
Abstract
Polymer brush patterns have a central role in established and emerging research disciplines, from microarrays and smart surfaces to tissue engineering. The properties of these patterned surfaces are dependent on monomer composition, polymer height, and brush distribution across the surface. No current lithographic method, however, is capable of adjusting each of these variables independently and with micrometer-scale resolution. Here we report a technique termed Polymer Brush Hypersurface Photolithography, which produces polymeric pixels by combining a digital micromirror device (DMD), an air-free reaction chamber, and microfluidics to independently control monomer composition and polymer height of each pixel. The printer capabilities are demonstrated by preparing patterns from combinatorial polymer and block copolymer brushes. Images from polymeric pixels are created using the light reflected from a DMD to photochemically initiate atom-transfer radical polymerization from initiators immobilized on Si/SiO2 wafers. Patterning is combined with high-throughput analysis of grafted-from polymerization kinetics, accelerating reaction discovery, and optimization of polymer coatings.
Similar content being viewed by others
Introduction
Controlling the morphology and chemical composition of interfaces with micrometer-scale precision is a key challenge in surface chemistry, with repercussions from biology to optics and material science. Polymer brushes1,2,3, and particularly polymer brush patterns with micrometer-scale features, are increasingly explored in the context of microarrays4,5,6, smart surfaces7,8,9, and tissue engineering10,11,12,13 because their interfacial properties can be manipulated by tailoring the degree of polymerization, the monomer composition, and the grafting density of the polymer brushes. Of the many grafted-from polymerization reactions14,15,16, living polymerizations are ideal for patterning because the degree of polymerization is dependent upon reaction time, which affords precise control over the polymer height and block length17,18,19,20. Several challenges, however, have precluded the realization of the full potential of polymer brush patterning technologies. Typically, polymer brush coatings are composed of identical polymers uniformly distributed across the surface because no available printing tool is capable of controlling simultaneously polymer height and polymer composition at each position, while producing features with micrometer-scale dimensions. Also because the brushes on a surface are generally grown under identical conditions, a new surface must be prepared and characterized to determine the properties arising from each new set of reaction conditions21,22,23,24. This makes optimization of grafted-from reactions and developing structure–activity relationships time-consuming and costly. Therefore, there is a pressing need to develop printing platforms that (1) combine the feature resolution of modern lithography strategies with the ability to spatiotemporally localize a stimulus (e.g., light or heat) that induces the polymerization reaction, (2) can accommodate the demanding reaction conditions required to carry out advanced polymerization chemistries (e.g., in solution and O2 and H2O free), and (3) have the capability to independently vary the monomer composition of each pixel across the surface and along each polymer chain.
Various lithographic approaches are being explored currently in an effort to create polymer brush patterns with micrometer-scale feature dimensions and vary the composition of polymer brushes across the surface. For example, microcontact printing has been used to pattern self-assembled monolayers of initiators, and polymer brushes of uniform height and identical composition were grown upon immersing the substrate in monomer solution25,26,27. An alternative approach involves uniformly functionalizing the surface with the initiators and subsequently irradiating through a photomask, in the presence of the monomer solution to create a pattern23. This strategy can create gradients, which are important for tissue engineering28,29 or to study cell adhesion and migration30,31,32, but patterns containing multiple different polymers are printed with difficulty because photolithography generates one pattern per photomask, and serially aligning photomasks is tedious. Hawker et al. for instance have recently prepared patterns with four different polymer brush compositions following a multistep process, consisting of creating a polymer brush coating, modifying it through a photomask using click chemistry, and repeating this process several times to create patterns composed of up to four different polymer brush compositions33. Scanning probe lithographies34,35 have successfully produced polymer brush patterns by using the tip to deposit the initiator36, or creating a pattern with a resist, where, following resist development, the initiator is subsequently deposited37. Features as small as 25 nm are obtained, however, these methods uniformly irradiate the surface, so the resulting polymers are identical across the substrate. Electron-beam lithography has been successfully used for direct writing of polymer brushes with 20 nm feature dimensions38. Other strategies, such as self-assembly-driven growth of high-density polymer brush nanoarrays can reach feature sizes as small as 15 nm (ref. 39). Using massively parallel beam pen arrays, a monomer mixture was deposited onto a surface. Subsequently, irradiating the droplets produced glycopolymer arrays, where the polymer height in each position could be varied by manipulating the irradiation time, but each polymer was composed of the same monomer40. In an effort to create multiplexed polymer brush patterns using scanning probe technologies, we recently integrated microfluidics with beam pen lithography, but could only print a pattern composed of two different polymer compositions41. While each of these efforts is a milestone in polymer brush patterning, the goal of independently controlling height, composition, and feature dimensions for each pixel across the entire pattern, with micrometer resolution, and over a large (>1 mm2) area has remained elusive.
