
Abstract
It is unclear why some tissues are refractory to the mitogenic effects of the oncogene Myc. Here we show that Myc activation induces rapid transcriptional responses followed by proliferation in some, but not all, organs. Despite such disparities in proliferative response, Myc is bound to DNA at open elements in responsive (liver) and non-responsive (heart) tissues, but fails to induce a robust transcriptional and proliferative response in the heart. Using heart as an exemplar of a non-responsive tissue, we show that Myc-driven transcription is re-engaged in mature cardiomyocytes by elevating levels of the positive transcription elongation factor (P-TEFb), instating a large proliferative response. Hence, P-TEFb activity is a key limiting determinant of whether the heart is permissive for Myc transcriptional activation. These data provide a greater understanding of how Myc transcriptional activity is determined and indicate modification of P-TEFb levels could be utilised to drive regeneration of adult cardiomyocytes for the treatment of heart myopathies.
Similar content being viewed by others


Introduction
Myc is a basic helix-loop-helix–leucine zipper (bHLH–LZ) transcription factor that binds preferentially to specific sequences in the genome, termed E-boxes1, via association with its bHLH–LZ heterodimerisation partner Max2,3,4. Myc functions principally as a transcriptional activator by potentiating transcription initiation via association with various cofactors, such as TRRAP and associated histone acetyl-transferases5,6,7 and facilitating productive transcriptional elongation by promoting RNA polymerase II (PolII) loading and via its association with the positive transcription elongation factor (P-TEFb)8,9,10,11. P-TEFb is comprised of CDK9 and Cyclin T1 that are stringently regulated by various transcriptional and post-transcriptional mechanisms12,13,14,15, and dynamically controlled by an association with an inactivation complex comprised of 7SK snRNA, Larp7, MEPCE and HEXIM16,17,18. P-TEFb phosphorylates DSIF, NELF and serine 2 of the C-terminal domain (CTD) of paused RNA PolII leading to productive elongation12,13,14,15.
Myc is a highly pleiotropic proto oncogene that coordinates multiple transcriptional programmes underpinning normal cell replication, differentiation, metabolism and apoptosis2,19,20,21,22. Together with the dedicated role in controlling the growth and division of normal cells, Myc also governs diverse cell extrinsic processes required for tissue regeneration in a manner that is tightly tailored to the tissue, in which Myc is activated23,24,25. Since deregulated and elevated Myc expression is a pervasive and causal attribute of most, perhaps all, tumours, understanding how tissue-specific responses to Myc are determined at a molecular level is imperative.
Comprehensive analysis of Myc transcriptional output in individual cell types indicates that Myc regulates the expression of thousands of genes, perhaps a third of the transcriptome26,27,28,29,30,31,32. Such studies show great diversity across experimental platforms29,32 and hint that components of the transcriptional repertoire of Myc are highly context specific. In particular, genome-wide analysis of Myc occupancy indicates the presence of Myc on virtually all promoters with open chromatin26,27,30,33,34, suggesting that tissue-specific variations in Myc activity arise by engagement of specific, pre-configured resident cellular transcriptional programmes. However, this large and diverse repertoire of potential Myc target genes, and the lack of comparative analysis of transcriptional responses to Myc in different tissues28,29,30, have together confounded reliable identification of common and tissue-specific Myc-dependent transcriptional programmes. Moreover, recruitment of Myc to a given gene does not always correlate with its level of transcription29,32, and binding efficiency and transcriptional outputs are influenced significantly by different levels of Myc expression31,35,36. Consequently, both the precise mechanism as to how Myc targets genes and what factors govern the transcriptional consequence of Myc binding in differing tissue types remains unclear.
We show that Myc transcription is dependent on the level of P-TEFb within a cell. Overexpression of both Myc and Cyclin T1 in the normally non-responsive heart facilitates Myc-driven transcription and proliferation.
Results
Myc-driven proliferation is tissue restricted
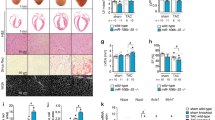
To compare the responses of different tissues to the acute activation of a similar level of Myc, we generated a mouse strain in which supraphysiological levels of the switchable Myc protein c-MycERT2 are ubiquitously expressed from a common promoter at comparable levels across tissues. In this knock-in mouse strain, the R26LSL-c-MycER locus (R26MER)36 is modified to include a CAG (chicken beta actin/CMV) enhancer that augments MycERT2 expression (Supplementary Fig. 1a). R26CAG-LSL-c-MycERT2 mice were crossed into the Tg(Zp3-cre)93Knw strain, in which Cre is active in the oocyte, efficiently excising the LSL stop cassette in all adult tissues of the resultant R26CMER mice37. Compared with the physiological level of Myc expressed in R26MER/+ cells (heterozygous for the R26MER allele)36, R26CMER/+ cells express around eightfold higher levels of MycERT2 RNA (Supplementary Fig. 1b) in each tissue tested. R26CAG-LSL-MER and R26LSL-MER mice were interbred to generate an allelic series of ascending levels of MycERT2 expression (R26+/+, R26LSL-MER/+, R26LSL-MER/LSL-MER, R26CAG-LSL-MER/+, R26CAG-LSL-MER/LSL-MER and R26CAG-LSL-MER/CAG-LSL-MER). To measure directly relative levels of MycERT2 in this allelic series, mouse embryonic fibroblasts (MEFs) were isolated from each strain and the LSL stop cassette excised by infection in vitro with a Cre-expressing adenovirus to engage constitutive MycERT2 expression. Western blot analysis of cell lysates confirmed an ascending allelic series of MycERT2 expression, with homozygous R26MER and R26CMER cells expressing twice the level of their respective heterozygous counterparts (Fig. 1a). This allelic expression series was precisely mirrored in tissues from heterozygous and homozygous R26CMER mice (Supplementary Fig. 1c).

a Immunoblot analysis of MycERT2 and endogenous c-Myc protein levels in wild-type (R26+/+) mouse embryonic fibroblasts (MEFs) maintained in serum-deprived media, and at the indicated type points (in hours) post addition of serum, all compared with asynchronous R26+/+, R26MER/+, R26MER/MER, R26CMER/+, R26MER/CMER and R26CMER/CMER MEFs. Expression of actin is included as a loading control. Image represents the results from six individual mice. b Immunohistochemical and immunofluorescence staining of Ki67 and BrdU in the brain, heart, kidney, lung, pancreas, liver, spleen and thymus isolated from wild-type (R26+/+) and R26CMER/+ mice 24 h post administration of tamoxifen. Representative images based on analysis of five independent mice. Scale bar represents 50 μm. c Quantification of p-H3-positive nuclei percentage from brain, heart, kidney, lung, pancreas, liver (hepatocytes), spleen (red pulp) and thymus isolated from oil-treated R26CMER/+ (n = 5 for heart, liver, lung, kidney, spleen and pancreas, and n = 3 for brain) mice or wild-type (R26+/+, n = 5 for heart, liver, lung, kidney and spleen, n = 4 for pancreas, and n = 3 for brain and thymus) and R26CMER/+ (n = 3) mice 24 h post administration of tamoxifen. Mean of five images per mouse; mean and s.e.m shown. d Immunoblot analysis of MycERT2 and endogenous c-Myc expression in the brain, heart, kidney, lung, pancreas, liver, spleen and thymus isolated from R26CMER/+ mice. Sample loading was normalised for equal protein content, as determined by a bicinchoninic acid assay. Expression of GAPDH is included as a confirmation of efficient protein isolation. Representative results based on analysis of four independent mice.
To rule out the possibility that high levels of Myc might modulate the Rosa26 promoter—and hence elicit artefactual feedback effects—we crossed R26CMER mice to mice carrying a R26mTmG (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo) reporter allele38. Systemic activation of MycERT2 in R26CMER/mTmG mice had no impact on expression of either Tomato or MycERT2 transcripts in any tested tissues (Supplementary Fig. 1d). Hence, elevated MycERT2 activity does not modulate activity of the Rosa26 promoter.
We next determined whether acute activation of MycERT2 elicits a proliferative response in tissues of R26CMER/+ mice. MycERT2 was activated for 24 h by systemic administration of tamoxifen39 and proliferation assessed by immunohistochemical staining of Ki67, bromo-2′-deoxyuridine (BrdU) and the mitotic marker phospho-histone H3 (p-H3). We observed a consistent pattern of proliferative responses to supraphysiological Myc in tissues (Fig. 1b, c) that fell into three general classes: (1) adult tissues, such as liver, lung and pancreas with normally low levels of endogenous Myc (Fig. 1d), but capable of significant regeneration after injury. Such tissues showed a marked induction of proliferation upon Myc activation. (2) Adult tissues, such as kidney, heart and brain, with normally low levels of endogenous Myc and a limited capacity to regenerate. In these, Myc elicited only a negligible rise in proliferation. (3) Tissues with constitutively high proliferative rates and substantial constitutive levels of endogenous Myc (Fig. 1d), such as thymus and spleen, in which activation of ectopic Myc elicited no significant additional proliferation above the already high basal level. Neither sustaining MycERT2 activation for 3 days nor increasing the expression level of MycERT2 from one to two copies of R26CMER changed the proliferative responses to Myc in any tissue (Supplementary Fig. 1e–h). Hence, different tissues harbour different inherent sensitivities to Myc-driven proliferation.
Myc binding does not correlate with efficient transcription
There are several plausible explanations for the failure of acute Myc activation to induce a proliferative response in tissues, such as heart and brain. First, non-responsive tissues might express high levels of one or more of the Mxd proteins, which could directly antagonise Myc’s transcriptional function by sequestering Max and/or competing for its binding to shared target genes. To investigate this possibility, we determined the levels of Mxd expression in various tissues. Although expression of Mxd family members varied across tissues (Supplementary Fig. 2a), we saw no correlation between the extent of Myc-induced proliferation and the levels of Mxd transcripts. Next, we determined whether Myc is still able to bind its target genes using ChIP-sequencing analysis of Myc binding sites 4 h after activation in exemplar responsive (liver) and non-responsive (heart) tissue. We observed a dramatic increase in gene occupancy upon MycERT2 activation in both tissues, from a few hundred sites in wild-type control, reflecting the low levels of endogenous Myc expression in these tissues, to ~30,000 after Myc activation (Fig. 2a). Hence, the failure of the heart to proliferate in response to Myc activation cannot be attributed to the inability of ectopic Myc to access its target genes. Around a quarter of Myc target binding sites were common to both tissues, with a significant overlap principally at promoter elements (Fig. 2b, c). Motif analysis demonstrated significant enrichment for the E-box consensus sequences at these regions (Fig. 2d) and gene ontology indicated that these overlapping promoter binding sites located to ‘classic’ core Myc-dependent genes implicated in control of gene expression, mRNA processing and mitotic cell cycle (Fig. 2d). In addition to these shared targets, however, Myc also bound discrete sets of promoter elements that differed between the two tissues (Fig. 2b). These tissue-specific sites were also enriched for E-box consensus sequences but demonstrated a generally lower affinity for Myc in comparison to the shared core elements (Fig. 2e, f). Interestingly, these tissue-specific promoter elements were associated with genes whose expression defines each tissue (i.e., hepatocytes vs cardiomyocytes respectively—Fig. 2e, f), consistent with the idea that Myc binds already active/open promoters26,27,30,32,33,34. Myc ChIP-seq profiles, RNA PolII ChIP-seq in normal liver and heart, ATAC-seq in purified cardiomyocytes40, publicly available data for chromatin marks of active transcription (H3K27 acetylation H3K4 tri-methylation and H3K4 mono-methylation) and DNAse-seq in heart and liver41 all concurred that Myc binding closely overlaps with transcriptionally active promoter and distal elements in each tissue. This holds true across both common and tissue-specific Myc-bound sites. For example, promoters bound in the heart but not liver are characterised by absence of active chromatin marks and RNA PolII binding in the liver. Conversely, promoters bound by Myc only in liver lack active chromatin marks or associated RNA PolII in heart (Fig. 3a, b, Supplementary Fig. 3a, b). To confirm Myc binding to genes implicated in the transcriptional control of cell cycle in the heart, liver and purified cardiomyocytes, we performed sub analysis of Myc binding and open chromatin marks in a mitotic cell cycle gene set (GO: 0000278). Over 60% of these sites were marked as open and Myc bound (Supplementary Fig. 3c, d). These data confirm that Myc binds “open” chromatin at both promoters and distal sites (enhancers), and that the disposition of chromatin accessibility to Myc is pre-configured in a tissue-specific way. Together, these data indicate that adult terminally differentiated heart retains an open chromatin architecture at sites required for Myc to drive transcriptional control of proliferation and that the regenerative capacity of the heart is lost due to the transcriptional control rather than an epigenetic block.

