
Abstract
p16INK4a (CDKN2A) is a central tumor suppressor, which induces cell-cycle arrest and senescence. Cells expressing p16INK4a accumulate in aging tissues and appear in premalignant lesions, yet their physiologic effects are poorly understood. We found that prolonged expression of transgenic p16INK4a in the mouse epidermis induces hyperplasia and dysplasia, involving high proliferation rates of keratinocytes not expressing the transgene. Continuous p16INK4a expression increases the number of epidermal papillomas formed after carcinogen treatment. Wnt-pathway ligands and targets are activated upon prolonged p16INK4a expression, and Wnt inhibition suppresses p16INK4a-induced hyperplasia. Senolytic treatment reduces p16INK4a-expressing cell numbers, and inhibits Wnt activation and hyperplasia. In human actinic keratosis, a precursor of squamous cell carcinoma, p16INK4a-expressing cells are found adjacent to dividing cells, consistent with paracrine interaction. These findings reveal that chronic p16INK4a expression is sufficient to induce hyperplasia through Wnt-mediated paracrine stimulation, and suggest that this tumor suppressor can promote early premalignant epidermal lesion formation.
Similar content being viewed by others


Introduction
Activity of tumor-suppressive cellular mechanisms is often associated with cell and organismal aging, yet cancer propensity substantially increases with age1,2,3. p16INK4a, encoded by the CDKN2A gene (p16 hereafter), represents an important link between cancer, cellular responses to stress, and aging. p16 is a central tumor suppressor, which is among the most commonly mutated genes in diverse human malignancies4,5. When activated, p16 binds and inhibits CDK4/6-Cyclin D complexes, leading to Rb activation, and thereby induces cell-cycle arrest and senescence4,6. This pathway represents one of the central mechanisms blocking the proliferation of damaged or oncogene-expressing cells.
Whereas p16 is not expressed in most embryonic and adult cells7, its levels increase in multiple tissues with age8,9,10,11. The specific stimuli underlying age-associated p16 activation have not been directly established. However, a variety of stresses, including radiation, DNA damaging agents, cigarette smoke, and oncogene activity, were shown to induce p1612,13,14,15. Aged animals lacking p16 show increased replicative and regenerative capacity in several tissues, indicating that it contributes to the aging-associated decline in these processes1. It was more recently shown that directed genetic elimination of p16-expressing senescent cells during mouse aging delays the functional deterioration of multiple organs and increases lifespan11. This finding and subsequent studies have highlighted the negative contribution of senescent cells to age-associated pathologies, and the therapeutic potential for their pharmacologic removal through senolytic drug treatment16,17. Whether senolytic treatments have potential benefit in cancer therapy is currently largely unknown.
The expression of p16 in aging tissues raises the question of whether its activity influences cancer development. Mice carrying an extra copy of Cdkn2a show increased resistance to cancer, consistent with the known tumor-suppressive role of p1618. In contrast, elimination of p16-expressing senescent cells reduces cancer mortality rates in mice, suggesting that such cells could contribute to tumor development11. The mechanisms underlying this are not fully known. It has been suggested that resident senescent cells can promote tumorigenesis during aging by generating inflammation mediated by cytokine secretion, a feature of senescence known as the senescence-associated secretory phenotype (SASP)3,19. It is, however, unclear whether all cells expressing p16 in vivo achieve a full senescence phenotype, and p16 activity itself appears to be insufficient to induce the SASP20,21. The functional contributions of p16 to age-associated changes in cancer propensity, therefore, remain poorly characterized.
Here we study the effects of prolonged p16 expression in the epidermis, in order to uncover its effects on tissue structure and cancer development. p16 levels and senescence were reported to increase with age in the skin dermis and epidermis22,23,24. UV radiation (UVR), the major cause of skin malignancies, activates p1613,25, and p16-expressing cells are detected in premalignant epidermal lesions such as actinic keratosis26,27,28. The high mutation rates of p16 in cutaneous squamous cell carcinoma and other skin malignancies5,29,30 indicate that it suppresses malignant progression. However, it is unknown whether the activity of p16 in the normal epidermis and in premalignant lesions influences the development of disease. Furthermore, whether p16-expressing cells in such early lesions can be targeted by senolytic therapy, and whether this may have therapeutic benefit, has not been tested.
Using transgenic mice allowing tissue-specific p16 activation, we demonstrate that the persistent expression of p16 in a subset of cells within the epidermis induces hyperplasia and dysplasia, and promotes tumor formation following mutagenesis. We show that p16 expression in mice and in cultured keratinocytes leads to Wnt-pathway activation, which contributes to epidermal hyperproliferation, and that senolytic elimination of p16-expressing cells inhibits hyperplasia. These findings reveal that chronic p16 activity is sufficient to induce premalignant tissue changes through a non-cell-autonomous mechanism, and uncover a potential tumor-promoting function of this gene during early tumorigenesis.
Results
Epidermal p16 induction causes partial senescence features
To study the effects of p16-expressing cells on the adult skin we crossed mice carrying a doxycycline-activated human p16 gene (tet-p16)21 with K5-rtTA mice31, allowing its inducible activation in the basal epidermis. Transgenic p16 protein was detected in ~40% of basal keratinocytes in the interfollicular epidermis (IFE) after 2 days of doxycycline (dox) treatment at 3 weeks of age (Fig. 1a–c). Tissues showed reduced phosphorylated Rb levels, consistent with expected Rb activation (Fig. 1d). Staining for Ki67 revealed a dramatic reduction in the number of proliferating IFE basal cells after 2 days of p16 activation (Fig. 1e, f). However, after 2 weeks of p16 activation, overall proliferation rates returned to those of control animals.

a Immunostaining of human p16 (brown) in back skin sections of K5-rtTA/tet-p16 and control tet-p16 mice treated with doxycycline (dox) for 2 days (2d) starting at 3 weeks of age. b Skin sections from the same mice stained for p16 (red) and K14 (green), which labels the basal epidermis. c Percentage of p16+ keratinocytes in the interfollicular epidermis (IFE) in mice treated with dox for 2 days (2d), 2 weeks (2w), or 6 months (6 m). Dots indicate individual mice, n = 5, 5, 4 in respective groups. d Skin sections from mice treated with dox for 2 days, stained for Rb phosphorylated on Thr-821/826 (p-Rb) (red) and K14 (green). e Immunostaining of the proliferation marker Ki67 (brown) in sections from mice treated with dox for 2 days (2d) or 2 weeks (2w). f Percentage of IFE keratinocytes expressing Ki67 in the same mice (dots). n = 4, 6, 4, 4. g SA-βGal staining (blue) of skin sections from indicated mice, treated with dox for 2 weeks. h Percentage of SA-βGal positive IFE cells from indicated mice (dots) treated with dox for 2 weeks. n = 9, 10. i Skin sections from indicated mice stained for E-Cadherin (brown) to indicate cell circumference. j Area of epidermal keratinocytes from indicated mice (dots). Shown are values for control mice (tet-p16), and for p16− and p16+ keratinocytes in K5-rtTA/tet-p16 mice. n = 3, 6, 6 mice, >60 cells were scored per mouse. k H&E-stained skin sections from the indicated mice treated with dox for 2 weeks, showing hair-follicle morphology. l Number of hair follicles per field in indicated mice (dots), following dox treatment for 6 months. n = 9 per group. m FACS analysis of epidermal cells from indicated mice treated with dox for 6 months, stained for CD34, CD49f, and Sca1. Charts show Sca1− (follicular) cells only. Gate indicates percentage of CD34+/CD49fhigh hair-follicle stem cells. Cells were pooled from three mice per group. All graphs indicate mean across mice ± S.E.M, all scores were conducted visually from images. Blue labels DNA in all fluorescence images. ***P < 0.0001, t test. ns non-significant. Scale bars—20 μm, except k—100 μm.
