
Abstract
Aldehyde is one of most synthetically versatile functional groups and can participate in numerous chemical transformations. While a variety of simple aromatic aldehydes are commercially available, those with a more complex substitution pattern are often difficult to obtain. Benzylic oxygenation of methylarenes is a highly attractive method for aldehyde synthesis as the starting materials are easy to obtain and handle. However, regioselective oxidation of functionalized methylarenes, especially those that contain heterocyclic moieties, to aromatic aldehydes remains a significant challenge. Here we show an efficient electrochemical method that achieves site-selective electrooxidation of methyl benzoheterocycles to aromatic acetals without using chemical oxidants or transition-metal catalysts. The acetals can be converted to the corresponding aldehydes through hydrolysis in one-pot or in a separate step. The synthetic utility of our method is highlighted by its application to the efficient preparation of the antihypertensive drug telmisartan.
Similar content being viewed by others
Introduction
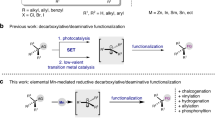
Benzylic oxygenation of alkylarenes provides crucial access to many industrial chemicals, such as terephthalic acid, phenol, and acetone, on a multimillion-ton scale1. Aldehyde is one of the most versatile synthetic handles and can be converted to numerous functionalities. As a result, aromatic aldehydes have been widely used in the manufacture of fine chemicals, nutraceuticals, performance materials, and pharmaceuticals. The oxygenation of methylarenes is a straightforward and attractive strategy for the preparation of aromatic aldehydes, especially considering that the starting materials are widely available and easy to handle. However, partial oxidation of methylarenes to aldehydes remains a largely unsolved challenge due to the strong propensity of product overoxidation under aerobic conditions (Fig. 1a)2,3, and unsatisfactory chemo- and regioselectivity with substrates bearing multiple oxidizable C–H bonds and/or functionalities4. Despite these difficulties, oxygenation of simple methylarenes to aldehydes has been achieved using stoichiometric chemical oxidants such as o-iodoxybenzoic acid (IBX)5, ceric ammonium nitrate (CAN)6, pyridinium chlorochromate7 or polyoxometalate H5PV2Mo10O408. Transition metal-catalyzed aerobic oxidation using hexafluoro-2-propanol as solvent9 or by adding polymethylhydrosiloxane as reagents to avoid overoxidation10 have also been reported (Fig. 1a). As an alternative to chemical oxidation, electrooxidation eliminates the use of stoichiometric chemical oxidants and is attracting increasing interests11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26. Notably, electrooxidation of electron-rich toluene derivatives to substituted benzaldehydes has been applied in the industrial production of p-anisaldehyde and 3,4,5-trimethoxybenzaldehyde27,28,29. Despite these accomplishments, the conversion of structurally complex methylarenes, including medicinally relevant benzoheterocycles in particular, has remained synthetically elusive because of selectivity issues and catalyst inhibition by the coordinating heteroatoms30.

a Examples of reported oxidation of relatively simple toluene derivatives to benzoic acids or benzaldehydes. b The present study focuses on site-selective electrooxidation of methyl benzoheterocycles to aromatic aldehydes.
We have been interested in electrochemical synthesis of heterocycles23,31,32 and recently reported intramolecular dehydrogenative cyclization reactions for the preparation of several types of benzoheterocycles33,34,35,36,37. Alternatively, we envision the synthesis of functionalized benzoheterocycles by modification of alkyl side chains of existing benzoheterocyclic scaffolds. Herein we report a generally applicable electrochemical strategy capable of oxidizing various methyl benzoheterocycles to aromatic acetals in a site-selective manner (Fig. 1b). These side chain oxidation reactions allow access to various functionalized benzoheterocycles difficult to obtain directly through cyclization processes.
Results
Reaction optimization
We began our study by first optimizing the electrooxidation of benzimidazole 1 bearing Me groups at positions 4 and 6 (Table 1). The best results were achieved in an undivided cell with refluxing methanol as solvent, Et4NPF6 (0.5 equiv) as electrolyte, a Pt plate cathode, and a reticulated vitreous carbon anode. Under these conditions, compound 1 reacted site-selectively at C6-Me group to give dimethyl acetal 2 in 72% yield (entry 1) without overoxidation to orthoester or unwanted oxidation of other potentially labile substituents such as C4-Me, N–Bn, or 3° C–H on the cyclohexyl moiety. Lowering the reaction temperature to RT dramatically decreased the yield of 2 to 25% (entry 2). Furthermore, moderate reduction in reaction efficiency was observed when the electrolysis of 1 was performed at a different current (entries 3 and 4), under air (entry 5), with a decreased amount of Et4NPF6 (entry 6), or with another electrolyte such as n-Bu4NPF6 (entry 8) or Et4NOTs (entry 9). Et4NBF4 (entry 7) was, however, equally effective as a supporting electrolyte. Product formation was also diminished with the use of a Pt (entry 10) or graphite anode (entry 11).