a The number of Myc peaks and their location (promoter, intragenic and intergenic). Replicates are derived from independent mice. b Venn diagram of the overlap of peaks called within promoter regions (−2 kb to +1 kb from the nearest TSS). c Venn diagram of the overlap of peaks called within distal elements. d Motif probability curves for common Myc-bound promoters show the probability of an E-box consensus sequence occurring at a given position relative to the Myc ChIP peak at each common promoter site, as determined by CentriMo (top left). Average read count of Myc peaks at common promoters containing 0, 1 or >2 E-box motifs within 1000 bp from the peak centre in liver and heart chromatin (bottom left). Selected significant GO Biological Process Gene sets that overlap with common Myc-bound promoter elements (right). e Motif probability curves for liver-specific promoters show the probability of an E-box consensus sequence occurring at a given position relative to the Myc ChIP peak at each liver-specific promoter site, as determined by CentriMo (left). Average read count of Myc peaks shown at common (black) and liver-specific (green) promoter sites (centre). Selected significant Mouse Gene Atlas Gene sets are shown that overlap with liver-specific Myc-bound promoter elements (right). f As for e but for heart-specific Myc-bound promoters bound by Myc only in the heart. Significant Mouse Gene Atlas Gene sets are shown that overlap with heart-specific Myc-bound promoter elements (right). All analysis determined by Myc ChIP sequencing performed from hearts and livers harvested from wild-type (R26+/+ n = 2) and R26CMER/+ (n = 2) mice at 4 h post administration of 4-OHT. MemeSuite Centrimo computed P-values are derived by a binomial test. Enrichr computed P-value (Fisher’s exact test), Z-score (modified Fisher’s exact test to determine deviation from an expected rank) and combined score (C=ln(P) × Z) are shown. Source data are provided as a Source Data file.

a Heatmap of peaks called by DNAse treated, acetylated H3K27 (H3K27ac), tri-methylated H3K4 (H3K4me3) and RNA polymerase II (PolII) ChIP sequencing at Myc-bound promoter elements that are common between both the liver and heart (common-black), or specific for an individual tissue (liver specific—green, heart specific—red). Myc ChIP sequencing was performed on the heart and livers isolated from R26CMER/+ mice 4 h post administration of 4-OHT, overlap of n = 2. PolII ChIP sequencing performed on the heart and livers isolated from wild-type (R26+/+) mice, overlap of n = 2. ChIP sequencing data for DNase treated, H3K27ac and H3K4me3 were taken from the ENCODE Project (accession numbers; GSM1014166, GSM1000093, GSM769017, GSM1014195, GSM1000140 and GSM769014). b As in a but for distal elements.
Since Myc efficiently binds target gene DNA in both heart and liver, we investigated other potential mechanism(s) that might limit proliferative output in the non-responsive heart. We first determined the overall transcriptional output in various tissues (heart, kidney, liver and lung) of R26CMER/+ and R26+/+ control mice, following acute Myc activation for 4 h. Differentially expressed genes (DEGs) were called in each tissue based on the fold change (FC) in mRNA levels between R26CMER/+ and R26+/+ control mice (Log2FC ≥ 0.5, q ≤ 0.05). In agreement with previous observations28,30,31,32, the total number of DEGs in each tissue (Fig. 4a, Supplementary Data 1) was much lower than the number of Myc-bound promoters, albeit with significant inclusive overlap (Fig. 4b). Comparison of DEGs in the liver, lung, kidney and heart indicated considerable commonality among up-regulated genes (Fig. 4c) implicated in prototypical core Myc-regulated processes, such as ncRNA, rRNA and tRNA, metabolic processes (Fig. 4d). By contrast, Myc-repressed genes showed very little overlap across tissues, although gene ontology analysis did suggest that genes overlapping in two or more organs favoured involvement in negative regulation of cell movement, locomotion and migration (Fig. 4e, f). Tissue-specific genes down-regulated by Myc showed significant overlap with genes normally expressed within that tissue, (Fig. 4g, Supplementary Data 2) and their functions were generally associated with differentiated tissue-specific processes (Fig. 4h, Supplementary Data 2).

a The number of differentially expressed genes (DEGs) between organs. b Venn diagram depicting overlap of peaks within promoter regions identified by c-Myc ChIP sequencing from heart (red) or liver (green) isolated from R26CMER/+ mice together with genes differentially expressed in response to Myc (UP DEGs—black, DOWN DEGs—grey). Areas are proportional to the numbers of promoters or DEGs within each cohort. c Venn diagram depicting numbers of genes showing increased expression in response to Myc (UP DEGs) in the kidney (yellow), liver (green), lung (blue) and heart (red); (FDR < 0.05 and abs(log2FC) > 0.5). d Six most significant GO Biological Process Gene sets that overlap with listed genes, showing a differential increase in expression (UP DEGs) in two or more tissues (kidney, liver, lung and heart). e Venn diagram depicting numbers of genes showing decreased expression in response to Myc (DOWN DEGs) in the kidney (yellow), liver (green), lung (blue) and heart (red). f Six most significant GO Biological Process Gene sets that overlap with listed genes that show a differential decrease in expression (DOWN DEGs) in two or more tissues (kidney, liver, lung and heart). g Three most significant Mouse Gene Atlas Gene sets that overlap with listed genes that show a differential decrease in expression (DOWN DEGs) only in the liver (liver specific). h Six most significant GO Biological Process Gene sets that overlap with listed genes that show a differential decrease in expression (DOWN DEGs) only in the liver (liver specific, absent in kidney, heart and lung). i Heatmap showing the union of DEGs called in each tissue; liver, lung, kidney and heart. Shown are mRNA expression fold changes (Log2) upon MycERT2 activation relative to wild type. j Hearts isolated from R26CMER/+ mice show enrichment of common Myc targets (exclusive to the liver, lung and kidney) in comparison to wild type (R26+/+). All analysis determined by RNA sequencing performed in the heart (n = 5), kidney, lung and liver (n = 3) of R26CMER/+ compared to wild-type (R26+/+) mice at 4 h post administration of 4-OHT. Enrichr computed P-value (Fisher’s exact test), Z-score (modified Fisher’s exact) and combined score (C = ln(P) × Z) are shown.
Remarkably, DEG numbers also correlated well with each tissue’s proliferative response to MycERT2 (Figs. 4a and 1b, c): tissues with the lowest overall transcriptional changes displayed the least proliferative responses. However, while activation of ectopic Myc induced significant transcriptional changes in a greater number of target genes in liver than in heart, quantitative analysis of the RNA showed that most of the mRNAs that were induced in liver also exhibited modest, if non-significant, responses in the heart (Fig. 4i). Indeed, gene-set enrichment analysis in the heart confirmed a significant trend in the increased expression of common Myc targets, even when defined as targets commonly up-regulated in response to Myc in all tissues, excluding the heart (Fig. 4j). Hence, the transcriptional response to activation of ectopic Myc in the heart is not absent but highly attenuated and below that required to initiate cell proliferation. This refractoriness of the heart transcriptome to Myc remained evident even after prolonged (24 h) Myc activation or in response to very high Myc levels (homozygous R26CMER mice; Supplementary Fig. 4a, b).
Myc transcriptional output is limited by P-TEFb
The most plausible explanation for our data is that the attenuated transcriptional and biological response to Myc in heart is due to either active transcriptional inhibition and/or insufficiency of requisite transcriptional cofactors or machinery. Consistent with this, both the total and phosphorylated levels of the CTD of RNA PolII (Rpb1) were much lower in non-responsive heart and kidney than in Myc-responsive tissues, such as the liver and lung (Fig. 5a). Since recruitment of P-TEFb is necessary for transcriptional elongation8,9,10, we next assessed the expression levels of the components of P-TEFb–CDK9 and Cyclin T1. Surprisingly, both CDK9 and Cyclin T1 proteins were present at considerably lower levels in the heart and kidney compared with lung or liver, along with components of the pause factors NELF and DSIF (Fig. 5a, Supplementary Fig. 5a). Conversely, both Larp7, a negative regulator of P-TEFb, and the 7SK short non-coding RNA that serves as the scaffold for the P-TEFb inactivation complex were relatively more abundant in the heart and kidney (Fig. 5a, Supplementary Fig. 5b). This intimated that the inability of Myc to elicit transcriptional changes in the heart arises from a tissue-specific deficiency in core transcriptional cofactors, notably P-TEFb and RNA PolII itself, that are required for efficient transcriptional initiation and elongation. Since paused RNA PolII is dependent upon P-TEFb for progression42, we directly tested whether transcriptional elongation is generally rate limiting for Myc-driven transcription using the specific CDK9 inhibitor AZ5576 (AstraZeneca) to inhibit P-TEFb in liver. Wild-type and R26CMER/+ mice were concurrently treated with 4-hydroxytamoxifen (4-OHT; to activate MycERT2) and AZ5576, and livers collected 4 h later. AZ5576 effectively inhibited CDK9 activity, as attested by a decrease in phosphorylated Rpb1 levels (Fig. 5b) and, notwithstanding the presence of active MycERT2, such inhibition of CDK9 significantly attenuated transcription of Smpdl3b, Cad, Gnl3 and Polr3g, genes regulated by Myc in multiple tissues (Fig. 5c). Hence, inhibition of P-TEFb, and consequential decreased phosphorylation of RNA PolII, renders liver non-permissive for Myc response. The same was observed in MEFs (Supplementary Fig. 5c, e). Conversely, ectopic overexpression of Cyclin T1 in adult R26CMER/+ cardiomyocytes in vitro efficiently abrogated their normal refractoriness to Myc and permitted efficient expression of Myc target genes previously shown to be Myc unresponsive in adult heart (Bzw2, Pinx1, Polr3d, St6 and Cdc25a; Fig. 5d, e, Supplementary Fig. 5f, g). Together, these data support the notion that transcriptional activity of Myc is limited by P-TEFb availability. Further experiments would be required to determine the mechanistic basis of this dependency.