Increased numbers of cells expressing the senescence markers SA-βGal and SenTraGor32 were detected in mice that have undergone 2 weeks of p16 activation (Fig. 1g, h and Supplementary Fig. 1a, b), yet those were lower than the numbers of p16-expressing cells, suggesting that not all transgene-expressing cells enter a fully senescent state. The average size of keratinocytes was increased in p16-expressing mice, specifically cells expressing the transgene, resulting in a thickening of the epidermis, with no increase in cell numbers (Fig. 1i, j and Supplementary Fig. 1c–e). Changes in other known markers of senescence such as p21, Lamin B1, DNA damage, or secreted factors were not detected. Together these findings indicate that p16 expression in epidermal keratinocytes induces cell-cycle arrest, cell hypertrophy, and some features of senescence.
p16 induction led to delayed growth of hair follicles (HF), reduced hair-follicle stem cell (HFSC) proliferation, and the formation of deformed HFs during the anagen stage initiated at 3 weeks of age (Fig. 1k and Supplementary Fig. 1f, g), reminiscent of the effect of p14ARF activation33. Upon prolonged p16 expression, mice displayed a decrease in the number of HFs and hair loss (Fig. 1l and Supplementary Fig. 1f, h), and the numbers of CD34+/CD49fhigh HFSCs were dramatically reduced (Fig. 1m). p16 expression thus blocks the proliferation of HFSCs and leads to their gradual depletion.
Prolonged p16 expression induces hyperplasia and dysplasia
To examine the effects of prolonged p16 expression on the epidermis, we activated the transgene for 6 months. At this timepoint we detected p16 protein in ~20% of IFE cells (Fig. 1c). Strikingly, p16-expressing mice exhibited a thickened epidermis, in which both cell number and average cell size were increased (Fig. 2a–c). We observed an increased proportion of suprabasal differentiated cells in p16-expressing skins, while basal layer marker expression (K14, K5) was maintained and expanded in some regions (Fig. 2a, d, e and Supplementary Fig. 2a, b). These changes contrast with the normal young and aged mouse epidermis, which contains only a thin suprabasal layer (Supplementary Fig. 2c). p16-induced mice also often showed an expansion of the dermis with dense collagen deposition regions, and a reduction of fat layer width (Supplementary Fig. 2d). Although we did not observe a prominent inflammatory reaction, numbers of epidermal and dermal T-cells, as well as of macrophages, were increased (Supplementary Fig. 2e, f). Control mice in which GFP was activated instead of p16 for the same period did not show these changes (Supplementary Fig. 2g).

a H&E-stained skin sections of K5-rtTA/tet-p16 and control tet-p16 mice treated with dox for 6 months (6m). Typical (middle) and pronounced (right) phenotype regions are shown for p16-induced mice. b IFE keratinocyte cell number per field (40×) in indicated mice (dots). n = 7, 12 in respective groups. c Average area of IFE cells from the same mice, visually measured from E-cadherin-stained sections. n = 7, 12. d Sections from the same mice stained for K10 (red) labeling suprabasal cells and K14 (green) labeling basal cells. e Percentage of K10+ epidermal cells isolated from dissociated mouse ear epidermis of indicated mice (dots) after 6 months of dox treatment, scored by FACS. n = 3, 4. f Skin sections from the indicated mice stained for Ki67 (brown). Right image shows a region with dysplasia. g Percentage of Ki67+ basal IFE cells in indicated mice (dots) after 6 months of dox treatment. n = 3, 8. h Skin sections from same mice co-stained for human p16 (red), the proliferation marker Mcm7 (white) and K14 (green). i Percentage of Mcm7+ basal IFE cells in control mice (tet-p16), and among p16− and p16+ cells in the epidermis of K5-rtTA/tet-p16 mice treated with dox for 6 months. n = 3, 7, 7. j Percentage of BrdU+ basal epidermal cells in same mice, scored from skin sections co-stained for BrdU and p16. n = 9 per group. k Serial sections of a human actinic keratosis lesion stained for p16 (middle) and Rb (right) (both brown). Staining for p16 in normal human skin is shown in left panel. Dotted lines label the basement membrane. l Percentage of p16+ cells within individual human actinic keratosis (AK) lesions (dots), among lesions showing p16 expression. n = 47. m AK lesion co-stained for p16 (red), Ki67 (white) and K14 (green). n Percentage of Ki67+ cells among p16− and p16+ cells in human AK lesions (dots). n = 47. All graphs indicate mean across mice ± S.E.M, except panels l, n which indicate mean across lesions. All scores were conducted visually on images, except e. Blue labels DNA in all images. ***P < 0.0001, t test. ns non-significant. Scale bars—20 μm.
Strikingly, proliferation rates in the epidermis of mice expressing p16 for 6 months were dramatically increased, with 81% of basal keratinocytes expressing Ki67 relative to 33% in control mice (Fig. 2f, g). Furthermore, some epidermal regions exhibited dysplasia (atypia), exhibiting changes in nuclear morphology and basal layer organization (Fig. 2f, right panel). Co-staining for p16 and the proliferation marker Mcm7 revealed that 93% of basal IFE keratinocytes in which p16 was not activated (p16–) were proliferating, as opposed to 26% of basal IFE cells in control mice (Fig. 2h, i). A corresponding increase in BrdU incorporation, marking cells in S-phase, was observed (Fig. 2j). These findings indicate that prolonged p16 activity in a subset of epidermal cells is sufficient to induce the proliferation of the majority of surrounding keratinocytes. Surprisingly, 30% of p16-expressing (p16+) cells also stained positive for Mcm7, and 5% stained positive for BrdU, suggesting that some of the transgene-expressing cells possessed the ability to proliferate (Fig. 2i, j). Chronic epidermal p16 expression thus leads to hyperplasia and regional dysplasia, consistent with paracrine stimulation emanating from the p16+ cells.
We tested whether p16-induced hyperplasia influenced the regenerative capacity of the skin following wounding. Healing was delayed in mice in which p16 was activated for 2 weeks prior to wounding, and a similar delay was observed in mice in which p16 was activated for 6 months (Supplementary Fig. 3), indicating that the p16-induced hyperplasia does not lead to enhanced regenerative capacity.
