 V for various catalysts in 0.1 M HClO4. d LSV curves of FeSA–N–C in 0.1 M HClO4 before (black line) and after (red line) the addition of SCN−. e Polarization and power density curves of PEMFCs with FeSA–N–C (red dots) and Pt/C (black dots) cathode catalysts. f Free energy diagrams of ORR on FeSA–N–C (red line) and FeNP–N–C (black line) in acidic media (pH = 1).
To better understand the extraordinary ORR activity of FeSA–N–C, DFT calculations have been performed to obtain the free energy diagrams of FeSA–N–C at equilibrium potential based on the four elementary steps of ORR in acidic media (Fig. 4f, Supplementary Figs. 33–35, Supplementary Tables 10–12). It is clear that FeSA–N–C and FeNP–N–C show different ORR rate-determining steps. For FeSA–N–C, the formation of OH* is the most sluggish step with the highest uphill free energy change of 0.55 eV. As for FeNP–N–C, the rate-determining step is the last electron transfer step, requiring the energy change of 1.25 eV. Obviously, the resistance of ORR on FeSA–N–C is much smaller than that of FeNP–N–C, which well explains the outstanding performance of FeSA–N–C.
Discussion
In summary, we have rationally developed a synthetic strategy toward high-loading SACs by nanocasting SiO2 into a MOF, PCN-222(Fe). The 3D skeleton of PCN-222(Fe) realizes the spatial isolation of Fe atoms that are bound by the N atoms in the linker. More importantly, the SiO2 nanocasted in the MOF mesochannels further offers anchoring effect, generating thermally stable FeN4/SiO2 interfaces and further inhibiting Fe agglomeration under pyrolysis. By integrating these dual protections, the SiO2@PCN-222(Fe) composite, as an ideal precursor, affords FeSA–N–C with a Fe loading (3.46 wt%) higher than almost all reported single-atom Fe in N-doped carbon materials. Moreover, the synthetic approach is readily extendable to other single metal atoms, such as CoSA and NiSA. Thanks to the high-content FeSA sites, hierarchical pores and high conductivity of carbon matrix, FeSA–N–C possesses excellent ORR performance in both alkaline and acidic media, far outperforming all other non-noble-metal catalysts and even the Pt/C. Furthermore, FeSA–N–C delivers excellent performance in acidic PEMFC, demonstrating the great potential of FeSA–N–C for PEMFC applications. We believe this nanocasting strategy might open up a fascinating avenue to the general fabrication of SACs with high loadings for broad applications.
Methods
Synthesis of PCN-222(Fe)
In a typical experiment, ZrOCl2 (108.6 mg), FeTCPPCl (32 mg), and CF3COOH (0.45 mL) were dissolved in DMF (10 mL), and ultrasonically dissolved in a 20 mL Pyrex vial. The mixture was heated in 120 °C oven for 18 h. After cooling down to room temperature, the obtained dark brown products were separated by centrifugation, and washed subsequently with DMF for thrice and acetone for twice. The as-obtained precipitates were activated in acetone and finally dried at 60 °C under vacuum overnight.
Synthesis of SiO2@PCN-222(Fe)
Typically, PCN-222(Fe) (30 mg) was transferred to a two-necked flask and degassed for 12 h at 130 °C. When the system was cooled down to room temperature, TEOS (1200 μL) was injected into the flask, and the mixture was sonicated under vacuum for 20 min. Then the obtained solution was centrifuged and the solid was heated at 60 °C under vacuum for 20 min to remove the TEOS on the external surface of the MOF sample. After that, the sample was exposed to 3 M HCl vapor for 9 h at 60 °C to induce the polycondensation of TEOS within the mesopores of the MOF to give SiO2@PCN-222(Fe) composite.
Synthesis of FeSA–N–C
Typically, SiO2@PCN-222(Fe) was heated from the room temperature to 800 °C with a heating rate of 5 °C min−1, then maintained at this temperature for 2 h in N2 atmosphere. The metal oxides were removed by immersing the sample in the HF (20 wt%) solution for 6 h at 80 °C (Caution! HF is highly toxic and should be handled very carefully). The black sample was collected by centrifuging, washed several times with distilled water and ethanol, and dried at 60 °C under vacuum overnight.
Synthesis of FeNP–N–C
Typically, FeNP–N–C was obtained from the direct pyrolysis of pure PCN-222(Fe) following the same procedure as FeSA–N–C.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Yang, X.-F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Liu, J. Catalysis by supported single metal atoms. ACS Catal. 7, 34–59 (2017).
Zhu, C., Fu, S., Shi, Q., Du, D. & Lin, Y. Single-atom electrocatalysts. Angew. Chem. Int. Ed. 56, 13944–13960 (2017).