Evaluation of substrate scope
With the optimized reaction conditions defined, we set out to explore the scope of the electrooxidation of methylarenes. Notably, the site-selectivity was not significantly affected by introducing a phenyl (3), bromo (4), or cyano group (5) at C5, or by varying the substituent on N1 (6–9) or C2 (11–29) of the C4,C6-dimethylated benzimidazole substrate (Fig. 2). However, the installation of a carbamoyl group on N1 resulted in a slight decrease of regioselectivity (10). The method showed broad compatibility with common functional groups or moieties such as alkyl bromide (7), alkyne (8), alkene (9), ester (15, 16), alcohol (17, 18), ketone (19), aldehyde (20), Boc-protected amine (21–23), ketal (24), azido (25), and aromatic heterocycles (12, 13, 26, and 27). Molecular fragments derived from natural products dehydroabietic acid (28) and lithocholic acid (29) were equally well tolerated. On the other hand, site-selective oxidation of the C6-Me group in C5,C6-dimethylated benzimidazoles bearing an aryl substituent at C2 (30–32) could also be achieved. The replacement of the aryl group with a cyclohexyl, however, resulted in a moderate site-selectivity (33). This reduction in site-selectivity for the 2-cyclohexyl substrate was probably caused by the increased reactivity of the corresponding radical cation compared with the 2-aryl counterparts.

Reaction conditions: heterocycle (0.2 mmol), MeOH (0.022 M), reflux, 2.2–4.5 h. All yields are isolated yields. Regioisomers were not observed unless otherwise mentioned.
Benzoxazoles (34–37) and benzothiazoles (38, 40–42) with multiple open benzylic positions were all found to undergo site-selective oxidation at the C6-Me group (Fig. 3). The site-selectivity was maintained even for substrates bearing an ethyl (37) or isopropyl group (41) at C5 that contained secondary or tertiary benzylic C–H bonds. Notably, the oxidation of 5,6,7-trimethyl benzothiazole proceeded site-selectively as intended despite the high steric hindrance of its C6-Me substituent. However, the resultant product mixture comprised mono-methoxylated 42 as the main product with a minor amount of acetal 43, because the steric environment was detrimental to the second C–H cleavage38. Meanwhile, oxidation of a C4,C7-dimethylated benzothiazole with a methylated phenyl group on C2 occurred preferentially on the C4-Me (44). The electrooxidation method was successfully extended to many other benzoheterocycles, including 2-benzimidazolidinone (45, 46), 2-benzoxazolone (47, 48), 2-oxindole (49), 3,4-dihydro-1H-quinolin-2-one (50), and quinoxalinone (51). Once again, probably due to the steric hinderance, monomethoxylated product 46′ could be obtained selectively with good yield when the electrolysis was stopped at 4.1 F mol−1. The electrochemical method was not limited to benzoheterocycles as demonstrated by the site-selective oxidation of methylated alkoxybenzenes (52–54). The relatively electron-deficient 3,4-dimethylphenylboronic acid (55), however, decomposed into intractable material and did not afforded any aldehyde product. The above results clearly suggested that the site-selectivity for the electrochemical benzylic oxidation reaction are not controlled by steric effects or bond dissociate energies (BDEs) of the C–H bonds.

Reaction conditions: methylarene (0.2 mmol), MeOH (0.022 M), reflux, 2.2–5.3 h. All yields are isolated yields. Regioisomers were not observed unless otherwise mentioned. aElectrolysis was followed by hydrolysis with aqueous HCl (2 N).
The Me oxidation reaction could be coupled with amidine cyclization that we previously described to construct functionalized benzimidazoles (56–65) and imidazopyridines (66–70) (Fig. 4)33. The reaction of an amidine containing a 3,4-disubstituted phenyl ring afforded two products 62 and 62′ because of unselective cyclization. The benzylic oxidation was, however, selective for both regioisomers. Compound 71 did not undergo further Me oxidation because its oxidation potential exceeded the decomposition potential of MeOH solvent. The tandem cyclization/Me oxidation process provided access to benzimidazoles with substitution patterns difficult for the existing methods33,39.

General reaction conditions: amidine (0.2 mmol), MeOH (0.022 M), reflux, 3.5–12.5 h. All yields are isolated yields. Regioisomers were not observed unless otherwise mentioned. aElectrolysis was followed by one-pot hydrolysis with HCl (2 N).