A subset of cells in human actinic keratosis express p16
The appearance of dysplastic regions in p16-expressing mice was reminiscent of early-stage human premalignant epidermal lesions. To establish whether p16+ cells are detected within such lesions, we stained sections of human actinic keratosis (AK) lesions, the common precursors to cutaneous squamous cell carcinoma (cuSCC)34. Previous studies reported varying percentages and staining patterns of p16 in these lesions26,27,28. We detected p16 protein in 47 of 72 lesions (65%), collected in three medical centers, with a mean of 11.3% p16+ cells per lesion in those that contained such cells (0.8–41.8%, n = 47) (Fig. 2k, l). p16+ cells were often interspersed among or adjacent to dividing, p16–, cells (Fig. 2m, n and Supplementary Fig. 4a). An average of 6% of the cells in AK lesions positively stained for SenTraGor, suggesting the presence of senescent cells in them (Supplementary Fig. 4b, c). Rb protein was also detected in these lesions, excluding Rb deletion or silencing, which would render p16 ineffective (Fig. 2k, right panel). Furthermore, as observed in the p16-induced mice, some p16+ cells were proliferating, based on Ki67 expression, yet in lower numbers than the surrounding cells (Fig. 2m, n). These findings are consistent with potential crosstalk between p16+ and p16– subpopulations in human AK lesions, as suggested by the mouse model.
p16 promotes formation of carcinogen-induced papillomas
The finding that p16 expression was sufficient to induce epidermal hyperplasia and dysplasia raised the possibility that cells carrying oncogenic mutations would be influenced by a p16-activated environment. We, therefore, tested whether chronic p16 expression would affect the rates of papilloma formation after carcinogen treatment, using the DMBA/TPA model35. We treated K5-rtTA/tet-p16 mice, as well as sibling control tet-p16 mice, with a single topical dose of the mutagen DMBA at 3 weeks of age, and 1 week later initiated p16 activation by dox treatment, and simultaneously began twice-weekly topical treatments with TPA. We tracked papilloma formation for 5 months. Strikingly, p16-expressing mice developed approximately double the number of papillomas than control mice (Fig. 3a, b). This indicates that p16 generates an environment that supports the ability of keratinocytes bearing an oncogenic mutation to initiate papillomas.

a Average number of papillomas per mouse formed over time (age in weeks) in K5-rtTA/tet-p16 and sibling control tet-p16 mice treated once with DMBA at 3 weeks, followed by continued TPA treatment and p16 activation by dox starting at 4 weeks. n = 14, 11 mice per group combined from four independent experiments. b Images of indicated mice at 27 weeks of age, showing papillomas formed on back skins. c H&E staining of papillomas from control and p16-induced mice. d Mean diameter of papillomas from the same mice at 27 weeks. n = 84, 136 papillomas per group. e Immunostaining for p16 (brown) in papillomas from same mice. Enlarged inset is shown on right. f Staining for Ki67 (brown) in papillomas from same mice. g Percentage of Ki67-stained area of the epithelial component of papillomas (dots) from indicated mice, scored by image analysis. n = 13,8 in respective groups. h Papillomas from control and p16-induced mice stained for K14 (green) and K10 (red), marking the basal and suprabasal layers, respectively. i Relative mRNA levels of epidermal differentiation-associated genes in whole papillomas from control and p16-expressing mice, measured by mRNA-seq. n = 4, 3. P < 0.05 for all genes. Graphs indicate mean values across mice (a) or across lesions (d, g, i) ± S.E.M. *P < 0.01, **P < 0.001, ***P < 0.0001, t test. Scale bars—200 μm, except f—100 μm.
Interestingly, the papillomas that developed in p16-expressing mice were smaller than those in control mice, and contained fewer proliferating cells, which were more restricted to the basal epidermal regions of the lesions (Fig. 3c–g and Supplementary Fig. 5a). p16+ cells were readily detected within these papillomas (Fig. 3e). SenTraGor staining indicated higher numbers of senescent cells within the p16-induced papillomas, relative to the adjacent non-transformed epidermis (Supplementary Fig. 5b,c), consistent with previous reports indicating that p16 exerts a stronger pro-senescent effect when activated in the context of a strong pro-mitotic signal36. Expression profiling of whole dissected papillomas revealed a dramatic elevation in markers of differentiated epidermal layers and reduced expression of stem cell-associated markers, in addition to reduced expression of cell cycle, signaling, and cytoskeletal genes (Fig. 3h, i and Supplementary Fig. 5d, e).
Together these findings indicate that p16-expressing cells in the non-transformed tissue stimulate neighboring mutated oncogene-expressing cells to form growing premalignant tumors. However, p16 expression within formed oncogene-expressing lesions restricts proliferation, increasing senescence and differentiation, and thereby reduces tumor growth, consistent with its known tumor-suppressive roles.
p16 expression activates Wnt in epidermal keratinocytes
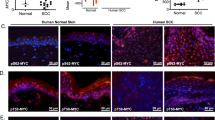
We next set out to uncover the mechanisms by which p16-expressing cells stimulate proliferation and induce hyperplasia. We generated triple-transgenic K5-rtTA/tet-p16/tet-GFP mice, in which p16 and GFP were co-activated, and treated the mice with dox for 6 months (Fig. 4a). Co-expression of GFP did not reduce the numbers of detected p16+ cells (Supplementary Fig. 6a, b). We then isolated GFP+ keratinocytes from these mice and from K5-rtTA/tet-GFP control mice expressing only GFP (Fig. 4b and Supplementary Fig. 6c). The transcriptome of GFP+ cells from p16-expressing mice revealed changes in multiple genes relative to GFP+ cells from control mice (624 upregulated and 750 genes downregulated, Supplementary Data 1), representing pathways known to be involved in senescence, including cell-cycle regulation, cytoskeletal structure, cell adhesion, protein turnover and metabolism (Fig. 4c). Several cytokine-encoding genes were upregulated, including Ccl20, Cxcl1, and Cxcl9, as well as Tgfβ ligands, yet these did not constitute a full SASP (Fig. 4d). Interestingly, we found that the levels of several genes encoding Wnt ligands, as well as known Wnt transcriptional target genes, were elevated, while Fzd receptor-encoding genes showed reduced mRNA levels (Fig. 4e). Tcf7, which encodes the Wnt-target-activating Tcf1 transcription factor (and a Wnt target itself)37,38 was upregulated; conversely, Tcf7l1 and Tcf7l2, which respectively encode the Tcf3 and Tcf4 factors and are known to often act as Wnt target repressors37,38, were downregulated, consistent with transcriptional target activation (Fig. 4e). The proliferation-associated Ccnd genes (encoding Cyclin D proteins) which also respond to Wnt, were among the upregulated genes, as was often previously observed in senescent cells39. Consistent with Wnt-pathway activation, we detected by immunohistology increased numbers of cells expressing nuclear β-catenin in the epidermis of p16-expressing mice, as well as cells expressing Tcf1, Cyclin D1 and CD44 (Fig. 4f, g).

a Diagram of transgenic mouse lines crossed for co-induction of GFP and p16. b FACS plots showing GFP levels and gates used for isolation of GFP+ epidermal cells from K5-rtTA/tet-GFP/tet-p16 (right) and control K5-rtTA/tet-GFP (left) mice, after 6 months of induction. Plots show the CD31−/CD140a−/CD45− cell fraction only. c Gene sets whose expression was preferentially upregulated (blue) or downregulated (grey) in GFP+ cells from p16-expressing mice relative to GFP+ cells from control mice. Values indicate –log10 (adjusted P value) by hypergeometric test. d mRNA levels of genes encoding cytokines and genes associated with the TGFβ pathway upregulated in p16-expressing GFP+ cells, measured by mRNA-seq. Values were normalized to the mean expression levels in GFP+ cells from control mice, defined as 1. P < 0.05 for all genes, n = 4 samples per group, each pooled from 3 mice. e Relative mRNA levels of genes associated with the Wnt pathway in the same samples. P < 0.05 for all genes, n = 4 samples per group, each pooled from 3 mice. f Skin sections from K5-rtTA/tet-p16 and control tet-p16 mice treated with dox for 6 months stained for β-Catenin, Tcf1, Cyclin D1 or CD44. Arrowheads indicate representative positively stained cells. g Percentages of nuclear β-catenin+, Tcf1+, and CyclinD1+ IFE cells in control and p16-expressing mice after 6 months of induction, as scored visually from images. P values calculated by t test. All graphs indicate mean across mice ± S.E.M. Scale bars—20 μm.