Zhang, H., Liu, G., Shi, L. & Ye, J. Single‐atom catalysts: emerging multifunctional materials in heterogeneous catalysis. Adv. Energy Mater. 8, 1701343 (2018).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Xu, H., Cheng, D., Cao, D. & Zeng, X. C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 1, 339–348 (2018).
Zhang, H. et al. Dynamic traction of lattice-confined platinum atoms into mesoporous carbon matrix for hydrogen evolution reaction. Sci. Adv. 4, eaao6657 (2018).
Gu, J., Hsu, C., Bai, L., Chen, H. & Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 364, 1091–1094 (2019).
Chen, Y. et al. Single-atom catalysts: synthetic strategies and electrochemical applications. Joule 2, 1242–1264 (2018).
Zhang, L. et al. Graphene defects trap atomic Ni species for hydrogen and oxygen evolution reactions. Chem 4, 285–297 (2018).
Liu, P. et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 352, 797–800 (2016).
Hülsey, M. J., Zhang, J. & Yan, N. Harnessing the wisdom in colloidal chemistry to make stable single‐atom catalysts. Adv. Mater. 30, 1802304 (2018).
Yin, P. et al. Single cobalt atoms with precise N‐coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016).
Sun, X. et al. Single cobalt sites in mesoporous N-doped carbon matrix for selective catalytic hydrogenation of nitroarenes. J. Catal. 357, 20–28 (2018).
Chen, Y.-Z., Zhang, R., Jiao, L. & Jiang, H.-L. Metal-organic framework-derived porous materials for catalysis. Coord. Chem. Rev. 362, 1–23 (2018).
Zhang, J., Chen, G., Müllen, K. & Feng, X. Carbon‐rich nanomaterials: fascinating hydrogen and oxygen electrocatalysts. Adv. Mater. 30, 1800528 (2018).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013).
Zhou, H.-C. & Kitagawa, S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 43, 5415–5418 (2014).
Ma, T. Y., Dai, S., Jaroniec, M. & Qiao, S. Z. Metal-organic framework derived hybrid Co3O4-carbon porous nanowire arrays as reversible oxygen evolution electrodes. J. Am. Chem. Soc. 136, 13925–13931 (2014).
Li, B. et al. Emerging multifunctional metal–organic framework materials. Adv. Mater. 28, 8819–8860 (2016).
Islamoglu, T. et al. Postsynthetic tuning of metal-organic frameworks for targeted applications. Acc. Chem. Res. 50, 805–813 (2017).
Jiao, L., Wang, Y., Jiang, H.-L. & Xu, Q. Metal-organic frameworks as platforms for catalytic applications. Adv. Mater. 30, 1703663 (2018).
Wang, Y.-R. et al. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of CO2. Nat. Commun. 9, 4466 (2018).
Li, G., Zhao, S., Zhang, Y. & Tang, Z. Metal-organic frameworks encapsulating active nanoparticles as emerging composites for catalysis: recent progress and perspectives. Adv. Mater. 30, 1800702 (2018).
Zhao, X., Wang, Y., Li, D.-S., Bu, X. & Feng, P. Metal–organic frameworks for separation. Adv. Mater. 30, 1705189 (2018).
Jiao, L. & Jiang, H.-L. Metal-organic-framework-based single-atom catalysts for energy applications. Chem 5, 786–804 (2019).
Jiao, L. et al. From metal–organic frameworks to single‐atom Fe implanted N‐doped porous carbons: efficient oxygen reduction in both alkaline and acidic media. Angew. Chem. Int. Ed. 57, 8525–8529 (2018).
Zhang, H. et al. Single atomic iron catalysts for oxygen reduction in acidic media: particle size control and thermal activation. J. Am. Chem. Soc. 139, 14143–14149 (2017).
Ye, Y. et al. Surface functionalization of ZIF-8 with ammonium ferric citrate toward high exposure of Fe-N active sites for efficient oxygen and carbon dioxide electroreduction. Nano Energy 38, 281–289 (2017).
Jiang, R. et al. Edge-site engineering of atomically dispersed Fe-N4 by selective C-N bond cleavage for enhanced oxygen reduction reaction activities. J. Am. Chem. Soc. 140, 11594–11598 (2018).
Wan, X. et al. Fe-N-C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat. Catal. 2, 259–268 (2019).
Chen, Y. et al. Isolated single iron atoms anchored on N‐doped porous carbon as an efficient electrocatalyst for the oxygen reduction reaction. Angew. Chem. Int. Ed. 56, 6937–6941 (2017).
Abdel-Mageed, A. M. et al. Highly active and stable single-atom Cu catalysts supported by a metal–organic framework. J. Am. Chem. Soc. 141, 5201–5210 (2019).
Zhang, X. et al. Silica‐protected ultrathin Ni3FeN nanocatalyst for the efficient hydrolytic dehydrogenation of NH3BH3. Adv. Energy Mater. 8, 1702780 (2018).