Synthesis of telmisartan
The synthetic utility of our electrooxidation reaction was demonstrated through the construction of the antihypertensive drug telmisartan (Fig. 5a). We first prepared benzimidazole 75 from a commercially available aniline 72 in four steps. Subsequently, site-selective electrooxidation of 75 afforded dimethyl acetal 76 in 46% yield on a decagram scale. In contrast, the oxidation of 75 by a stoichiometric amount of chemical oxidant such as CAN6 or IBX5, in the presence of a Co catalyst under aerobic conditions9, or iron catalyzed oxidation10 with K2S2O8 afforded 80 in <10% yield despite the success of these methods with toluene derivatives (Fig. 5b). Compound 76 was then converted to telmisartan by treating with aqueous HCl to hydrolyze its acetal group to aldehyde and remove its tBu, followed by condensation with o-phenylenediamine 78. Notably, the starting material 72 employed in this synthetic route are much less expensive than ester 79 used in a previously published 8-step method40.

a Our synthetic route employing site-selective benzylic electrooxidation as a key step. b Oxidation of 75 under reported conditions for benzylic oxidation. aMe ester was recovered instead of the original tBu ester 75 because of transesterification. NHPI N-Hydroxyphthalimide, Fe(II)Pc iron(II) phthalocyanine.
Mechanistic investigation
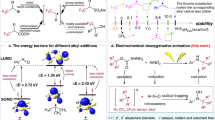
The reaction regioselectivity that we observed in this study suggested that the mechanism likely involved single electron transfer oxidation of the benzene nucleus to a radical cation, followed by benzylic C–H cleavage28. This hypothesis is further supported by the finding that bromination of benzoxazole 81 with NBS, known to proceed through hydrogen atom transfer, afforded a regioisomeric mixture of 82 (50%) and 83 (17%) along with dibrominated 84 (10%) (Fig. 6a). Density functional theory calculations were also performed to provide a plausible rationale for the origin of the observed site-selectivity (Fig. 6b). We first analyzed the distributions of the lowest unoccupied molecular orbitals (LUMO) of the radical cations derived from benzimidazoles (I–III), benzoxazoles (IV, V), benzothiazoles (VI, VII), and 2-benzoxazolone (VIII) that bear multiple Me groups. As shown in Fig. 6b, the LUMOs are delocalized throughout the carbon skeletons of the benzoheterocycles with the distributions on C6 atoms being higher than other carbon atoms attached with a Me group. Furthermore, the natural population analysis shows that the charges of C6 are also more positive than those of other atoms bearing a Me group, indicating deprotonation of the C6-Me groups is preferred41.

a Radical bromination of 81 with NBS. b Computed LUMOs and NPA charges of radical cations derived from various methylated benzoheterocycles. N, C, O, S, and H atoms are colored in blue, gray, red, yellow and white, respectively. The LUMOs are visualized by light blue and yellow isosurfaces. The NPA charges of the atoms bearing a Me group are indicated by black (C6) and blue numbers. NBS N-bromosuccinimide, AIBN azobisisobutyronitrile.
In summary, we have shown that electrooxidation of methyl benzoheterocycles occurs in a site-selective manner to afford a wide range of structurally diverse aromatic acetals. The site-selectivity is governed by the innate electronic properties of the benzo ring instead of BDEs of the C(sp3)–H bonds. The benzylic oxidation takes place efficiently in a simple undivided cell and employs traceless electric current as the reagents without need for stoichiometric chemical oxidants. These features render the reactions scalable and attractive for industrial scale applications.
Methods
Representative procedure for the electrooxidation of methylarenes
A 10 mL three-necked round-bottomed flask was charged with 1 (0.20 mmol, 1.0 equiv) and Et4NPF6 (0.10 mmol, 0.5 equiv). The flask was then equipped with a condenser, a reticulated vitreous carbon (100 PPI, 1.2 cm × 1.0 cm × 0.8 cm) anode and a platinum plate (1.0 cm × 1.0 cm) cathode, and flushed with argon. MeOH (9.0 mL) was then added. The electrolysis was carried out at 80 °C (oil bath temperature) using a constant current of 10 mA until complete consumption of the substrate (2.3 h, 4.3 F mol–1). The reaction mixture was cooled to RT and concentrated under reduced pressure. The residue was chromatographed through silica gel eluting with ethyl acetate/hexanes containing 1% triethylamine to give the desired product 2 in 72% yield as a white solid. All new compounds were fully characterized (See the Supplementary methods).