Isolation and transcriptome analysis of the GFP– cell fraction from p16-expressing mice revealed that, relative to the GFP+ cells from the same mice, this fraction was enriched for proliferation-associated gene sets (as expected from their hyperproliferative state), and showed reduced expression of senescence-related sets, such as oxidative stress response, protein translation, and cytoskeletal reorganization (Supplementary Fig. 6c–e). Keratinocyte differentiation genes were somewhat elevated, reflecting the presence of diverse differentiation states in the expanded epidermis (Supplementary Fig. 6f). We found that, similar to the GFP+ p16-expressing cells, the GFP– fraction showed elevation in Wnt-associated genes relative to cells from control mice, and that the Tcf7l1 gene was further silenced in these cells (Supplementary Fig. 6g). Together these findings indicate that p16 expression leads to activation of Wnt-pathway genes in both the p16-expressing cells and the surrounding IFE cells. The activation of the Wnt pathway by p16 appeared to occur gradually after prolonged p16 expression, as we did not observe elevation in Wnt-associated genes after 2 days or 2 weeks of p16 induction (Supplementary Fig. 6h).
p16-induced Wnt secretion drives keratinocyte proliferation
To better dissect p16-induced Wnt activity, we overexpressed p16 in cultured primary mouse keratinocytes by lentiviral transduction. p16 overexpression led to rapid cell-cycle arrest, acquisition of a senescent morphology, and expression of senescence-associated markers, yet, interestingly, with no evidence of SA-βGal activity (Fig. 5a, b). Within 10 days of p16 expression, the levels of multiple Wnt ligands and targets were upregulated in the p16-expressing cells (Fig. 5c). ELISA measurement indicated that Wnt3a levels in conditioned medium (CM) of p16-expressing cells was elevated, reaching 3 ng/ml (Fig. 5d). Treatment of the cells with XAV-939, a Tankyrase1/2 inhibitor that causes β-catenin degradation40, suppressed the upregulation of Wnt target genes, including the genes encoding Cyclin D1 and D2, as well as of Wnt ligands (Fig. 5c). The activator Tcf7 gene was upregulated in p16-expressing cells, whereas the repressors Tcf7l1 and Tcf7l2 were both downregulated by p16 overexpression, as was observed in the transgenic mice, and XAV-939 treatment reversed this effect (Fig. 5c). These findings indicate that p16 is sufficient to activate the Wnt pathway in keratinocytes, driving both target and ligand activation in a β-catenin-dependent manner.

a Primary mouse keratinocytes infected with a p16-expressing or control lentivirus, co-stained for K14 (green), p16 (red) and BrdU (indicating S-phase, white), or for SA-βGal, 10 days after infection. b Relative mRNA levels of the indicated genes in the same keratinocytes, measured by qRT-PCR. n = 3 replicates. P < 0.05 for all genes. c Relative mRNA levels of the indicated Wnt-associated genes measured by qRT-PCR in control and p16-expressing keratinocytes. Values are also shown for the same cells treated with the β-catenin inhibitor XAV-939 (5 μM) for 24 h. n = 3 replicates. P < 0.05 for all comparisons indicated by lines: p16 versus control (dark blue versus dark grey, short lines), and p16 versus p16+XAV-939 (light blue versus dark blue, long lines). d Secreted Wnt3a protein levels in medium of control and p16-expressing cells, measured by ELISA. n = 3 replicates. ***P < 0.0001 e Fold increase over 4 days in numbers of primary keratinocytes cultured with conditioned media from p16-expressing (CM-p16) or from control (CM-Cont) keratinocytes, or with 10 ng/ml recombinant Wnt3a, in the absence or presence of XAV-939 (darker and lighter columns, respectively). n = 6 replicates. ***P < 0.0001. f Relative mRNA levels of the indicated Wnt-associated genes measured by qRT-PCR in keratinocytes treated for 24 h with conditioned media from p16-expressing or control cells, or with Wnt3a, in the presence or absence of XAV-939. n = 3 replicates. P < 0.05 for all comparisons indicated by lines: CM-p16 versus CM-Cont (dark blue versus dark grey, short lines), and CM-p16 versus CM-p16+XAV-939 (light blue versus dark blue, long lines). All graphs indicate mean across replicates ± S.E.M., t test. Scale bars—50 μm.
To test whether Wnt activation and secretion by p16-expressing keratinocytes can influence naive cells, we treated proliferating primary keratinocytes with conditioned media from p16-expressing or control (empty vector-expressing) keratinocytes, or, as a positive control, with recombinant Wnt3a. While control-cell CM had no effect on the numbers of treated cells, the CM of p16-expressing cells led to enhanced proliferation of the treated cells, resulting in a 1.7-fold increase in cell numbers after 4 days of treatment, similar to the effect of Wnt3a (Fig. 5e). This stimulatory effect was almost completely lost when XAV-939 was administered together with CM or Wnt3a, indicating that proliferation is β-catenin mediated (Fig. 5e). Treated cells showed upregulation of Wnt target genes and downregulation of Tcf71,2, as well as increased levels of genes encoding Wnt ligands (Fig. 5f), mirroring the changes observed in the p16-expressing mice. These changes were blocked by XAV-939 (Fig. 5f). Together, these findings indicate that Wnt secretion by p16-expressing cells can stimulate the proliferation of naive keratinocytes, and that Wnt signals act on both the p16-expressing cells themselves and on the non-expressing proliferating cells, activating transcriptional targets as well as Wnt ligands, in a β-catenin-dependent manner (Supplementary Fig. 7).
The Wnt-pathway mediates p16-induced hyperplasia
We next tested whether Wnt activation contributes to the hyperplasia observed in p16-expressing mice. We activated p16 for 6 months, and then treated the mice systemically with XAV-939 for 1 week. XAV-939 treatment led to a significant reduction in epidermal proliferation rates, as well as a reduction in the expression of Wnt ligand and target genes (Fig. 6a–c). Wnt inhibition was therefore sufficient for a partial reversal of the ongoing hyperplastic effects of p16 expression.

a Skin sections from control tet-p16 mice (left) and K5-rtTA/tet-p16 mice treated with dox for 6 months, and subsequently treated for 1 week with XAV-939 (right) or with vehicle (middle), stained for Ki67 (brown). b Percentage of Ki67+ basal IFE cells in sections from same mice (dots). n = 7, 8, 8, combined from two independent experiments. c Relative mRNA levels of the indicated Wnt-associated genes in isolated epidermis from the same mice, measured by qRT-PCR. n = 8 mice per group. P < 0.05 for all comparisons between p16-expressing mice (blue) and XAV-939-treated mice (green). d Skin sections from control (tet-p16), K5-rtTA/tet-p16 and K5-rtTA/tet-p16/tet-Tcf3 mice treated with dox for 6 months, stained for p16 (red) and Tcf3 (white). e Skin sections from the same mice as in d stained for Ki67 (brown). f Percentage of Ki67+ basal IFE cells in mice expressing p16 (p16) or p16 together with Tcf3 (p16+Tcf3) for 6 months, and from controls (Cont). n = 7, 4, 5. g Relative mRNA levels of the indicated Wnt-associated genes in isolated epidermis from the same mice, measured by qRT-PCR. n = 7, 4, 5. Scores were done visually from images. All graphs indicate mean values across mice ± S.E.M. P < 0.05 for all comparisons between p16-expressing mice (blue) and mice co-expressing p16 and Tcf3 (green). ***P < 0.0001. t test was used for all significance tests. Scale bars—20 μm.