a Immunoblot analysis of the C-terminal domain (CTD) of RNA Polymerase II—total (Rpb1) and phosphorylated (p-Rpb1(S5) and p-Rpb1(S2)), CDK9, Cyclin T1 and Larp7 expression in the heart, liver, lung and kidney isolated from wild-type (R26+/+) mice. The composite figure is generated from the same samples loaded across multiple blots and a representative image for GAPDH is shown. b Immunoblot analysis of the CTD of RNA Polymerase II, phosphorylated (p-Rpb1(S2)), and MycERT2 protein expression in wild type (R26+/+) or R26CMER/+ livers, isolated 4 h post administration of 4-OHT either alone (4-OHT) or in combination with 60 mg/kg AZ5576 (4-OHT + AZ5576). c Quantitative RT-PCR analysis of Smpdl3b, Cad, Gnl3 and Polr3g in wild type (R26+/+) and R26CMER/+ livers, isolated 4 h post administration of 4-OHT (n = 6 R26+/+, n = 3 R26CMER/+) either alone or in combination with 60 mg/kg AZ5576 (4-OHT + AZ5576, n = 4). Expression is relative to the respective wild type (R26+/+). Mean and s.d shown. Two-way ANOVA with Tukey’s multiple comparisons test; R26+/+ vs 4-OHT: ***P = 0.001 (Smpdl3b, Cad, Gnl3 and Polr3g), R26CMER/+ 4-OHT vs 4-OHT + AZ5576: ***P = 0.001 (Smpdl3b, Cad and Gnl3), **P ≤ 0.01 (Polr3g). d Immunoblot analysis of Cyclin T1 and phosphorylated CTD of RNA polymerase II (p-Rpb1(S2)) in R26CMER/+ primary cardiomyocytes infected with an adenovirus encoding either GFP (Ad-GFP) or Ccnt1 (Ad-Ccnt1). Replicate samples are derived from independent primary cardiomyocyte isolations. e Quantitative RT-PCR analysis of Cad, Bzw2, Pinx1, Polr3d, St6 and Cdc25a in wild type (R26+/+, n = 5 except Pinx1 where n = 4 for Ad-GFP control) and R26CMER/+ (n = 5 except St6 where n = 4 for Ad-GFP control) primary cardiomyocytes infected with an adenovirus encoding either GFP (Ad-GFP) or Ccnt1 (Ad-Ccnt1), 4 h post addition of 100 nM 4-OHT. Expression is relative to an individual wild type (R26+/+) Ad-GFP control. Mean and s.d shown. One-way ANOVA with Tukey’s multiple comparisons test; Ad-GFP R26+/+ vs R26CMER/+: *P = 0.05 (Cad), Ad-Ccnt1 R26+/+ vs R26CMER/+: *P = 0.05 (Cad, Pinx1, Polr3d and St6) **P = 0.01 (Bzw2 and Cdc25a). Replicate samples are derived from independent primary cardiomyocyte isolations and independent mice. Source data are provided as a Source Data file.
Enabling Myc transcription in heart restores proliferation
Expression of CDK9, Cyclin T1 and RNA PolII all progressively decrease during post-natal cardiac maturation43. We therefore first determined the extent of Myc-dependent transcriptional responsiveness in juvenile heart tissue. Unlike the adult heart, 15-day-old juvenile hearts express levels of P-TEFb, RNA PolII comparable to those in adult liver (Fig. 6a, Supplementary Fig. 6a, b). Acute (4 h) MycERT2 activation in R26CMER/+ juvenile heart induced expression of a panel of common Myc transcriptional target genes (Bzw2, Pinx1, Polr3d, St6 and Cdc25a) normally up-regulated by Myc in the adult liver but not in adult heart (Fig. 6b). RNA sequencing indicated a proficient global transcriptional response in juvenile heart (1601 DEGs up-regulated and 1133 down-regulated; Fig. 6c, Supplementary Data 3), with significant enrichment for common Myc targets (Supplementary Fig. 6c), defined from the overlap of genes up-regulated in response to the activation of Myc in all four tissues (Fig. 4c). Following 24 h of tamoxifen treatment, juvenile R26CMER/+ mice heart exhibited a marked proliferative response as indicated by substantial induction of both G1 (Cdk4) and G2 (Cdk1/Ccnb1) cell cycle genes (Supplementary Fig. 6d), enrichment of pro-cell cycle progression gene sets (Supplementary Fig. 6e) and significant levels of mitosis (Fig. 6d). The extent of Myc-induced cardiomyocyte proliferation was comparable to that observed in tamoxifen-activated R26CMER/+ adult liver (Fig. 6d). To confirm that Myc was acting to induce proliferation directly in cardiomyocytes themselves, we restricted expression of MycERT2 to cardiomyocytes using the Myh6Cre allele (Tg(Myh6-cre)2182Mds/J44, in which Cre recombinase is driven from the Myh6 promoter that directs expression of the Myosin heavy chain α isoform solely in cardiomyocytes. Myh6Cre; R26LSL-CMER/+ mice were generated and at 15 days of age given a single bolus of tamoxifen to activate MycERT2. Within 48 h, Myh6Cre; R26LSL-CMER/+ hearts displayed both p-H3 and Ki67 positivity specifically in cardiac troponin T and PCM1-positive cardiomyocytes, displayed aurora B kinase positivity at centrally located mid-bodies (Fig. 7a, Supplementary Fig. 7a) and had doubled in size compared with R26LSL-CMER/+ controls (Fig. 7b, c). Overt cardiomyocyte division was evident from a 1.8-fold increase in cardiomyocyte number without any discernible change in cardiomyocyte size (Fig. 7c, d, Supplementary Fig. 7b–d). To further confirm cytokinesis, transmission electron micrographs (TEM) from Myh6Cre; R26LSL-CMER/+mice 48 h post tamoxifen treatment were generated and 3D rendering used to identify cardiomyocytes in mitosis and cytokinesis (Fig. 7e, Supplementary Movies 1 and 2). Over the longer term, transient elevated MycERT2 activity in 15-day-old Myh6Cre; R26LSL-CMER/+ mice caused a large increase in heart size and increased p-H3 immunoreactivity. Furthermore, a large proportion of cardiomyocytes displayed disassembled sarcomeres and mice did not survive beyond 4 days post MycERT2 activation (Supplemental Fig. 7e).

a Immunoblot analysis of the C-terminal domain of RNA polymerase II, total (Rpb1) and phosphorylated (p-Rpb1(S2)), Cyclin T1 and CDK9 protein expression in wild-type heart and liver isolated from 15-day-old (15d) and 60-day-old (60d) mice. Replicate samples are derived from independent mice. b Quantitative RT-PCR analysis of Cad, Bzw2, Pinx1, Polr3d, St6 and Cdc25a expression in wild-type (R26+/+) and R26CMER/+ heart isolated from 15-day-old (15d, Bzw2 and Cad n = 6, Cdc25a, Pinx1, Polr3d and St6 n = 7) mice vs heart (Bzw2, R26+/+ Pinx1 and R26CMER/+ Cad n = 9, Cdc25a, Polr2d, St6, R26+/+ Cad and R26CMER/+Pinx1 n = 8, R26CMER/+ St6 n = 7, R26CMER/+ Cdc25a, Polr3d n = 6) and liver (R26+/+ Bzw2, Pinx1, Cad, Cdc25a and St6 n = 9, R26+/+ Polr2d n = 8, R26CMER/+ Cad n = 10, R26CMER/+ Bzw2 n = 8, R26CMER/+ St6 n = 7, R26CMER/+ Cdc25a, Pinx1, Polr3d n = 6) isolated from 60-day-old (60d) mice 4 h post administration of 4-OHT. Expression is relative to the respective wild type (R26+/+). Mean and s.d shown. One-way ANOVA with Tukey’s multiple comparisons test; 15 day R26+/+ 4-OHT vs R26CMER/+ 4-OHT: ***P = 0.001 (Cad, Bzw2, Pinx1, St6 and Cdc25a), 15 day R26+/+ 4-OHT vs R26CMER/+ 4-OHT: **P = 0.001 (Polr3d), adult heart R26+/+ 4-OHT vs R26CMER/+ 4-OHT: ***P = 0.001 (Cad), adult liver R26+/+ 4-OHT vs R26CMER/+ 4-OHT: ***P = 0.001 (Cad, Bzw2, Pinx1, St6 and Cdc25a). Replicate samples are derived from independent mice. c Heatmap showing the union of DEGs called in each tissue; adult liver (n = 3), adult heart (n = 5) and 15-day-old (15d) heart. mRNA expression fold changes (Log2) upon MycER activation are shown relative to wild type, as determined by RNA sequencing of R26CMER/+ (n = 3) tissues relative to wild type (R26+/+, n = 3) at 4 h post administration of 4-OHT. d Quantification of p-H3-positive nuclei percentage in heart (positive cardiomyocyte nuclei only) and liver (positive hepatocyte nuclei only) isolated from 15-day-old (15d) and 60-day-old (60d) wild-type (R26+/+, n = 10 15d, n = 8 60d) and R26CMER/+ (n = 4) mice 24 h post administration of tamoxifen. Means are taken from five images per mouse; mean and s.d. shown. One-way ANOVA with Tukey’s multiple comparisons test; R26+/+ 4-OHT vs R26CMER/+ 4-OHT: ***P = 0.001 (15-day heart and adult liver). Source data are provided as a Source Data file.