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 1964756. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The data supporting the findings of this study are available within the article and its Supplementary Information files. Any further relevant data are available from the authors on request.
References
Sterckx, H., Morel, B. & Maes, B. U. W. Catalytic aerobic oxidation of C(sp3)−H bonds. Angew. Chem. Int. Ed. 58, 7946–7970 (2019).
Ishii, Y., Sakaguchi, S. & Iwahama, T. Innovation of hydrocarbon oxidation with molecular oxygen and related reactions. Adv. Synth. Catal. 343, 393–427 (2001).
Yoshino, Y., Hayashi, Y., Iwahama, T., Sakaguchi, S. & Ishii, Y. Catalytic oxidation of alkylbenzenes with molecular oxygen under normal pressure and temperature by N-hydroxyphthalimide combined with Co(OAc)2. J. Org. Chem. 62, 6810–6813 (1997).
White, M. C. & Zhao, J. Aliphatic C–H oxidations for late-stage functionalization. J. Am. Chem. Soc. 140, 13988–14009 (2018).
Nicolaou, K. C., Baran, P. S. & Zhong, Y. L. Selective oxidation at carbon adjacent to aromatic systems with IBX. J. Am. Chem. Soc. 123, 3183–3185 (2001).
Fehr, C., Chaptal-Gradoz, N. & Galindo, J. Synthesis of (−)-vulcanolide by enantioselective protonation. Chem. Eur. J. 8, 853–858 (2002).
Hosseinzadeh, R., Tajbakhsh, M. & Vahedi, H. Selective oxidation of methylarenes with pyridinium chlorochromate. Synlett 2005, 2769–2770 (2005).
Sarma, B. B., Efremenko, I. & Neumann, R. Oxygenation of methylarenes to benzaldehyde derivatives by a polyoxometalate mediated electron transfer–oxygen transfer reaction in aqueous sulfuric acid. J. Am. Chem. Soc. 137, 5916–5922 (2015).
Gaster, E., Kozuch, S. & Pappo, D. Selective aerobic oxidation of methylarenes to benzaldehydes catalyzed by N-hydroxyphthalimide and cobalt(II) acetate in hexafluoropropan-2-ol. Angew. Chem. Int. Ed. 56, 5912–5915 (2017).
Hu, P. et al. Bio-inspired iron-catalyzed oxidation of alkylarenes enables late-stage oxidation of complex methylarenes to arylaldehydes. Nat. Commun. 10, 2425 (2019).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B. & Kampf, C. J. Electrochemical arylation reaction. Chem. Rev. 118, 6706–6765 (2018).
Yoshida, J.-i, Shimizu, A. & Hayashi, R. Electrogenerated cationic reactive intermediates: the pool method and further advances. Chem. Rev. 118, 4702–4730 (2018).
Sauermann, N., Meyer, T. H., Qiu, Y. & Ackermann, L. Electrocatalytic C–H activation. ACS Catal. 8, 7086–7103 (2018).
Yang, Q. L., Fang, P. & Mei, T. S. Recent advances in organic electrochemical C—H functionalization. Chin. J. Chem. 36, 338–352 (2018).
Kärkäs, M. D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 47, 5786–5865 (2018).
Feng, R., Smith, J. A. & Moeller, K. D. Anodic cyclization reactions and the mechanistic strategies that enable optimization. Acc. Chem. Res. 50, 2346–2352 (2017).
Ye, Z. & Zhang, F. Recent advances in constructing nitrogen-containing heterocycles via electrochemical dehydrogenation. Chin. J. Chem. 37, 513–528 (2019).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Jiang, Y., Xu, K. & Zeng, C. Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 118, 4485–4540 (2018).
Nutting, J. E., Rafiee, M. & Stahl, S. S. Tetramethylpiperidine N-oxyl (TEMPO), phthalimide N-oxyl (PINO), and related N-oxyl species: electrochemical properties and their use in electrocatalytic reactions. Chem. Rev. 118, 4834–4885 (2018).
Xiong, P. & Xu, H. C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Sauer, G. S. & Lin, S. An electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal. 8, 5175–5187 (2018).
Das, A., Nutting, J. E. & Stahl, S. S. Electrochemical C–H oxygenation and alcohol dehydrogenation involving Fe-oxo species using water as the oxygen source. Chem. Sci. 10, 7542–7548 (2019).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C-H oxidation. Nature 533, 77–81 (2016).
Cardoso, D. S. P., Sljukic, B., Santos, D. M. F. & Sequeira, C. A. C. Organic electrosynthesis: from laboratorial practice to industrial applications. Org. Process Res. Dev. 21, 1213–1226 (2017).