We next tested whether co-activation of the repressive Tcf3 (Tcf7l1) together with p16 would influence Wnt-pathway activation and hyperplasia. We crossed K5-rtTA/tet-p16 mice with mice carrying an inducible Tcf3 transgene (tet-Tcf3)41 to generate triple-transgenic mice. Co-activation of Tcf3 together with p16 for 6 months resulted in significantly reduced epidermal hyperplasia, as well as in reduced levels of Wnt ligand and target genes (Fig. 6d–g). Tcf3 expression thus largely blocks the pro-proliferative effects of p16. Together these results indicate that activation of the Wnt pathway upon chronic p16 expression contributes to the induction of epidermal hyperplasia.
Senolytic elimination of p16+ cells suppresses hyperplasia
To study whether the continued presence of p16-expressing cells in the epidermis is required for maintenance of hyperplasia, even after prolonged stimulation, we tested whether these cells can be eliminated by senolytic treatment. The Bcl-2 protein-family inhibitor ABT-737 and its analog ABT-263 (navitoclax) inhibit Bcl-2, Bcl-xl, and Bcl-w, and were shown to have senolytic activity in various settings16,42,43. We previously showed that ABT-737 eliminates epidermal cells in which the p53 activator, p14ARF, was overexpressed42. However, it was unclear, in light of the partial induction of senescence by p16, whether ABT-737 would be effective in eliminating p16-expressing cells. Following six months of p16 induction, we treated mice with ABT-737 six times over nine days and then sacrificed the mice. ABT-737-treated mice showed a significant reduction in the percentage of p16+ cells in the IFE (an approximate 60% reduction), and a corresponding reduction in basal cell proliferation and epidermal thickness, indicating partial reversal of the hyperplasia (Fig. 7a–d). Furthermore, the expression of Wnt ligands and targets was significantly reduced in the ABT-737-treated mice (Fig. 7e). p16-expressing cells are thus sensitive to senolytic treatment, and their residence in the epidermis is necessary for the ongoing maintenance of Wnt stimulation and consequent hyperplasia.

a Skin sections from K5-rtTA/tet-p16 and control tet-p16 mice treated with dox for 6 months and subsequently treated with ABT-737 (6 injections over 9 days), stained by H&E, for p16, or for Ki67. b Percentage of p16+ IFE cells in ABT-737-treated and untreated p16-induced and control mice (dots). c Percentage of Ki67+ basal IFE cells in same mice. d Epidermal thickness in same mice. e Relative mRNA levels of the indicated Wnt-pathway-associated genes in whole skins isolated from the same mice, measured by qRT-PCR. P < 0.05 for all comparisons indicated by lines: p16 versus control (dark blue versus dark grey, long lines) and p16+ABT-737 versus p16 (light blue versus dark blue, short lines). n = 4, 4, 4, 5 in respective groups in all panels. All graphs indicate mean values across mice ± S.E.M. Scores were conducted visually from images. ***P < 0.0001, t test. Scale bars—20 μm.
Discussion
The function of p16 as one of the most important tumor suppressor genes is well established, and is supported by its recurrent loss-of-function alterations in a wide variety of human malignancies and by numerous studies in which it was shown to block immortalization and tumorigenesis4. An important additional dimension to p16 function is its activation, often linked to cellular senescence, in a subset of cells within aging tissues. Recent studies revealed that p16-expressing cells contribute to aging processes and to associated diseases, and have highlighted the question of whether such resident cells influence the propensity of tissues to develop cancer11,16. Our findings reveal that the continuous expression of p16 itself in the epidermis has pro-proliferative and pro-tumorigenic effects. p16 expression, when activated after carcinogen treatment, promoted the initiation of papillomas, indicating that it generates an environment that supports the ability of cells carrying an oncogenic mutation to initiate tumors. p16 may thus function in some settings as a double-edged sword, in the short-term blocking proliferation of damaged cells, but, over time, stimulating the proliferation of neighboring cells and thereby contributing to the formation of premalignant lesions.
Our findings indicate that Wnt signaling plays a central role in mediating p16-induced hyperplasia. Wnt and β-catenin are known to regulate the acquisition of stem cell traits, particularly of bulge hair-follicle stem cells, and also to promote the proliferation of interfollicular epidermal cells and the formation of epidermal tumors44,45,46,47. p16 overexpression, both in vivo and in cultured primary keratinocytes, led to Wnt ligand secretion and Wnt-pathway activation, stimulating the hyperproliferation of target cells that do not express p16. Interestingly, the recipient cells not only activated Wnt transcriptional targets, but also upregulated Wnt ligand expression, thereby generating a positive feedback loop. In both the p16-expressing and the naive cells, Wnt ligand activation was β-Catenin dependent. This mode of combined autocrine and paracrine stimulation is consistent with previous dissection of Wnt activity in the IFE, in which it was shown that basal cells expressing the Wnt target Axin2 also secrete Wnt ligands, in a β-Catenin-dependent manner44. Wnt over-activation in the p16-expressing epidermis occurred gradually over a prolonged period. In contrast, in cultured keratinocytes, Wnt-pathway genes were activated within days of p16 overexpression. The reasons for this difference are unclear, but may be due to more stringent negative feedback loops active in the living tissue.
Wnt activation could be expected to also promote stem cell identity and function in p16-expressing skins. Recent studies have shown that senescence can promote stem cell-like traits in some settings48,49,50. However, we found that p16 expression blocked the proliferation of hair-follicle stem cells, and led to their gradual depletion from the tissue, results similar to those observed in a previous study51. Wound healing, a stem cell-dependent process52, was delayed in the p16-expressing mice, even during epidermal hyperplasia. It is therefore possible that additional actions of p16 counteract the stimulation of stem cell function by Wnt, particularly in the days immediately following p16 activation, in which the proliferation of HFSCs is arrested and Wnt levels are not yet elevated.
p16 expression induced only partial features of senescence. Numbers of SA-βGal+ and SenTraGor+ cells were elevated, but were lower than those of p16+ cells. We observed an increase in keratinocyte size, a hallmark of senescence, and changes in senescence-associated gene sets, such as genes involved in cytoskeletal structure and protein translation. Yet various other known markers of senescence53 were unchanged. Several cytokine-encoding genes were upregulated in p16-expressing cells, and while these could potentially contribute to the observed phenotypes, they did not constitute a full SASP response, which typically involves high levels of multiple cytokines. This is consistent with previous observations that p16 does not activate the SASP20. While several past studies have linked Wnt ligand activation to senescence, the paracrine actions of Wnt were not addressed, and in some settings Wnt activity was reported to counteract senescence50,54,55,56. Our work thus reveals a tumor-promoting contribution of p16 that does not necessitate a broad SASP but acts mainly through Wnt.
Accumulation of p16-expressing cells may preferentially occur in epidermal regions exposed to damage. p16 is known to be activated by UVR, the major cause of human skin malignancies13,25. UV-irradiated mouse skins show a thickening reminiscent of that induced by p16 overexpression57. Expanding previous observations26,27,28, we show that p16 is indeed often expressed in actinic keratosis lesions, which are among the most common human premalignant growths and are induced by UVR. Furthermore, we show that p16+ cells in these lesions are in close proximity to p16– dividing cells, in a pattern similar to that observed in our mouse model. This pattern is consistent with paracrine interaction between these cell subsets, yet such crosstalk between them remains to be directly proven.