a Immunofluorescent staining of cardiac troponin, p-H3, Ki67, PCM1, wheat germ agglutinin and aurora B-positive mid-body in the heart of 15-day-old control (no cre) and Myh6-Cre; R26LSL-CMER/+ (cre+) mice. Representative images based on analysis of five independent mice. Scale bar represents 10 μm. b Image of the whole heart and a tibia isolated from a 15-day-old control (no cre) and Myh6-Cre; R26LSL-CMER/+ (cre+) mice. Scale bar represents 10 mm. Representative images based on analysis on 21 independent mice. c The weight (mg) of hearts (left, R26LSL-CMER/+; no cre, vehicle = 9, no cre tamoxifen = 11; R26LSL-CMER/+; cre+ vehicle n = 7, cre+ tamoxifen n = 10) and quantification of the number of cardiomyocytes (right) isolated from 15-day-old control (no cre, vehicle = 3, no cre tamoxifen = 4) and Myh6-Cre;R26LSL-CMER/+ (cre+ vehicle n = 6, cre+ tamoxifen n = 7) mice post administration of tamoxifen or oil (veh), expressed as fold change over the length (mm) of a tibia isolated from the same mouse. Mean and s.e.m. shown. One-way ANOVA with Tukey’s multiple comparisons test; vehicle cre vs tamoxifen cre+, vehicle no cre vs tamoxifen cre+, tamoxifen no cre vs tamoxifen cre+ ***P = 0.001 (weight). Vehicle cre vs tamoxifen cre+ **P = 0.01, vehicle no cre vs tamoxifen cre+, tamoxifen no cre vs tamoxifen cre+ ***P = 0.001 (cardiomyocyte number). Replicate samples are derived from independent mice. d Flow cytometry quantification of p-H3 in cardiac troponin T-positive cardiomyocytes (left) and immunofluorescent staining (right) of cardiac troponin T (green), p-H3 (red) and DNA (blue) from 15-day-old control (no cre, n = 4) and Myh6-Cre; R26LSL-CMER/+ (cre+, n = 7) mice. Mean and s.d. shown. Unpaired t-test; no cre vs cre+ ***P = 0.0006. Replicate samples are derived from independent mice. Scale bar represents 50 μm. e Three-dimensional render (second from left) generated from 500 image z-stack (50 µm) of transmission electron micrographs from a Myh6-Cre; R26LSL-CMER/+ heart. Green shows outline of cells, blue shows nuclei, pink line shows site of cross section through cells, outlined in green (second from right) and expanded (right). Imaging performed on a single heart. Left image shows mitotic nuclei, arrow. Structures are indicated; f, myofibrils; n, nucleus; m, mitochondria; bv, blood vessel. Scale bars represent 5 μm. All analysis 48 h post administration of tamoxifen. Source data are provided as a Source Data file.
To directly test if increasing P-TEFb levels within cardiomyocytes of the adult heart in vivo is permissive for efficient Myc transcriptional activation and proliferation, we infected wild type, R26CMER/+, Myh6Cre; R26LSL-CMER/+ and R26LSL-CMER/+ mice with an adeno-associated virus directing ectopic expression of Cyclin T1 (encoded by Ccnt1) or RFP under the troponin T promoter specifically in cardiomyocytes. After 4 weeks, we confirmed viral Cyclin T1 overexpression in 34% of cardiomyocytes by immunohistochemistry (Fig. 8a). As described previously43,45 and confirmed here, elevated expression of Cyclin T1 in cardiomyocytes led to an increase in CDK9 and phosphorylated RNA PolII (S2; Fig. 8b). Acute MycER activation in the presence of overexpressed Cyclin T1 induced the transcription of a panel of common Myc target genes normally up-regulated by Myc in the liver but not in the adult heart (Fig. 8c, Supplementary Fig. 8a). RNA sequencing indicated a proficient global transcriptional response to the activation of Myc in the presence of ectopic Cyclin T1 in the adult heart or purified cardiomyocytes (Supplementary Data 4 and 5), with positive enrichment for common Myc targets and pro-cell cycle progression gene sets (Supplementary Fig. 8b). Positive enrichment of common Myc targets was also observed in cardiomyocytes purified from Ccnt1-infected Myh6Cre; R26LSL-CMER/+ hearts vs uninfected Myh6Cre; R26LSL-CMER/+ hearts 4 h post activation of Myc (Fig. 8d). A short pulse of MycERT2 activation in combination with Cyclin T1 overexpression resulted in a marked proliferative response in hearts collected at 48 h post a single dose of tamoxifen, cardiomyocytes displaying markers of cell cycle, mitosis, cytokinesis (Fig. 9a, b, Supplementary Fig. 9a, b), expression of mitosis and cytokinesis gene sets (Supplementary Fig. 9c, Supplementary Data 6), increased heart size (Fig. 9c, d, Supplementary Fig. 9d). At 72 h post MycERT2 activation in combination with Cyclin T1 overexpression heart size was further increased along with increased cardiomyocyte number, but without any measurable change in cardiomyocyte size (Fig. 9c–e) or nucleation (Supplementary Fig. 9e). A single transient burst of MycERT2 activity in Cyclin T1 expressing Myh6Cre; R26LSL-CMER/+ adult cardiomyocytes was compatible with long-term survival, with no histological abnormalities detected at 28 days after MycERT2 activation (Supplementary Fig. 9f). Collectively, these data illustrate that Myc transcriptional activity can be facilitated in adult cardiomyoctes by co-activation of the transcriptional co-factor P-TEFb, thus enabling adult heart tissue to re-enter the cell cycle.

a Immunohistochemistry (left) and quantification (right) of Cyclin T1 in the liver isolated from adult R26CMER/+ or heart isolated from Myh6-Cre; R26LSL-CMER/+ mice systemically infected with adeno-associated virus (serotype 9, 1 × 1011 vg/mouse) encoding either RFP (AAV9-RFP, n = 5) or Ccnt1 (AAV9-Ccnt1, n = 4) 4 weeks post infection. Mean and s.e.m shown. Unpaired t-test; AAV9-RFP vs AAV9-Ccnt1: ***P < 0.0001. Representative images based on analysis of five independent mice. b Immunoblot analysis of the C-terminal domain of phosphorylated RNA polymerase II (p-Rpb1(S2)), CDK9 and Cyclin T1 expression in the heart isolated from adult Myh6-Cre; R26LSL-CMER/+ mice systemically infected with adeno-associated virus encoding either RFP (AAV9-RFP) or Ccnt1 (AAV9-Ccnt1) 4 weeks post infection. Replicate samples are derived from independent mice. c Quantitative RT-PCR analysis of Cad, Bzw2, Pinx1, Polr3d, St6 and Cdc25a expression in control (R26LSL-CMER/+; no cre, n = 5) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+, n = 7) adult mouse heart isolated 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 4 h post administration of 4-OHT. Expression is relative to the respective wild type (R26LSL-CMER/+). Mean and s.e.m shown. Unpaired t-test; no cre vs cre+: ***P < 0.0005 (Cad, Bzw2, Pinx1 and St6), **P = 0.0013 (Cdc25a), **P = 0.0035 (Polr3d). Replicate samples are derived from independent mice. d Enrichment of common Myc targets (liver, lung, kidney and heart) in cardiomyocytes isolated from the heart of AAV-CCNT1 infected Myh6-cre; R26LSL-CMER/+ (Myc+Cyclin T1) mice in comparison to uninfected Myh6-cre; R26LSL-CMER/+ mice (Myc) at 4 h post administration of 4-OHT, as determined by RNA sequencing. Including normalised enrichment score (NES) and FDR q-value (FDR).

a Immunofluorescent staining of cardiac troponin, p-H3,Ki67, PCM1, wheat germ agglutinin (WGA) and aurora B in control (R26LSL-CMER/+; no cre) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+) adult mouse heart isolated 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 48 h post administration of tamoxifen. Representative images based on analysis of five independent mice. Scale bars represents 10 μm (aurora B) and 50 μm (all others). b Quantification of p-H3-positive nuclei percentage in control (R26LSL-CMER/+; no cre, n = 9) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+, n = 7) adult mouse heart isolated 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 48 h post administration of tamoxifen. Means are taken from five images per mouse; Mean and s.e.m shown. Unpaired t-test; no cre vs cre+ P < 0.0001. c The weight (mg) of hearts isolated from control (R26LSL-CMER/+; no cre, n = 9 48 h, n = 5 72 h) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+, n = 7 48 h, n = 6 72 h) adult mouse heart 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 48 and 72 h post administration of tamoxifen, expressed as fold change over the length (mm) of a tibia isolated from the same mouse. Mean and s.e.m shown. One-way ANOVA with Tukey’s multiple comparisons test; no cre vs cre+: *P = 0.05 (48 h), ***P = 0.001 (72 h). Replicate samples are derived from independent mice. d Image of the whole heart and a tibia from control (R26LSL-CMER/+; no cre) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+) adult mouse heart isolated 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 72 h post administration of tamoxifen. Scale bar represents 10 mm. Representative images based on analysis of 11 independent mice. e Quantification of the number and size of cardiomyocytes from control (R26LSL-CMER/+; no cre, n = 5) and Myh6-Cre; R26LSL-CMER/+ (R26LSL-CMER/+; cre+, n = 6) adult mouse heart isolated 4 weeks post systemic infection with an adeno-associated virus encoding Ccnt1 (AAV9-Ccnt1) and 72 h post administration of tamoxifen. Mean and s.d shown. One-way Mann–Whitney test; no cre vs cre+ **P = 0.0043. Replicate samples are derived from independent mice. Source data are provided as a Source Data file.
Discussion
Myc is widely acknowledged to play a pivotal coordinating role in the transcriptional control of somatic cell proliferation and tissue regeneration, and its activity is deregulated and typically elevated in the majority of aggressive cancers2,21,22. Past studies have largely focused on identifying Myc transcriptional programmes that are common across cell types. However, Myc exhibits great cell type variability, both with respect to how efficiently it can drive cell proliferation and the phenotypes its activities elicit in different tissues23,25,46,47. We developed a unique mouse model whereby a switchable variant of Myc is driven at a comparable and defined oncogenic level from an augmented version of the endogenous Rosa26 promoter in target cells. The variant, augmented Rosa26 promoter incorporates an additional CAG (CMV and β-actin promoter elements) that increases the expression level of Myc to supraphysiological levels frequent in many cancers. Unlike most tissue-specific promoters used to drive transgenic oncogenes, neither Rosa26 nor RosaCAG promoters are repressed by Myc activation, obviating a major confounding artefact in most conventional transgenic models. Replacing Myc with the well validated, rapid and reversible switchable 4-OHT-dependent MycERT2 variant allows the analysis of the immediate and direct impact of Myc on its target genome48,49. Inclusion of tissue-specific regulation via a Cre recombinase-excisable transcriptional STOP element provides the option for tissue restricted MycERT2 expression.
Using proliferation as the phenotypic signature of Myc output, we identified three general classes of response to activation of oncogenic levels of Myc: tissues that proliferated in response to MycERT2 activation; tissues that did not; and tissues that exhibited innately high endogenous Myc and proliferative indices. The correlation between proliferative response to ectopic Myc in these tissues (Fig. 1b, c) and the innate regenerative potential of each tissue is striking and suggests an underlying mechanistic connection50.
ChIP-seq analysis 4 h post MycERT2 activation in exemplar Myc-responsive vs non-responsive tissues (respectively, liver and heart) showed a dramatic increase in gene occupancy by Myc in both tissues. Hence, tissue non-responsiveness to Myc is not due to failure of ectopic Myc to access its chromatin targets. On the contrary, we found striking similarities between the numbers of sites to which Myc is bound across the different tissues analysed, underscoring that the phenotypic outcome effected by Myc is not solely dictated at the level of recruitment to chromatin. The significant overlap in Myc-bound promoter regions across both the liver and heart affirms the widely held notion that Myc binds a discrete set of target genes common to multiple cell types and implicated in governing core processes required for cell growth and cell cycle progression. Nonetheless, Myc also binds a set of promoter elements specific to each tissue, indicating that the overall Myc-dependent cistrome is cell type specific. We also note that this tissue-specific Myc binding repertoire is characterised by the presence of chromatin marks indicative of open chromatin, in keeping with the findings of others26,27,30,32,33,34.