Wendt, H. & Bitterlich, S. Anodic synthesis of benzaldehydes-i. voltammetry of the anodic-oxidation of toluenes in nonaqueous solutions. Electrochim. Acta 37, 1951–1958 (1992).
Zhu, Y. H. et al. A promising electro-oxidation of methyl-substituted aromatic compounds to aldehydes in aqueous imidazole ionic liquid solutions. J. Electroanal. Chem. 751, 105–110 (2015).
Lumb, J. P. Stopping aerobic oxidation in its tracks: chemoselective synthesis of benzaldehydes from methylarenes. Angew. Chem. Int. Ed. 56, 9276–9277 (2017).
Cai, C.-Y., Shu, X.-M. & Xu, H.-C. Practical and stereoselective electrocatalytic 1,2-diamination of alkenes. Nat. Commun. 10, 4953 (2019).
Cai, C.-Y. & Xu, H.-C. Dehydrogenative reagent-free annulation of alkenes with diols for the synthesis of saturated O-heterocycles. Nat. Commun. 9, 3551 (2018).
Zhao, H.-B. et al. Amidinyl radical formation through anodic N−H bond cleavage and its application in aromatic C−H bond functionalization. Angew. Chem. Int. Ed. 56, 587–590 (2017).
Xu, F., Long, H., Song, J. & Xu, H.-C. De novo synthesis of highly functionalized benzimidazolones and benzoxazolones through an electrochemical dehydrogenative cyclization cascade. Angew. Chem. Int. Ed. 58, 9017–9021 (2019).
Hou, Z.-W., Yan, H., Song, J.-S. & Xu, H.-C. Electrochemical synthesis of (Aza)indolines via dehydrogenative [3+2] annulation: application to total synthesis of (±)-hinckdentine A. Chin. J. Chem. 36, 909–915 (2018).
Wu, Z.-J., Li, S.-R., Long, H. & Xu, H.-C. Electrochemical dehydrogenative cyclization of 1,3-dicarbonyl compounds. Chem. Commun. 54, 4601–4604 (2018).
Qian, X.-Y., Li, S.-Q., Song, J. & Xu, H.-C. TEMPO-catalyzed electrochemical C–H thiolation: synthesis of benzothiazoles and thiazolopyridines from thioamides. ACS Catal., 2730–2734 (2017).
Baciocchi, E., Bietti, M. & Lanzalunga, O. Mechanistic aspects of β-bond-cleavage reactions of aromatic radical cations. Acc. Chem. Res. 33, 243–251 (2000).
Brasche, G. & Buchwald, S. L. C-H functionalization/C-N bond formation: copper-catalyzed synthesis of benzimidazoles from amidines. Angew. Chem. Int. Ed. 47, 1932–1934 (2008).
Ries, U. J. et al. 6-Substituted benzimidazoles as new nonpeptide angiotensin II receptor antagonists: synthesis, biological activity, and structure-activity relationships. J. Med. Chem. 36, 4040–4051 (1993).
Margrey, K. A., McManus, J. B., Bonazzi, S., Zecri, F. & Nicewicz, D. A. Predictive model for site-selective aryl and heteroaryl C–H functionalization via organic photoredox catalysis. J. Am. Chem. Soc. 139, 11288–11299 (2017).
Acknowledgements
The authors acknowledge the financial support of this research from MOST (2016YFA0204100), NSFC (No. 21672178, 21971213), and Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
P.Xiong and H.B.Z. contributed equally to this work. P.Xiong, H.B.Z., L.H.J., H.L., P.Xu, Z.J.L., and Z.J.W. performed the experiments and analyzed the data, X.T.F. and J.C. conducted the theoretical studies. H.C.X. designed and directed the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks David Cardoso and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiong, P., Zhao, HB., Fan, XT. et al. Site-selective electrooxidation of methylarenes to aromatic acetals. Nat Commun 11, 2706 (2020). https://doi.org/10.1038/s41467-020-16519-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16519-8
This article is cited by
-
Paired electrocatalysis unlocks cross-dehydrogenative coupling of C(sp3)-H bonds using a pentacoordinated cobalt-salen catalyst
Nature Communications (2024)
-
Electrochemical synthesis of peptide aldehydes via C‒N bond cleavage of cyclic amines
Nature Communications (2024)
-
Electrochemical oxidative C(sp3)–H cross-coupling with hydrogen evolution
Nature Synthesis (2023)
-
Electrochemically driven regioselective C−H phosphorylation of group 8 metallocenes
Nature Communications (2022)
-
Electrochemical aromatic C–H hydroxylation in continuous flow
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