Mutations in p53 occur very early in skin tumorigenesis, as a direct consequence of UVR34. Interestingly, this may result in further enhanced compensatory p16 expression and senescence in such lesions9,58, possibly providing a proliferation-promoting environment. Upon progression to cuSCC p16 is often mutated29,30, suggesting that its counter-proliferative functions outweigh the potential benefit of its activity. Our observation that the expression of p16 restricts proliferation within growing papillomas suggests that its activity following tumor initiation blocks further growth, as is expected from its tumor-suppressive function (Supplementary Fig. 7). Interestingly, the major effect of p16 within the papillomas was restriction of proliferation and maintenance of a differentiated state, together with increased senescence. Unlike the untransformed tissue, p16-expressing papillomas did not show overall increased Wnt activation. The actions of p16 in cells that express an active oncogenic driver may thus be distinct from its effects in normal cells.
Pharmacologic elimination of senescent cells through drugs with senolytic activity has been shown in recent studies to be beneficial in a variety of preclinical disease models, and some of these drugs are currently being tested in clinical trials16,17,59. Our findings suggest that senolytic treatment may be beneficial for early premalignant epidermal lesions.
Altogether, we reveal in this study a novel dimension of the functions of p16 in skin tumorigenesis, suggesting a non-cell-autonomous contribution to the formation of premalignant lesions, and a potential avenue for blocking this function through senolytic treatment.
Methods
Mice
The following transgenic mouse lines were crossed as indicated: tet-p16 (mixed C57Bl6 and 129sv), K5-rtTA (FVB), tet-GFP (mixed C57Bl6 and 129sv) and tet-Tcf3 (FVB). For transgene induction mice received 2 mg/mL doxycycline (Bio Basic) in their drinking water starting at the age of 3 weeks, and treatment was maintained for the indicated durations. In all experiments, the experimental and control mouse groups were siblings grown and treated together. For short-term inductions (up to 2 weeks), both males and females were used; for inductions of 6 months and wound-healing assays all presented results are from male mice, whereas for papilloma formation assays only data of females is shown, due to higher average lesion formation rates. For bromodeoxyuridine (BrdU) labeling mice were injected intraperitoneally with 100 mg/Kg BrdU 2 h prior to sacrifice. For Wnt-pathway inhibition, mice that received dox for 6 months were injected intraperitoneally with 200 μg XAV-939 (Sigma-Aldrich) in 10% DMSO/90% 0.9% NaCl twice a day for seven consecutive days. ABT-737 (WuXi AppTec), was prepared at 75 mg/kg in 30% propylene glycol, 5% Tween 80, 3.3% dextrose in water pH 4–5, and mice were injected intraperitoneally 6 times over 9 days after 6 months of induction. Control mice were injected with vehicle solutions. Upon sacrifice, mice were shaved and their dorsal skins were paraffin-embedded for immunohistology or frozen in OCT solution for SA-βgal staining. Chemically-induced skin carcinogenesis was conducted as described35. Three-week-old K5-rtTA/tet-p16 and control tet-p16 mice were shaved, and one day later received a single topical treatment of 50 nmole DMBA (7,12-Dimethylbenz[a]anthracene) in 200 μl acetone. After one week dox was added to the drinking water and the mice began twice-weekly topical treatments with 4 μg TPA (12-O-tetradecanoyl phorbol-13-acetate) in 200 μl acetone. Mice were examined for 23 additional weeks for papilloma detection and measurement and were sacrificed 10 days after the last TPA treatment. Papilloma numbers were scored weekly, with diameters measured by caliper. Only papillomas with ≥1 mm diameter were scored. Individual whole papillomas were excised and processed for RNA extraction and profiling, or paraffin embedded. For wound-healing assays dorsal skins were shaved and treated with depilating cream two days prior to wound formation; mice were anesthetized, their back skin was sterilized with povidone-iodine solution, and 1 cm2 full-thickness excision wounds were introduced to the upper paravertebral area using sterile scissors. Wounds were measured daily with a caliper. Animals were housed in specific pathogen-free conditions, at 20–24 °C, 30–70% humidity and a 12:12 h light-dark cycle. The Hebrew University is an AAALAC International accredited institute. All experiments were conducted with approval from the Hebrew University Animal Care and Use Committee.
Cultured keratinocytes
Epidermal keratinocytes were isolated following published procedures60. Dorsal skin was removed from newborn mice, and was incubated with dispase (Sigma-Aldrich) followed by isolation of the epidermis and treatment with trypsin (Biological Industries). Keratinocytes were plated on fibroblast feeder cells for four passages and then plated in regular tissue culture dishes. Passage 16 cells were infected with a lentivirus expressing human p16 (pLV-hCDKN2a-GFP-Puro, Vector Builder) or a control empty lentiviral vector, and selected with puromycin (2 μg/mL). 10 days after infection cells were fixed and stained as indicated, or lysed for RNA extraction. Conditioned medium (CM) was collected from cells after 3 days of culture and filtered through a 0.45 μm filter. Wnt3a levels in CM were measured using Wnt3a ELISA kit (R&D Systems, DY1324B-05). Naive keratinocytes were treated with CM in a 1:1 ratio with fresh media, or with 10 ng/ml recombinant Wnt3a (R&D Systems). RNA was collected from treated cells one day after treatment. For proliferation measurements, 104 naive keratinocytes were plated in 24-well plates, treated with CM from p16-expressing or control cells, or with Wnt3a (6 replicates for each), and cell numbers were scored after 4 days. XAV-939 (Sigma-Aldrich, 5 μM) was added where indicated and control samples were treated with DMSO vehicle.
Human actinic keratosis samples
A total of 72 human actinic keratosis lesions, surgically resected, were collected in three medical centers: 19 patient samples were obtained from the laboratory of Histology-Embryology, Athens Medical School under approval of the Bio-Ethics Committee of Athens Medical School, in accordance with the Declaration of Helsinki and local laws and regulations, following written consent from patients; 42 lesion samples from 35 patients were obtained from the University of Arizona, USA, approved by the University of Arizona Institutional Review Board protocol, with patient informed consent; 11 patient samples were obtained from the Hadassah-Hebrew University Medical Center, Jerusalem, Israel under approval of the Hadassah Medical Center Helsinki Committee for usage of fully anonymized archival specimens, and approved waiver of patient consent. All slides underwent pathologist review to verify lesion type. Patients were aged 38 to 92, with equal representation of males and females. All available patient samples were included in the analysis, with no selection.