Kang, X. et al. Synthesis of supported ultrafine non‐noble subnanometer‐scale metal particles derived from metal-organic frameworks as highly efficient heterogeneous catalysts. Angew. Chem. Int. Ed. 55, 1080–1084 (2016).
Wang, Q. et al. Structural evolution of solid Pt nanoparticles to a hollow PtFe alloy with a Pt‐skin surface via space‐confined pyrolysis and the nanoscale kirkendall effect. Adv. Mater. 28, 10673–10678 (2016).
Malonzo, C. D. et al. Thermal stabilization of metal-organic framework-derived single-site catalytic clusters through nanocasting. J. Am. Chem. Soc. 138, 2739–2748 (2016).
Sa, Y. J. et al. A general approach to preferential formation of active Fe-Nx sites in Fe-N/C electrocatalysts for efficient oxygen reduction reaction. J. Am. Chem. Soc. 138, 15046–15056 (2016).
Feng, D. et al. Zirconium‐metalloporphyrin PCN‐222: mesoporous metal-organic frameworks with ultrahigh stability as biomimetic catalysts. Angew. Chem. Int. Ed. 51, 10307–10310 (2012).
Morris, W. et al. Synthesis, structure, and metalation of two new highly porous zirconium metal-organic frameworks. Inorg. Chem. 51, 6443–6445 (2012).
Chen, Y., Hoang, T. & Ma, S. Biomimetic catalysis of a porous iron-based metal-metalloporphyrin framework. Inorg. Chem. 51, 12600–12602 (2012).
Liang, H. W., Zhuang, X., Brüller, S., Feng, X. & Müllen, K. Hierarchically porous carbons with optimized nitrogen doping as highly active electrocatalysts for oxygen reduction. Nat. Commun. 5, 4973 (2014).
Zhang, Z., Sun, J., Wang, F. & Dai, L. Efficient oxygen reduction reaction (ORR) catalysts based on single iron atoms dispersed on a hierarchically structured porous carbon framework. Angew. Chem. Int. Ed. 57, 9038–9043 (2018).
Meng, J. et al. General oriented formation of carbon nanotubes from metal–organic frameworks. J. Am. Chem. Soc. 139, 8212–8221 (2017).
Liang, Y. et al. Covalent hybrid of spinel manganese-cobalt oxide and graphene as advanced oxygen reduction electrocatalysts. J. Am. Chem. Soc. 134, 3517–3523 (2012).
Jiang, W.-J. et al. Understanding the high activity of Fe-N-C electrocatalysts in oxygen reduction: Fe/Fe3C nanoparticles boost the activity of Fe-Nx. J. Am. Chem. Soc. 138, 3570–3578 (2016).
Wang, Q. et al. Phenylenediamine-based FeNx/C catalyst with high activity for oxygen reduction in acid medium and its active-site probing. J. Am. Chem. Soc. 136, 10882–10885 (2014).
Acknowledgements
L.J., R.Z., and G.W. contributed equally to this work. This work is supported by the NSFC (21725101, 21871244, 21673213, and 21521001), China Postdoctoral Science Foundation (2019TQ0298 and 2019M660151), International Partnership Program of CAS (211134KYSB20190109), Fundamental Research Funds for the Central Universities (WK2060030029), BSRF, SSRF, National Synchrotron Radiation Laboratory Foundation (KY2060000160) and Fujian Institute of Innovation (CAS). Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners. The calculations in this work are supported by the supercomputing system in the Supercomputing Center of University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
H.-L.J. conceived the idea and supervised the project. L.J. and R.Z. performed the experiments, characterizations, and electrochemical measurements. G.W. and H.Z. performed the X-ray absorption spectroscopy measurements and analyses. W.Y. performed the theoretical calculations. X.W. and J.S. performed the fuel cell measurements. H.-L.J. and L.J. analyzed the data and co-wrote the paper. H.Z., J.S., and S.-H.Y. discussed the results and commented the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiao, L., Zhang, R., Wan, G. et al. Nanocasting SiO2 into metal–organic frameworks imparts dual protection to high-loading Fe single-atom electrocatalysts. Nat Commun 11, 2831 (2020). https://doi.org/10.1038/s41467-020-16715-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16715-6
This article is cited by
-
An in situ exploration of how Fe/N/C oxygen reduction catalysts evolve during synthesis under pyrolytic conditions
Nature Communications (2024)
-
Monosymmetric Fe-N4 sites enabling durable proton exchange membrane fuel cell cathode by chemical vapor modification
Nature Communications (2024)
-
Dual single atomic Ni sites constructing Janus hollow graphene for boosting electrochemical sensing of glucose
Microchimica Acta (2024)
-
Recent advances in Pt catalysts and membrane electrode assemblies fabrication for proton exchange membrane fuel cells
Rare Metals (2024)
-
Silicon dioxide-protection boosting the peroxidase-like activity of Fe single-atom catalyst for combining chemo-photothermal therapy
Nano Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