Interestingly, as early as 4-h post Myc activation, we clearly see a significant number of genes to be transcriptionally repressed. These repressed genes principally articulate differentiated cell type functions and show very little overlap between tissue types. Moreover, around a third (34%) of the repressed genes appear to be indirect targets, since Myc is not detected at their promoters (Fig. 4b). Suppression of differentiation-specific genes by oncogenic Myc has been widely observed irrespective of cell or tissue type and is an example of the robust inverse generic relationship between cell proliferation and differentiation51.
It has been proposed that stringent normalisation strategies employed in the analysis of bulk RNA sequencing data can mask the more global impact of Myc on transcription that might be predicted from its promiscuous binding to open chromatin26,27. Such general amplification is in no way excluded by our analyses. Nonetheless, it is clear that the magnitude of transcriptional change that the activation of ectopic Myc can instruct is significantly limited in tissues that lack regenerative potential, like the heart.
A simple hypothetical explanation for the pervasive suppression of differentiation by Myc is that certain essential components of the transcriptional machinery, such as P-TEFb or even total levels of Rbp1, available for loading onto promoters are limiting and that repression of differentiation-specific genes is a mere consequence of these limiting factors’ being redeployed to Myc target genes, as recently proposed in 3T3 fibroblasts11. Analogous scarcities in transcriptional machinery could also explain why some tissues fail to respond transcriptionally to Myc. We observed that both total and phosphorylated levels of Rpb1 are significantly lower in the adult heart in comparison to the liver (Supplemental Fig. 6b). However, we presume that the levels of Rpb1 are sufficient to allow the transcription of genes required to maintain heart function. Indeed, when we performed RNA PolII ChIP sequencing in the adult heart, we detected heart-specific genes loaded with significant amounts of RNA PolII. There are several potential mechanisms that may concentrate the available RNA PolII at specific locations. First, it is possible that the generation of “transcriptional hotspots” might concentrate essential factors at the relevant genes52. Second, transcription in the presence of low Rpb1 may be aided by tissue-specific super enhancers that recruits a large portion of the enhancer-associated RNA PolII and its associated cofactors and chromatin regulators53, unfortunately, testing of these hypotheses lies outside the scope of this manuscript. Myc has been shown to modify both RNA PolII loading and its phosphorylation by P-TEFb (refs. 8,9,10,11,12,42,54). We showed that responsiveness of individual tissues to Myc correlates with the levels of expression of the basal transcription factors, P-TEFb and RNA PolII, the pause factors, DSIF and NELF, and, inversely, with the expression of the P-TEFb repression complex.
Adult mammalian heart is a prototypical, terminally differentiated55, Myc non-responsive tissue in which Cyclin T1 is known to be a limiting transcriptional regulator, and whose chronic ectopic overexpression elicits promiscuous RNA polymerase activity and cardiac hypertrophy43,56. While P1 neonatal heart retains proliferative potential, this rapidly declines after birth and is largely lost by P7 (refs. 57,58,59). This decline in neonatal cardiac proliferative potential correlates with down-regulation of multiple genes involved in cell cycle40,55,60,61,62,63, including endogenous Myc, which is high at P1 but significantly reduced by P5 (Supplementary Fig. 6a). Although changes in microRNAs (refs. 59,64), modulation of the Hippo pathway65,66, and wholesale metabolic switching from glycolysis to oxidative phosphorylation67,68 have all been implicated, the mechanism underlying post-natal cardiac maturation and loss of regenerative potential remains unknown. Interestingly, both neonatal mouse heart and the zebrafish larval heart express high levels of P-TEFb. However, whereas the zebrafish adult heart retains 70% of its larval levels of P-TEFb and remains competent to regenerate throughout development, P-TEFb levels in the non-regenerative adult mouse heart drop by 85% (refs. 43,69). Little is known regarding the regulation of P-TEFb during mammalian cardiac development. Levels of Cyclin T1 are regulated at both transcriptional and post-transcriptional levels12,13,14,15, with mitogens and cytokine signalling known to increase Cyclin T1 protein stability70,71. We and others have shown that the level of Cyclin T1 is the key factor regulating the level and activity of P-TEFb within cardiomyocytes43,45. Oncogenic Ras signalling increases Cyclin T1 in the heart, leading to cardiac hypertrophy56. It is tempting to speculate that the noted oncogenic cooperation between Ras and Myc may, in part, arise through Ras-dependent induction of P-TEFb, so promoting the efficiency of Myc-driven transcription.
Re-establishing regenerative proliferation in the adult heart has proven very difficult. Inhibition of Hippo signalling, enforced expression of both G1 (CDK4 and Ccnd1) and G2/M (CDK1 and Ccnb1) factors, and hypoxia all elicit modest regeneration66,72,73,74, but at levels well below those of P1 neonatal heart. While the adult heart is refractory to Myc-induced proliferation, enforced ectopic Myc expression during neonatal life did extend the window of neonatal cardiomyocyte proliferation up to around P15, a period that that coincides with high endogenous cardiac P-TEFb expression. The large increase in cardiomyocyte number and aurora B localisation to centrally located mid-bodies suggests that cardiomyocytes isolated from juvenile mice can progress through the cell cycle and many can complete cytokinesis. Remarkably, we show that an increase in the level of P-TEFb in the adult heart is sufficient to support ectopic Myc-dependent transcription, proliferation and cytokinesis of cardiomyocytes in vivo, resulting in increased heart size and cardiomyocyte number within a very short timeframe. Importantly, this transient wave of proliferation induced in a smaller fraction of total cardiomyocytes than in juvenile mice is compatible with long-term survival. It will be of great interest to determine if this genetic combination will prove beneficial in models of cardiac injury.
Despite the significant changes in regenerative capacity and responsiveness to Myc that attend cardiac development from embryonic stem cells through mesoderm to precursor cardiomyocyte to cardiomyocyte, the chromatin status at promoters for genes involved in basic cellular function, including cell cycle regulation, show very little variation75,76. Similarly, the Myc binding sites of the adult liver (proliferative) and the adult heart (non-proliferative) exhibit significant overlap, indicating that even after its maturation, adult, terminally differentiated heart retains an open chromatin architecture at sites required to drive transcriptional control of proliferation. Hence, if Myc could be induced by mitogenic stimulus in the heart, cardiac epigenome architecture does not preclude heart regenerative capacity: rather, it is the inability of Myc to drive transcriptional output from regenerative target genes (due to lack of the P-TEFb) that thwarts cardiomyocyte proliferation.
Overall, our data indicate that tissue regenerative capacity is tightly linked to the capacity of that tissue to respond to Myc and that tissue Myc responsiveness is governed principally by availability of key components of the core transcriptional machinery, which Myc co-opts to drive its regenerative biological output. However, in addition to its core target genes, which are common across tissues, a significant number of its targets are expressed in a tissue-specific manner. By contrast, tissue-specific accessibility of target genes to Myc appears to be dictated principally by hard-wired, tissue-specific epigenome organisation. Our current study focuses in the main on liver and heart as, respectively, prototypical examples of regenerative and Myc-responsive vs non-regenerative and Myc-unresponsive tissues. However, it seems reasonable that our findings could be extrapolated more generally to offer insights into both regenerative medicine and cancer susceptibility.
Methods
Mice and mouse maintenance
All experimental procedures received ethical approval and were conducted in accordance with the Home Office UK guidelines, under project licences 70/7586, 80/2396 (G.I.E.), which were evaluated and approved by the Animal Welfare and Ethical Review Body at the University of Cambridge. All animals were kept under SPF conditions. Mice were maintained on regular diet in a pathogen-free facility on a 12 h light/dark cycle with continuous access to food and water. Mouse strains, Tg(Zp3-cre)93Knw, Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo and Tg(Myh6-tTA)6Smbf/J were obtained from the Jackson Laboratory. For generation of the R26CAG-LSL -c-MycER mice, MycERT2-IRES-eGFP cDNA was cloned into pBigT, and then into the pROSA26CAG-PAS vector77 to create the final R26CAG-LSL-c-MycER targeting vector. This vector was then linearised and introduced into mouse embryonic stem cells. Germ-line transmission was confirmed by PCR and verified by Southern analysis. Adult animals requiring 4-OHT were either i.p. injected with 1 mg (Z)-4-OHT (Sigma, H7904) in 10% ethanol and vegetable oil (5 mg/ml), and collected 4 h post injection or with 1 mg tamoxifen (Sigma, T5648) in 10% ethanol and vegetable oil (10 mg/ml) twice over a 24-h period and collected at 24–72 h post initial i.p. injection. Adolescent animals were given 0.5 mg of either 4-OHT or tamoxifen per injection. For 48 or 72-h collection points, mice were i.p. injected once with either 0.5 mg (adolescent) or 1 mg (adult) tamoxifen in 10% ethanol and vegetable oil. 5-BrdU (Sigma, B9285) was administered by i.p. injection (100 µl, 10 mg/ml) 2 h before culling. AZ5576 was administered to mice at 60 mg/kg in 20% Captisol® Solution by oral gavage.
Mouse genotyping
Ear biopsies were collected from 2–5-week-old mice and digested overnight at 55 °C in 10% Chelex 100 Resin (Bio-Rad Catalogue #142-1253), 0.1 % Tween-20 and 0.25 mg/ml Proteinase K (Sigma P8044). A typical PCR reaction was performed using GoTaq (Promega) with 1 µl of Chelex extracted gDNA following manufacturers’ instructions. The following PCR conditions were applied: 5 min, 95 °C initial denaturation; 35 cycles of 30 s at 95 °C, 30 s at 60–65 °C and 1.5 min at 72 °C, followed by a final 5 min at 72 °C. PCR amplification products were analysed by agarose gel electrophoresis. General primers for Rosa26CAG were used to detect R26CMER and R26mTmG alleles, were universal forward: 5′-CTCTGCTGCCTCCTGGCTTCT-3′ wild-type reverse: 5′-CGAGGCGGATCACAAGCAATA-3′ and CAG reverse: 5′-TCAATGGGCGGGGGTCGTT-3′. General primers for Rosa26 used to detect the R26MER allele were R26F: 5′-AAAGTCGCTCTGAGTTGTTAT-3′ R523: 5′- GGAGCGGGAGAAATGGATATG-3′ and R1295: 5′-GCGAAGAGTTTGTCCTCAACC-3′. Generic primers to recognise Cre were Cre1: 5′-GCTGTTTCACTGGTTATGCGG -3′ and Cre2: 5′-TTGCCCCTGTTTCACTATCCAG -3′.
Mouse embryonic fibroblasts
MEFs were generated from embryos 13.5 d.p.f. and cultured in DMEM (Thermo Fisher 41966052) supplemented with penicillin–streptomycin (Thermo Fisher, 15140-122), L-glutamine (Thermo Fisher, 25030-024) and 10% BGS (Hyclone SH30541.03HI). All experiments were performed between passage 3 and 5. For experiments, MEFs were plated at 5 × 105 cells per 6 cm culture dish in 10% BGS (where appropriate, plus adenovirus). To render cells quiescent, the next day cells were washed twice with PBS and cultured in media containing 0.1% BGS for 48 h. After 48 h serum deprivation, cells were treated with 100 nM (Z)-4-OHT (Sigma, H7904) in ethanol to activate MycERT2 and 1 µM AZ5576 (AstraZeneca) in DMSO to block CDK9 and RNA, or protein harvested 4 h later or cell cycle analysis performed after 16 h.