Immunohistology and immunocytology
Immunohistologic stains were performed on 5 μm paraffin sections of collected mouse back skins, individual papillomas, or human AKs, according to standard protocols, using Peroxidase Substrate Kit (Vector) or fluorescently-labeled secondary antibodies (Jackson). The following primary antibodies were used: human p16INK4a (Abcam, ab108349), human p16INK4a (BD Pharmingen, 551153), p-Rb (Santa Cruz, sc-16669-R), Ki67 (Abcam, ab16667), BrdU (Bio-Rad, MCA2060), Mcm7 (Santa Cruz, sc-9966), E-Cadherin (BD Pharmingen, 610182), K14 (Progen, GP-CK14), K10 (Santa Cruz, sc-23877), K5 (Progen, GP-CK5), Rb (Santa Cruz, sc-102), CyclinD1 (Abcam, ab16663), β-catenin (BD Pharmingen, 610153), Tcf1 (Invitrogen, MA5-14965), Tcf3 (Santa Cruz, sc-8635), CD44 (eBioscience, 14-0441-82), Vimentin (Cell Signaling, 5741), CD3 (Bio-Rad, MCA1477), and F4/80 (Bio-Rad, MCA497). Masson’s Trichrome stain was conducted using staining kit (Sigma-Aldrich, HT15) according to manufacturer instructions. SenTraGor stain was conducted as described32. For SA-βGal stains, 12 μm frozen sections of OCT-embedded mouse skins, or cultured keratinocytes, were fixed in 0.5% glutaraldehyde for 15 min and stained using standard procedures33. For co-stains of cultured keratinocytes for BrdU, p16, and K14, cells were treated for 2 h with 10 μg/mL BrdU and then fixed with cold ethanol, treated with 1 M HCl followed by 0.1 M sodium tetraborate (Na2B4O7) for neutralization, and then stained, as indicated, using PBS + 10% fetal calf serum (FCS) for antibody suspensions. Brightfield images were collected using an Olympus CX41 and DS-Fi1 camera and processed using NIS-Elements software (Nikon). Fluorescent images were collected using an Olympus FV1000 confocal microscope and processed with the FV10-ASW software. All mouse skin images shown are of back skin. Percentages of SA-βGal+, SenTraGor+, p16+, Ki67+, Mcm7+, BrdU+, CycD1+, nuclear β-Catenin+, Tcf1+ and epidermal CD3+ cells, out of total IFE or basal IFE cells as indicated, were visually scored from 5 or more microscopic field images per mouse, and 2 or more fields from human AK lesions, with 100–1000 total cells scored per sample. Absolute numbers per microscopic fields of hair follicles, IFE cells, dermal CD3+ and dermal F4/80+ cells were scored from 8 or more fields per mouse (10× magnification for HFs, 40× for others). Percentage of Ki67+ area in papillomas out of epithelial cell area was measured using NIS-elements software (Nikon). Epidermal thickness was measured by NIS elements on 36 or more measurements per mouse. Keratinocyte area calculation was performed on skin sections co-stained for E-Cadherin and p16 by measuring cell circumference using NIS-Elements, with 60 cells or more measured per mouse.
FACS analysis
Adult epidermal cells were isolated following published procedures61. Mouse back skins were shaved and excised, and adipose and muscle tissues were removed. The skin was then incubated with 0.25% trypsin solution overnight at 4 °C, and the epidermis was scraped off, minced and suspended in a trypsin/PBS with 0.5% BSA solution, followed by filtration through a 70 μm cell strainer. Antibody stains were performed using standard procedures. For hair-follicle stem cell analysis live cells were stained for Sca-1, CD34, and CD49f, with three mice pooled in each group. Isolation of GFP+ keratinocytes for mRNA-seq profiling was conducted as described above, and live cells were stained for CD140a, CD45 and CD31 to gate out fibroblasts, immune and endothelial cells, respectively. For scoring of K10+ epidermal cell percentage, adult epidermal cells were isolated from ear skin samples to obtain improved dissociation. Ear epidermal layers were obtained by digestion with 2 mg/mL Dispase II (Roche Diagnostics) in PBS solution for 40 min at 37 °C, and treated with 2 mg/mL collagenase type II (Worthington Biochemicals) and 1 mg/mL DNase I (Roche) in PBS with 2% fetal calf serum for 25 min at 37 °C. Samples were then incubated with 0.5 M EDTA for 10 min and filtered through a 70 μm strainer. Cells were treated with fixation and permeabilization solution (BD Pharmingen) for 15 min and stained for K10 and secondary fluorescent-conjugated antibody. Samples were sorted on a FACS Aria III system using FACSDiva software (BD Biosciences), or were analyzed on a MACSQuant Analyzer (Miltenyi Biotec). Post-acquisition analysis was conducted with FCS Express (De Novo Software). Antibodies used: CD34 (BD Bioscience, 562608), Sca-1 (Biolegend, 108113), CD49f (eBioscience, 17-0495-80), K10 (Santa Cruz, sc-23877), CD45 (eBioscience, 25-0451-82), CD140a (eBioscience, 17-1401-81), and CD31 (eBioscience, 17-0311-80).
RNA extraction, qRT-PCR, and expression profiling
Total RNA from whole skin samples and whole papillomas was extracted using the Tissue RNA purification kit (Norgen Biotek). RNA from isolated epidermis was extracted following epidermal separation using the RNeasy Minikit (Qiagen). RNA extraction from FACS-sorted cells was done using the Single Cell RNA Purification Kit (Norgen Biotek). RNA was extracted from primary keratinocytes using Direct-zol kit (Zymo Research, #R2051). cDNA was prepared using iScript cDNA synthesis kit (Bio-Rad), and qRT-PCR was conducted using iTaq Universal SYBR Green Supermix (Bio-Rad) in triplicates, with expression values normalized to Hprt and Ppia expression levels for mouse and primary keratinocyte samples, respectively. Primer lists are provided in Supplementary Table 1. All RT-PCR graphs conducted on mouse samples indicate values across individual mice. We conducted two mRNA-seq profilings of isolated keratinocytes. In the first, we analyzed 80,000 GFP+ keratinocytes from K5-rtTA/tet-p16/tet-GFP and control K5-rtTA/tet-GFP mice, after exclusion of CD45+, CD31+ and CD140a+ cells. Cells were pooled from three mice in each replicate, with four replicates used for each group, obtained in three independent experiments. In the second experiment, we similarly isolated 80,000 GFP+ cells from the same mouse groups (after CD45+, CD31+ and CD140a+ exclusion), as well as 20,000 GFP− cells from K5-rtTA/tet-p16/tet-GFP mice, pooling 3 mice per each sample. n = 4, 4, 5 for these groups, respectively. mRNA-Seq transcriptional profiling was conducted using an adapted CEL-Seq2 protocol62: 3′ cDNA was synthesized and barcoded, followed by RNA synthesis, amplification by in vitro transcription, and library generation for paired-end sequencing. Reads were demultiplexed, quality filtered, and trimmed for adapters and poly-A tail using Cutadapt and aligned with the mouse genome (GRCm38) using Tophat2. Differentially expressed genes were determined by DESeq2, using a P < 0.05 cutoff. Quality control and principal component analyses were conducted using R: packages ‘RColorBrewer_1.1-2’, ‘pheatmap_1.0.10’, ‘ggplot2_3.1.0’ and ggrepel_0.8.0. Enrichment of gene sets was determined using the hypergeometric test through Metascape63, with adjusted P values calculated using the Benjamini-Hochberg procedure. Profiling of papillomas was done with the same method, using RNA from whole isolated lesions, 4 control and 3 p16-expressing. Analysis of gene sets in these comparisons was done for up- or downregulated genes with Log2fold change > 0.6 at P < 0.05. We excluded in this analysis genes not expressed in keratinocytes, based on our profiling of FACS-sorted populations, to reduce biases introduced by differential inclusion of non-epithelial cell types within the lesions. Gene sets representing epidermal keratinocyte differentiation states were compiled from Joost et al.64.
Statistical analysis
Two-tailed unpaired Student’s t test was used to derive P values in all comparisons stating usage of t test. Other methods were used as indicated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The mRNA-seq expression profiles were deposited in GEO database, under accession number GSE146979 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi]. All the other data supporting the findings of this study are available within the article and its supplementary information files and from the corresponding author upon reasonable request. A reporting summary for this article is available as a Supplementary Information file.
References
Sharpless, N. E. & DePinho, R. A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 8, 703–713 (2007).
de Magalhaes, J. P. How ageing processes influence cancer. Nat. Rev. Cancer 13, 357–365 (2013).
Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Kim, W. Y. & Sharpless, N. E. The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275 (2006).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337 (2018).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Zindy, F., Quelle, D. E., Roussel, M. F. & Sherr, C. J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 15, 203–211 (1997).
Krishnamurthy, J. et al. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114, 1299–1307 (2004).
Yamakoshi, K. et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J. Cell Biol. 186, 393–407 (2009).
Burd, C. E. et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152, 340–351 (2013).
Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Sorrentino, J. A. et al. p16INK4a reporter mice reveal age-promoting effects of environmental toxicants. J. Clin. Invest. 124, 169–173 (2014).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Matheu, A. et al. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 18, 2736–2746 (2004).
Faget, D. V., Ren, Q. & Stewart, S. A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 19, 439–453 (2019).
Coppe, J. P. et al. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396–36403 (2011).
Helman, A. et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 22, 412–420 (2016).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
Ressler, S. et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5, 379–389 (2006).
Velarde, M. C., Flynn, J. M., Day, N. U., Melov, S. & Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 4, 3–12 (2012).
Pavey, S., Russell, T. & Gabrielli, B. G2 phase cell cycle arrest in human skin following UV irradiation. Oncogene 20, 6103–6110 (2001).
Hodges, A. & Smoller, B. R. Immunohistochemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod. Pathol. 15, 1121–1125 (2002).
Salama, M. E. et al. p16INK4a expression in actinic keratosis and Bowen’s disease. Br. J. Dermatol. 149, 1006–1012 (2003).
Nilsson, K., Svensson, S. & Landberg, G. Retinoblastoma protein function and p16INK4a expression in actinic keratosis, squamous cell carcinoma in situ and invasive squamous cell carcinoma of the skin and links between p16INK4a expression and infiltrative behavior. Mod. Pathol. 17, 1464–1474 (2004).
Pickering, C. R. et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 20, 6582–6592 (2014).
Li, Y. Y. et al. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 21, 1447–1456 (2015).
Diamond, I., Owolabi, T., Marco, M., Lam, C. & Glick, A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J. Invest. Dermatol. 115, 788–794 (2000).
Evangelou, K. et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 16, 192–197 (2017).
Tokarsky-Amiel, R. et al. Dynamics of senescent cell formation and retention revealed by p14ARF induction in the epidermis. Cancer Res. 73, 2829–2839 (2013).
Ratushny, V., Gober, M. D., Hick, R., Ridky, T. W. & Seykora, J. T. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest. 122, 464–472 (2012).
Abel, E. L., Angel, J. M., Kiguchi, K. & DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat. Protoc. 4, 1350–1362 (2009).
Takahashi, A. et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 8, 1291–1297 (2006).
Yi, F. et al. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 13, 762–770 (2011).
Lien, W. H. et al. In vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nat. Cell Biol. 16, 179–190 (2014).
Lucibello, F. C., Sewing, A., Brusselbach, S., Burger, C. & Muller, R. Deregulation of cyclins D1 and E and suppression of cdk2 and cdk4 in senescent human fibroblasts. J. Cell Sci. 105(Pt 1), 123–133 (1993).
Huang, S. M. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009).
Nguyen, H., Rendl, M. & Fuchs, E. Tcf3 governs stem cell features and represses cell fate determination in skin. Cell 127, 171–183 (2006).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Ovadya, Y. et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435 (2018).
Lim, X. et al. Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Science 342, 1226–1230 (2013).
Lien, W. H. & Fuchs, E. Wnt some lose some: transcriptional governance of stem cells by Wnt/beta-catenin signaling. Genes Dev. 28, 1517–1532 (2014).
Clevers, H., Loh, K. M. & Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346, 1248012 (2014).
Sherwood, V. & Leigh, I. M. WNT signaling in cutaneous squamous cell carcinoma: a future treatment strategy? J. Invest. Dermatol. 136, 1760–1767 (2016).
Mosteiro, L., et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354, aaf445 (2016).
Ritschka, B. et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183 (2017).
Milanovic, M. et al. Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018).
Boquoi, A. et al. Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16Ink4a. Aging Cell 14, 139–147 (2015).
Ito, M. et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 11, 1351–1354 (2005).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Ye, X. et al. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol. Cell 27, 183–196 (2007).
Adams, P. D. & Enders, G. H. Wnt-signaling and senescence: A tug of war in early neoplasia? Cancer Biol. Ther. 7, 1706–1711 (2008).
Basu, D., Reyes-Mugica, M. & Rebbaa, A. Role of the beta catenin destruction complex in mediating chemotherapy-induced senescence-associated secretory phenotype. PLoS ONE 7, e52188 (2012).
Tavana, O. et al. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 9, 3328–3336 (2010).
Leong, W. F., Chau, J. F. & Li, B. p53 deficiency leads to compensatory up-regulation of p16INK4a. Mol. Cancer Res. 7, 354–360 (2009).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Nowak, J. A. & Fuchs, E. Isolation and culture of epithelial stem cells. Methods Mol. Biol. 482, 215–232 (2009).
Jensen, K. B., Driskell, R. R. & Watt, F. M. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat. Protoc. 5, 898–911 (2010).
Hashimshony, T., Wagner, F., Sher, N. & Yanai, I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666–673 (2012).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Joost, S. et al. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 3, 221–237 (2016).
Acknowledgements
We thank Safa Hirbawi for technical assistance, Norma E. Kidess-Bassir for histological preparation, Sharona Elgavish, and Hadar Benyamini for bioinformatics assistance, Dan Lehmann of the Core Research Facility at Hebrew University for FACS assistance, and Yaron Fuchs for advice. We thank Elaine Fuchs for the tet-Tcf3 mice. This study was supported by grants from the Israel Science Foundation (1009/13, 2092/18) and from the DKFZ-MOST Fund (CA-161) (I.B.), by a fellowship from the Israel Council for Higher Education (N.A.), and by the Welfare Foundation for Social & Cultural Sciences (KIKPE) Greece, and NKUA-SARG Greece, grants No 70/3/8916, 70/3/12128 (V.G.G.).
Author information
Authors and Affiliations
Contributions
N.A. and I.B.-P. designed the study and wrote the manuscript, N.A. conducted the experiments, B.A. assisted in experimentation, E.W., S.R, and Y.N. conducted bioinformatic analysis, A.M., K.M., E.P., A.K.W., and V.G.G. conducted pathological analyses and collected human lesion samples. S.V.R. conducted senescence stains and quantification. J.C. and C.L. generated and characterized cultured primary keratinocytes.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no conflict of interest.
Additional information
Peer review information Nature Communications thanks Adam Glick, Eiji Hara and the other, anonymous, reviewer for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Azazmeh, N., Assouline, B., Winter, E. et al. Chronic expression of p16INK4a in the epidermis induces Wnt-mediated hyperplasia and promotes tumor initiation. Nat Commun 11, 2711 (2020). https://doi.org/10.1038/s41467-020-16475-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16475-3
This article is cited by
-
RSK3 switches cell fate: from stress-induced senescence to malignant progression
Journal of Experimental & Clinical Cancer Research (2023)
-
Targeting chemoresistant senescent pancreatic cancer cells improves conventional treatment efficacy
Molecular Biomedicine (2023)
-
Cellular senescence and frailty: a comprehensive insight into the causal links
GeroScience (2023)
-
Nociceptin/orphanin FQ opioid receptor (NOP) selective ligand MCOPPB links anxiolytic and senolytic effects
GeroScience (2022)
-
Algorithmic assessment of cellular senescence in experimental and clinical specimens
Nature Protocols (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