Cardiomyocyte purification and culture
Cardiomyocytes CMs were isolated from adult mouse hearts (8–10 weeks of age) using a simplified Langendorff-free method78 and cultured in M199 (Sigma, M4530) supplemented with 0.1% BSA (supplier, number), 1X ITS Liquid Media Supplement (Sigma, I3146), 10 mmol/l BDM (Sigma, B0753), 1X Chemically Defined Lipid Concentrate (Fisher, 11548846) and penicillin–streptomycin (Thermo Fisher, 15140-122). When needed, adenovirus was administered to CMs in culture media immediately after removal of plating media. Medium was then replaced after 24 h with culture media together with 100 nM (Z)-4-OHT (Sigma, H7904) in ethanol and collected after 4 h for RNA extraction.
Adenovirus-mediated infections
Ad5CMVeGFP (VVC-U of Iowa-1174) was obtained from the University of Iowa, Viral Vector Core and supplied at 3 × 101 pfu/ml. For MEFs, 0.5 µl of virus was used per 5 × 105 cells. For CMs, 0.25 µl of virus was used per 2 × 104 cells. Ad-RFP-mCCNT1 (ADV-223024) was obtained from Vector Biolabs and supplied at 5.6 × 1010 pfu/ml. For MEFs, 20 µl of virus was used per 5 × 105 cells. For CMs, 10 µl of virus was used per 2 × 104 cells. AAV9-cTnT-3xFLAG-mCCNT1-WPRE and AAV-cTnT-RFP-WPRE (cat. no: VB5443) were obtained from Vector Biolabs and supplied at 2.6 × 1013 GC/ml and 6.0 × 1013 GC/ml, respectively. For systemic infection, AAV viruses were diluted in PBS, each mouse received 1 × 1011 GC in 100 µl via tail vein injection at 4–7 weeks of age.
Quantitative reverse transcription PCR
Total RNA was isolated using TRIzol Reagent (Thermo Fisher, 15596-018) following manufacturers instructions. cDNA was synthesised from 1 µg of RNA using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher, 4374966) following manufacturers instructions. Quantitative reverse transcription PCR (qRT-PCR) reactions were performed on a Applied Biosystems QuantStudio 5 Real-Time PCR System using Fast SYBR Green Master Mix (Thermo Fisher, 4385612), following manufacturers instructions. Primer sequences in Supplementary Table 1. Expression is normalised to Gapdh and Actin.
Flow cytometry analysis of DNA content
For MEFs: Cells were trypsinised, washed in PBS and fixed in 70% ethanol overnight. Fixed cells were rehydrated, washed in PBS, treated with RNase A (10 µg) and PI (5 µg), and analysed by Flow cytometry using a BD Accuri C6 flow cytometer. For each sample, at least 10,000 events were recorded and analysed using FlowJo software (Tree Star).
Fix-dissociation of cardiomyocytes
Whole hearts were isolated from mice and washed in PBS followed by fixation in 1% PFA overnight at room temperature with agitation. The following day hearts were washed four times in PBS. Hearts were cut into 1–2 mm3 pieces and incubated with 0.5 U/mL collagenase B (Roche #11088807001) in 0.2% NaN3/PBS and left to oscillate at 1000 rpm at 37 °C. Every 12 h, the cardiomyocyte supernatant was collected and stored at 4 °C in 0.2% NaN3/FBS. Once dissociation was complete (~8 days) cells were centrifuged at 1000 × g for 3 min, washed twice in PBS and stored in 0.2% NaN3/PBS at 4 °C. All of the following incubations were carried out at room temperature. A total of 500,000 cardiomyocytes were incubated with blocking buffer (4% BSA, +0.2% Triton X-100, +1 mM EDTA and +0.02% sodium azide) and agitated (1000 rpm) at room temperature for 3 min. Cells were then re-suspend in primary antibodies (Cardiac Troponin T (13-11; Thermo Fisher, MA5-12969; 1:100, p-H3 (Millipore, 1:400))) and incubated with agitation for 1 h. After two washes in blocking buffer, secondary antibodies (Alexa Fluor 488 Goat Anti-mouse IgG (H+L; Life Technologies, A11008)) were incubated together with Hoechst and agitated for 1 h at room temperature. Following two washes in blocking buffer, cells were re-suspend in blocking buffer, and either spun onto slides and mounted for imaging or analysed by flow cytometry. Cardiomyocyte length and width was measured using ImageJ software on images over-exposed for Cardiac Troponin T. A mean cell size for each heart was determined from measurements of >13 individual cells. Flow cytometry was performed on a BD LSRFortessa and analysed using FlowJo software (Tree Star).
Immunoblotting
Snap frozen animal tissues were ground on liquid nitrogen. Isolated cells were directly lysed on cell culture dishes and proteins extracted in buffer containing 1% SDS, 50 mM Tris pH 6.8 and 10% glycerol on ice for 10 min. Lysates were boiled for 10 min, followed by sonication (Bioruptor, Diagenode) for 15 min on ice. Total protein (50 µg) was electrophoresed on an SDS–PAGE gel and transferred onto immobilon-P (Millipore) membranes. These were then blocked in 5% non-fat milk and incubated with primary antibodies overnight at 4 °C. Secondary antibodies were applied for 1 h followed by chemiluminescent visualisation (Thermo Scientific, 32106 or Millipore, WBKLS0500). Immunoblots were developed on Fuji RX X-ray film, scanned on an Epson Perfection V500 Photo flatbed scanner. Images were cropped using Adobe Photoshop but otherwise unprocessed. Primary antibodies: GAPDH (D16H11) XP® (Cell Signaling Technology, 5174, used at 1:5000), β-actin (AC-15; Santa-Cruz Biotechnology, sc-69879, used at 1:5000), α-tubulin antibody (DM1A; Santa-Cruz Biotechnology, sc-32293, used at 1:5000), Myc (Y69; Abcam, ab32072, used at 1:10,000), Rpb1 CTD (4H8; Cell Signaling Technology, 2629, used at 1:2500), phospho-Rpb1 CTD (Ser2; E1Z3G; Cell Signaling Technology, 13499, used at 1:2500), phospho-Rpb1 CTD (Ser5; D9N5I; Cell Signaling Technology, 13523, used at 1:2500), CDK9 (C12F7; Cell Signaling Technology, 2316, used at 1:1000), Cyclin T1 (D1B6G; Cell Signaling Technology, 81464, used at 1:1000), LARP7 (Abcam, ab134746, used at 1:1000), anti-rabbit IgG HRP (Sigma, A0545, used at 1:10,000) and anti-mouse IgG HRP (Sigma, A0944, used at 1:10,000). Sample loading was normalised for equal protein content. Expression of GAPDH or α-tubulin is included as a confirmation of efficiency of protein isolation and comparable loading between individual tissue samples. Where necessary a composite figure was generated from the same samples loaded across multiple blots and a representative image of GAPDH is shown, Fig. 5a and Supplementary Fig. 5a.
Immunohistochemistry
Immunohistochemistry was performed on 4 µm sections. Sections were de-paraffinized and rehydrated. Antigens were retrieved by boiling in 10 mM citrate buffer (pH 6.0) for 10 min. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide for 30 min. Sections were then treated with rabbit VECTASTAIN Elite ABC horseradish peroxidase kit (Vector Laboratories, PK-6101) following the manufacturers protocols with all incubations were carried out at room temperature. Briefly, sections were blocked in normal goat serum for 20 min, followed by 1 h incubation in primary antibody. Sections were then washed three times in PBST, and incubated for 30 min in secondary antibody, sections were then washed again in PBST and then incubated in ABC complex. Sections were developed in DAB (3,3′-diaminobenzidine) for 5 min, counterstained in haematoxylin, dehydrated and mounted in DPX. Staining was imaged on a Zeiss Axio Imager using the Zeiss AxioVision 4.8 software using the AutoLive setting and interactive white balance. Quantification was performed by counting number of cells per field of view for five images per organ/mouse, the mean of five raw counts was calculated and is represented by each data point per graph.
Primary antibodies; anti-Ki67 (SP6; Thermo Scientific, RM-9106; 1:200), anti-phospho-Histone H3 (Ser10; Merck Millipore, 06-570; 1:1250) and anti-Cyclin T1 (AbCam, ab238940; 1:500).
Immunoflorescence
Immunoflorescence was performed on 4 µm sections. Sections were de-paraffinized and rehydrated. Antigen retrieval was performed by boiling in 10 mM citrate buffer (pH 6.0) for 10 min. For BrdU staining, DNA was denatured with 2N HCL, 0.2% triton X for 10 mins at 37 °C. Sections were blocked in 2.5% goat serum (from number), 1% BSA in PBST for 20 mins at room temperature. Primary antibody, made up in blocking buffer, was added for 1 h at room temperature, followed by PBST washes and secondary antibodies added for 1 h at room temperature. Nuclei were visualised using Hoechst (Sigma, 861405) and sections mounted in prolong gold (Thermo Fisher, P36930). Antibodies: anti-phospho-Histone H3 (Ser10; Merck Millipore, 06-570: Antibody; 1:500), cardiac troponin T (13-11; Thermo Fisher, MA5-12969; 1:100), anti-BrdU (BU1/75 (ICR1)) antibody (Abcam, ab6326, 1:200), anti-aurora B antibody (Abcam, ab2254; 1:200), anti-PCM1 (SigmaAldrich, HPA023370, 1:100), anti-Ki67 (SolA15; Thermo Fisher, 1:100), Alexa Fluor 555 goat anti-rabbit IgG (H+L; Life Technologies, A21428), Alexa Fluor 555 goat anti-mouse IgG (H+L; Life Technologies, A21422), Alexa Fluor 555 goat anti-rat IgG (H+L; Life Technologies, A21434), Alexa Fluor 488 goat anti-rabbit IgG (H+L; Life Technologies, A11008), Alexa Fluor 488 goat anti-rat IgG (H+L; Life Technologies, A11006), Alexa Fluor 350 goat anti-mouse (H+L; Life Technologies A11045), wheat germ agglutinin, Alexa Fluor™ 488 conjugate (Thermo Fisher, W11261).
Southern blotting
DNA (≥10 µg) was digested overnight with appropriate restriction enzymes and run on a 0.8% agarose gel to separate fragments. Gels were subject to depurination and neutralisation before transferring DNA to Hybond-XL membrane (RPN203S, GE Healthcare Amersham) overnight by capillary action. The membrane was then washed and dried at 80 °C for 2 h. αP32–dCTP probes were labelled using Prime-It® II Random Primer Labelling Kit (300385, Agilent) and purified with Illustra ProbeQuant™ G-50 Micro columns (28-9034-08, GE Healthcare Amersham). Following pre-hybe for 1–2 h in Rapid-hyb™ Buffer (RPN1636, GE Healthcare Amersham), the membrane was hybridised with the probe for at least 5 h, washed, exposed to Fuji RX X-ray film and the film scanned on an Epson Perfection V500 Photo flatbed scanner.
Transmission electron microscopy
Day 15 R26CMER/+ mouse heart was cut into 1 mm2 pieces and fixed overnight in fresh 2.5% glutaraldehyde/PBS solution (Sigma, G7651), followed by three washes in PBS. Samples were then placed in fresh fix and irradiated for 6 min at 80 W under vacuum in a Pelco Biowave (Ted Pella In, Redding, CA). Fixed tissue was then stained with 3% potassium ferricyanide and 2% osmium tetroxide in 0.1 M cacodylate buffer for 30 min, then washed three times in distilled water and incubated in 1% thiocarbohydrazide for 30 min at room temperature. Samples were then washed three times in distilled water and immersed in 2% osmium tetroxide for 30 min at room temperature, then again washed three times in distilled water. Samples were contrasted with 1% aqueous uranyl acetate for 30 min at 4 °C and washed three times in distilled water. A solution of 0.06% lead nitrate was prepared by dissolving in aspartic acid (pH 5.5) at 60 °C, which was then filtered and added to samples for 20 min at 60 °C before washing three times with distilled water at room temperature. Pieces of tissue underwent serial dehydration by immersing in each increasing ethanol concentration twice (30%, 50%, 70%, 90% and 100%) and irradiating for 40 s at 250 W in a Pelco microwave. Samples were then infiltrated with increasing concentrations of epon LX112 resin (25%, 50% and 75%) in ethanol for 3 min at 250 W under vacuum in a Pelco microwave, then twice in 100% resin before polymerising at 60 °C overnight79. Samples were analysed on a Apreo FESEM equipped with a VolumeScope serial block face system and MAPS software (Thermo Fisher) with a slice thickness of 100 nm. Datasets were aligned, segmented and visualised in Imod (Bio3D) and Chimera (UCSF) software.
3View EM stacks were assembled and 3D modelling of structures was performed in IMOD 3D reconstruction and visualisation software (University of Colorado, Boulder).
ChIP sequencing
Mouse tissue was harvested from mice, washed in PBS and cut into small 2–3 mm pieces with a scalpel blade. Tissue was then fixed in 1% formaldehyde/PBS for 15 min, washed in PBS three times, snap frozen and stored at −80 °C until further processing. Upon thawing, tissue was incubated in 125 mM glycine (5 min, room temperature), washed in PBS and nuclei prepared on ice using a dounce homogeniser. All following steps were performed on ice. Nuclei were washed in PBS, resuspended in ChIP buffer (4 ml of buffer per 0.5 ml of tissue; 100 mM NaCl, 133 mM Tris pH 8.0, 5 mM EDTA, 0.02% NaN3, 0.67% SDS and 1.67% Triton X-100) supplemented with protease inhibitors (Complete Protease Inhibitor Cocktail Tablets, Roche) and phosphatase inhibitors (50 mM NaF, 1 mM β-GP and 1 mM NaV). Chromatin was sonicated to an average length of 300 bp, pre-cleared twice with protein G-sepharose beads, pre-blocked with 0.5% Escherichia. coli tRNA (Sigma) and 0.5% BSA (ref. 80). In this study, the following antibodies were used: Myc N262 (Santa Cruz, sc-764×), RNA PolII N20 (Santa Cruz, sc-899×). A total of 10 µg of antibodies were incubated overnight with 500 µl of chromatin (+500 µl of ChIP buffer to each a volume of 1 ml per reaction). Blocked protein G-sepharose beads were added, incubated for 2–3 h and washed as follows: three times with mixed micelle buffer (150 mM NaCl, 20 mM Tris pH 8.1, 5 mM EDTA, pH 8.0, 5.2% sucrose, 0.02% NaN3, 1% Triton X-100 and 0.02% SDS), 2x with buffer 500 (50 mM HEPES pH 7.5, 0.1% sodium deoxycholate, 1 mM EDTA, 500 mM NaCl, 1% Triton X-100 and 0.02% NaN3), two times with LiCl/detergent buffer (10 mM Tris pH 8.0, 0.5% sodium deoxycholate, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40 and 0.02% NaN3) and once with TE. DNA was de-crosslinked overnight (2% SDS in TE, 65 °C), purified using QiaQuick columns (Qiagen) and quantified using Qbit (Thermo Fisher). A total of 2–5 ng of ChIP DNA were used for ChIP-seq library preparation, using the TruSeq ChIP Library Preparation Kit (Illumina), following manufacturers instructions.
RNA sequencing
Total RNA from tissues was purified using TRIzol (Invitrogen). Briefly, tissue was homogenised in Trizol (Invitrogen) and chloroform was added. After centrifugation, RNA was purified using RNeasy Mini columns (Qiagen) according to the manufacturer’s protocol, including the on column DNaseI digestion step. Alternatively, total RNA was purified using PureLink RNA Mini Kit (Thermo Scientific) together with on column DNA digestion using PureLink DNase (Thermo Scientific). Then 1–5 µg of purified RNA was treated with Ribozero rRNA removal kit (Illumina) and ethanol precipitated. RNA quality and removal of rRNA were checked with the Agilent 2100 Bioanalyser (Agilent Technologies). Libraries for RNA-seq were then prepared with the TruSeq RNA Sample Prep Kits v2 (Illumina) or NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (NEB), following manufacturers instructions starting from the RNA fragmentation step.
RNA-seq sample processing and analysis
Adult tissues: Sequence reads were aligned to the mm9 mouse reference genome using TopHat aligner (version 2.0.8). In case of duplicated reads, only one read was kept. FeatureCounts software was used to associate read counts to genes (http://bioinf.wehi.edu.au/featureCounts/). Exon reads per kilobase per million mapped reads (eRPKM) defined gene expression using total library size as the number of reads aligned on exons. We applied a filter for a minimum expression level average eRPKM > 3.5 and Log2FC > 0.5, DEGs were identified using the Bioconductor package DESeq2 (ref. 81).
AAV infected and day 15 heart + mouse samples: Sequence reads were adaptor trimmed using Cutadapt82 (version 1.11) and aligned using BWA aln for short reads83 (version 2.5.2a) to the GRCm38 (mm10) assembly with the gene, transcript and exon features of the Ensembl (release 70) gene model. Expression was estimated using RSEM84 (version 1.2.30). Transcripts with zero read counts across all samples were removed prior analysis. Normalisation of RSEM expected read counts was performed by dividing by million reads mapped to generate counts per million, followed by the trimmed mean of M-values method from the edgeR package85,86. Differential expression analysis was performed using edgeR.
ChIP-seq data analysis
Data were generated and processed by aligning sequence reads to the mm9 genome with the BWA aligner using default settings. Peaks were called using the MACS software (v2.0.9)87. Normalised read counts within a genomic region were determined as the number of reads per million of library reads (total number of aligned reads in the sequencing library). Peak enrichment was determined as log2 (Peakw/Nc—inputw/Ni), where Peakw is the read count on the enriched region and inputw the read count on the same region in the corresponding input sample, Nc is the total number of aligned reads in the ChIP sample and Ni is the total number of aligned reads in the input sample. Promoter peaks were defined as all peaks with at least one base pair overlapping with the interval between −2 kb to +1 kb from the nearest TSS. Myc-binding peaks called in the heart but not in the liver were annotated as heart specific, and peaks called in the liver but not in the heart were annotated as liver specific. Myc-binding peaks identified in both tissues, heart and liver, were annotated as common Myc binding sites.
For the analysis of chromatin accessibility, regulatory elements and transcriptional activity, genomic regions of promoter Myc binding sites in liver and heart were extracted 6 kb around the peak centre. Read coverage of the 6 kb Myc-bound genomic regions, were extracted from the alignment bam files using the Genomation R package88. To overlap Myc peaks with chromatin accessibility and active promoter histone marks, we extracted publicly available data from 8 week adult C57BL/6 liver and heart tissue from ENCODE (DNaseI: http://genome.ucsc.edu/cgi-bin/hgFileUi?db=mm9&g=wgEncodeUwDnase, ChIP: http://genome.ucsc.edu/cgi-bin/hgFileUi?db=mm9&g=wgEncodeLicrHistone, Download: 01/08/2018). Specifically, chromatin accessibility was ascertained using a DNaseI hypersensitivity assay, and active promoters were identified based on H3K27ac and H3K4me3 ChIP sequencing. Read coverage of the 6 kb Myc-bound genomic regions were extracted using the genomation R package from bigwig files generated by ENCODE. For each ChIP (Myc, PolII, H3K27ac, H3K4Me1 and H3K4me3) and DNaseI experiment, read coverage of the 6 kb regions were binned into 100 equally sized bins with the maximum counts used for smoothing windows. Each experiment’s smoothed read coverage was visualised in a heatmap with independent colour scales to account for differences in experimental protocols, processing and tissue. Genomic regions were clustered based on whether Myc binding was specific for heart, liver or common to both tissues.
Statistical analysis
Pipelines for primary analysis (filtering and alignment to the reference genome of the raw reads) and secondary analysis (expression quantification, differential gene expression and peak calling) have been integrated in the HTS-flow system89. Bioinformatic and statistical analysis were performed using R with Bioconductor packages and comEpiTools packages90,91. Motif analysis was performed using CentriMo software92. Intron analysis was performed with the INSPEcT tool93,94. Gene lists from RNA and ChIP sequencing were analysed in Enrichr95. Statistical analyses for IHC and qRT-PCR were performed using GraphPad Prism v6.0d (GraphPad Software, Inc., San Diego, CA, USA) as indicated with P ≤ 0.05 considered to be statistically significant. Venn diagrams were draw in the interactive tool Venny [https://bioinfogp.cnb.csic.es/tools/venny/index.html].
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All datasets generated in this study, including Myc ChIP sequencing, RNA PolII ChIP sequencing and RNA sequencing, have been deposited in ArrayExpress (www.ebi.ac.uk/arrayexpress) under accession codes; E-MTAB-7592, E-MTAB-7593, E-MTAB-7595, E-MTAB-8462, E-MTAB-8515 and E-MTAB-7636. The source data underlying Figs. 1a, c d, 4a, 5a–e, 6a, b, d, 7c, d, 8a–c, 9b, c, e and, Supplementary Figs. 1b–d, f, h, 2a, 4a, b, 5a–f, 6a, b, d, 7c, 8a and 9a, d, e are provided as a Source Data file. Further information and requests for resources and reagents should be directed to, and will be fulfilled by, Gerard Evan (gie20@cam.ac.uk). Requests regarding AZ5576 should be directed to AstraZeneca PLC.
Code availability
No previously unreported custom computer code or algorithm used to generate results.
References
Blackwood, E. M. & Eisenman, R. N. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217 (1991).
Amati, B., Littlewood, T. D., Evan, G. I. & Land, H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 12, 5083–5087 (1993).
Blackwood, E. M., Luscher, B. & Eisenman, R. N. Myc and Max associate in vivo. Genes Dev. 6, 71–80 (1992).
Amati, B. et al. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359, 423–426 (1992).
McMahon, S. B., Van Buskirk, H. A., Dugan, K. A., Copeland, T. D. & Cole, M. D. The novel ATM-related protein TRRAP is an essential cofactor for the c- Myc and E2F oncoproteins. Cell 94, 363–374 (1998).
Bouchard, C. et al. Regulation of cyclin D2 gene expression by the Myc / Max / Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 15, 2042–2047 (2001).
Frank, S. R. et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 4, 575–580 (2003).
Rahl, P. B. et al. C-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
Kanazawa, S., Soucek, L., Evan, G., Okamoto, T. & Peterlin, B. M. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22, 5707–5711 (2003).
Eberhardy, S. R. & Farnham, P. J. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J. Biol. Chem. 277, 40156–40162 (2002).
De Pretis, S. et al. Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res. 10, 1658–1664 (2017).
Jonkers, I. & Lis, J. T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 167–176 (2015).
Peterlin, B. M. & Price, D. H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 (2006).
Zhou, Q., Li, T. & Price, D. H. RNA polymerase II elongation control. Annu. Rev. Biochem. 81, 119–143 (2012).
Zhou, Q. & Yik, J. H. N. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 70, 646–659 (2006).
Barboric, M., Price, P., Blazek, D., Price, D. H. & Peterlin, B. M. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition. EMBO J. 24, 4291–4303 (2005).
Yik, J. H. N. et al. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 12, 971–982 (2003).
He, N. et al. Article a la-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol. Cell 29, 588–599 (2008).
Dang, C. V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 3, a014217 (2013).
Evan, G. I. et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, 119–128 (1992).
Eilers, M., Schirm, S. & Bishop, J. M. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 10, 133–141 (1991).
Roussel, M. F., Cleveland, J. L., Shurtleff, S. A. & Sherr, C. J. Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature 353, 361–363 (1991).
Shchors, K. et al. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1β. Genes Dev. 20, 2527–2538 (2006).
Sodir, N. M. et al. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 25, 907–916 (2011).
Kortlever, R. M. et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315 (2017).
Lin, C. Y. et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67 (2012).
Nie, Z. et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79 (2012).
Zeller, K. I. et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl Acad. Sci. USA 103, 17834–17839 (2006).
Patel, J. H., Loboda, A. P., Showe, M. K., Showe, L. C. & McMahon, S. B. Analysis of genomic targets reveals complex functions of MYC. Nature Rev. Cancer 4, 562–568.
Sabò, A. et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492 (2014).
Walz, S. et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487 (2014).
Kress, T. R., Sabò, A. & Amati, B. MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015).
Guccione, E. et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 8, 764–770 (2006).
Soufi, A., Donahue, G. & Zaret, K. S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012).
Lorenzin, F. et al. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 5, e15161 (2016).
Murphy, D. J. et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457 (2008).
De Vries, W. N. et al. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 26, 110–112 (2000).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, N. & Luo, L. A global double-fluorescent cre reporter mouse. Genesis 45, 593–605 (2007).
Wilson, C. H. et al. The kinetics of ER fusion protein activation in vivo. Oncogene 33, 4877–4880 (2014).
Quaife-Ryan, G. A. et al. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation 136, 1123–1139 (2017).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Shao, W. & Zeitlinger, J. Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet. 49, 1045–1051 (2017).
Sano, M. et al. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 8, 1310–1317 (2002).
Agah, R. et al. Gene recombination in postmitotic cells: targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 (1997).
Sano, M. et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 23, 3559–3569 (2004).
Pelengaris, S., Littlewood, T., Khan, M., Elia, G. & Evan, G. Reversible activation of c-Myc in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 3, 565–577 (1999).
Pelengaris, S., Khan, M. & Evan, G. I. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109, 321–334 (2002).
Littlewood, T. D., Hancock, D. C., Danielian, P. S., Parker, M. G. & Evan, G. I. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23, 1686–1690 (1995).
Eilers, M., Picardt, D., Yamamotot, K. R. & Bishop, J. M. Chimaeras of Myc oncoprotein transformation of cells. Nature 1276, 1987–1989 (1989).
Kotton, D. N. & Morrisey, E. E. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat. Med. 20, 822–832 (2014).
Ruijtenberg, S. & van den Heuvel, S. Coordinating cell proliferation and differentiation: antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 15, 196–212 (2016).
Hadzhiev, Y. et al. A cell cycle-coordinated polymerase II transcription compartment encompasses gene expression before global genome activation. Nat. Commun. 10, 1–14 (2019).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Herold, S. et al. accumulation of stalled RNA polymerase. Nature 567, 545–549 (2019).
Porrello, E. R. & Olson, E. N. A neonatal blueprint for cardiac regeneration. Stem Cell Res. 13, 556–570 (2014).
Abdellatif, M. et al. A Ras-dependent pathway regulates RNA polymerase II phosphorylation in cardiac myocytes: implications for cardiac hypertrophy. Mol. Cell. Biol. 18, 6729–6736 (1998).
Li, F., Wang, X., Capasso, J. M. & Gerdes, A. M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746 (1996).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Porrello, E. R. et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl Acad. Sci. USA 110, 187–192 (2013).
Walsh, S., Pontén, A., Fleischmann, B. K. & Jovinge, S. Cardiomyocyte cell cycle control and growth estimation in vivo-An analysis based on cardiomyocyte nuclei. Cardiovasc. Res. 86, 365–373 (2010).
Ye, L. et al. Early regenerative capacity in the porcine heart. Circulation 138, 2798–2808 (2018).
Zhu, W. et al. Regenerative potential of neonatal porcine hearts. Circulation 138, 2809–2816 (2018).
Jopling, C. et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464, 606–609 (2010).
Eulalio, A. et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381 (2012).
Heallen, T. et al. Hippo signaling impedes adult heart regeneration. Development 140, 4683–4690 (2013).
Monroe, T. O. et al. Article YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo yap partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev. Cell 48, 765–779 (2019).
Mills, R. J. et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl Acad. Sci. USA 114, E8372–E8381 (2017).
Puente, B. N. et al. The oxygen rich postnatal environment induces cardiomyocyte cell cycle arrest through dna damage response. Cell 157, 565–579 (2014).
Matrone, G. et al. CDK9 and its repressor LARP7 modulate cardiomyocyte proliferation and response to injury in the zebrafish heart. J. Cell Sci. 128, 4560–4571 (2015).
Marshall, R. M. et al. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J. Immunol. 175, 6402–6411 (2005).
Fujita, T., Ryser, S., Piuz, I. & Schlegel, W. Up-Regulation of P-TEFb by the MEK1 – extracellular signal-regulated kinase signaling pathway contributes to stimulated transcription elongation of immediate early genes in neuroendocrine cells. Mol. Cell. Biol. 28, 1630–1643 (2008).
Leach, J. P. et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 550, 260–264 (2017).
Mohamed, T. et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173, 104–116 (2018).
Nakada, Y. et al. Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227 (2017).
Wamstad, J. A. et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206–220 (2012).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Gamper, I. et al. Determination of the physiological and pathological roles of E2F3 in adult tissues. Sci. Rep. 7, 9932 (2017).
Ackers-Johnson, M. et al. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 119, 909–920 (2016).
Takasato, M. et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015).
Kress, T. R. et al. Identification of MYC-dependent transcriptional programs in oncogene-addicted liver tumors. Cancer Res. 76, 3463–3472 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Bioinformatics 28, 1838–1844 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Akalin, A., Franke, V., Vlahoviček, K., Mason, C. E. & Schübeler, D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 31, 1127–1129 (2015).
Bianchi, V. et al. Integrated systems for NGS data management and analysis: open issues and available solutions. Front. Genet. 7, 1–8 (2016).
Gentleman, R. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Kishore, K. et al. methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics 16, 313 (2015).
Bailey, T. L. & MacHanick, P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 40, 1–10 (2012).
De Pretis, S. et al. Gene expression INSPEcT: a computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA- and 4sU-seq time course experiments. Bioinformatics 31, 2829–2835 (2015).
Tesi, A. et al. An early Myc-dependent transcriptional program orchestrates cell growth during B-cell activation. EMBO Rep. 20, 1–14 (2019).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Acknowledgements
The authors thank the support staff in the Cambridge University Biomedical Services at the Gurdon Institute and CRUK Cambridge Institute. We would like to thank AstraZeneca and, specifically, Lisa Drew and Justin Cidado for AZ5576 supply and guidance. We would like to thank Steven Lane for laboratory support and critical advice on the manuscript. The authors acknowledge the use of the Microscopy Australia Research Facility at the Center for Microscopy and Microanalysis at The University of Queensland. We would like to thank Charles Ferguson, Robyn Webb and Marcel Sayre (CMM, UQ), for assistance with electron microscopy. We also thank Roger Foo and Peter Li from the National University Heart Centre, Singapore for their technical advice on the isolation and infection of adult cardiomyocytes. This work was primarily supported by CRUK (Programme Grant A19013 to GIE). M.J.B. was funded by an EMBO long-term fellowship and an Australian NHMRC Early Career Fellowship (APP1072477). This work was supported by funding from the European Research Council (grant agreement no. 268671-MYCNEXT), the Italian Health Ministry (RF-2011- 02346976) and the Italian Association for Cancer Research (AIRC, IG 2015-16768 and IG 2018-21594) to B.A., and from Worldwide Cancer Research (15-1260) to A.S. This work was also supported by the National Health and Medical Research Council of Australia (grants APP1140064 and APP1150083 and fellowship APP1156489 to R.G.P.). R.G.P. is supported by the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology.
Author information
Authors and Affiliations
Contributions
C.H.W., M.J.B., A.S., D.L.B., G.I.E. and B.A. conceptualised the work. Experiments were performed by C.H.W., M.J.B., A.S. and D.L.B. C.H.W. and M.J.B. wrote the manuscript. A.S., V.P., T.R.K., B.A. and J.S. performed and analysed the global sequencing analysis. J.E.H., E.R.P. and G.A.Q. performed the cardiomyocyte counting and analysed global sequencing analysis. J.R. and R.G.P. performed the TEM. G.I.E., B.A. and T.D.L. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests, except G.I.E. is a member of AstraZeneca’s Oncology External Advisory Panel. M.J.B. and C.H.W. are named inventors on a provisional patent relating to data in this manuscript.
Additional information
Peer review information Nature Communications thanks Martin Eilers and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bywater, M.J., Burkhart, D.L., Straube, J. et al. Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat Commun 11, 1827 (2020). https://doi.org/10.1038/s41467-020-15552-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-15552-x
This article is cited by
-
MYC activity at enhancers drives prognostic transcriptional programs through an epigenetic switch
Nature Genetics (2024)
-
Regeneration of the heart: from molecular mechanisms to clinical therapeutics
Military Medical Research (2023)
-
MDIG-mediated H3K9me3 demethylation upregulates Myc by activating OTX2 and facilitates liver regeneration
Signal Transduction and Targeted Therapy (2023)
-
Oncogenic drivers dictate immune control of acute myeloid leukemia
Nature Communications (2023)
-
Ploidy-stratified single cardiomyocyte transcriptomics map Zinc Finger E-Box Binding Homeobox 1 to underly cardiomyocyte proliferation before birth
Basic Research in Cardiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