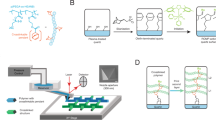
Here, we report an approach toward printing combinatorial grafted-from polymer brush patterns, where the monomer composition and feature height of each pixel in a pattern can be controlled independently and with ~5 µm pixel edge length, while circumventing the need for expensive photomasks. We refer to these patterns as hypersurfaces—borrowing from its mathematical namesake to indicate a pattern where more than three properties of each pixel can be controlled independently (i.e., x- and y-position, height, and chemical composition along the chain). In addition, we use this terminology because four-dimensional printing has been adopted already to indicate additive manufacturing of objects whose shapes change over time in response to an external stimulus42. To create these hypersurfaces, we integrated a digital micromirror device (DMD), microfluidics, and an oxygen-free reaction chamber that was mounted onto a piezoelectric stage (Fig. 1). DMD-based printers have been combined already with microfluidics for oligonucleotide43 and oligopeptide44 microarray fabrication, and to prepare scaffolds for tissue engineering45,46,47. Our printer was built upon a TERA-Print E-series instrument, which coordinates the DMD (1024 × 768 independently controllable mirrors), light source (405 nm LED, 32 mW cm−2), and the piezoelectric stage with a CPU interface to project patterns taken from an uploaded image file onto a substrate. The inert atmosphere chamber is composed of a hermetically sealed polystyrene cell, with a glass window for passing light from the DMD to the surface, and inlet and outlet apertures for tubing that introduces the monomer solutions to the reactive substrate. An additional glass plate over the functionalized substrate forms a ~50 µL reaction cell, where the solutions are drawn over the surface by capillary forces48. Reactive solutions composed of monomers, solvent, and photosensitizer are introduced and withdrawn using syringe pumps that control flowrate within the reaction cell. A microfluidic chaotic mixer can be incorporated upstream to mix different proportions of components48.

The printer uses microfluidics to introduce monomer solutions into the reaction chamber, a computer-controlled DMD to illuminate the surface, and a reaction cell mounted onto a piezoelectric stage to generate multicomponent polymer brush patterns by surface-initiated atom-transfer radical polymerization (inset).
Results
Optimization of surface-initiated ATRP printing
We first sought to demonstrate the ability to generate polymer brush patterns with independent control over height at each pixel using photoinduced surface-initiated atom-transfer radical polymerization (SI-ATRP). This reaction was selected for these proof-of-concept studies because of its broad monomer scope and narrow molecular weight distribution49, and, because of these advantageous attributes, it is increasingly used to generate grafted-from polymer coatings and patterns. Typically, to study the kinetics of a grafted-from polymerization, each data point requires printing and analyzing a separate surface, which is time-consuming and subject to substantial experiment-to-experiment variation23,50. Here, we study multiple different polymerization conditions in a single print to rapidly determine the relationship between irradiation time (t) and polymer height (h). First, the reactive substrate was prepared by immersing a freshly cleaned Si/SiOx wafer in an aminopropyltriethoxysilane solution to generate amine-functionalized wafers that were subsequently reacted with α-bromoisobutyryl bromide overnight to create a surface uniformly coated with the SI-ATRP initiator (Supplementary Fig. 1). Each step in this process was characterized by X-ray photoelectron spectroscopy (XPS) and contact angle measurements, and the data were consistent with the proposed surface reactions (Supplementary Fig. 2). The substrate was introduced into the inert chamber inside a glovebox, where the solutions are prepared with distilled dimethylformamide (DMF) and degassed monomers. Approximately 50 µL of a DMF solution containing methyl methacrylate (MMA), fluorescein O-methacrylate (FMA), and Ir(ppy)3 was deposited onto the surface and a cover glass was placed over the droplet. The chamber was then sealed, removed from the glovebox, and mounted onto the piezostage of the printer. FMA was added to MMA in a molar ratio 1:300 so that the patterns could be analyzed via fluorescence microscopy. A pattern of 15 features, irradiated at times ranging from 2–22 min, repeated 121 times across the 4.4 × 3.3 mm printing area, was sequentially projected onto the surface with the DMD. Following printing, the surfaces were washed with EtOH and sonicated for 5 min in DMF to remove any polymer that was not grafted to the surface, and the resulting polymer brush patterns were analyzed by fluorescence microscopy and atomic force microscopy (AFM; Fig. 2). Both h and normalized florescence (NF = feature fluorescence/background fluorescence) increase with increasing t. The feature h varied from 2.8 ± 0.4 nm to 10.7 ± 0.6 nm for 2 min and 22 min, respectively. The NF ranges from 2.2 ± 0.2 to 6.3 ± 0.4. The linear evolution of the polymer brush height with time is consistent with the kinetics measured in other studies of surface-initiated ATRP polymerizations23,51. Interestingly NF shows a logarithmic evolution, which may be the result of increasing adsorption of emitted light as the polymers grow (Fig. 2c). The printing was repeated under continuous flow at 5 µL/min to determine how flow affected printing, using the microfluidics to control the flowrate across the substrate, and these prints behave in a similar way (Supplementary Fig. 3).

a Fluorescence microscopy image (λex = 451.5‒486.5 nm, λem = 540–580 nm) of 3 × 5 patterns consisting of polymer brush features printed at 15 different t to study the effect of t on NF and h. Inset is a magnified image of one of the arrays with t from 2‒22 min. b Composite of eight AFM height images corresponding to one of the arrays shown in a. c Dependence of NF (circles) and h (squares) with t. Error bars correspond to the standard deviation of five measurements for NF and three measurements for h. d Original black and white picture of Statue of Liberty (This image is not covered by the article CC BY license. Image credit to Oliver Kuehl. All rights reserved, used with permission.), inverted image, and converted to five gray levels, from top to bottom, respectively. e Threshold images uploaded to the CPU controller corresponding to the five different t required to obtain a polymer brush pattern (300:1 MMA:FMA) with five different NF and h levels shown in f. g, h AFM height measurements from the areas marked with white boxes in f.
To confirm whether these features were the result of poly(methyl methacrylate) (PMMA) polymerization, substrates prepared under the same conditions were analyzed with XPS (Supplementary Fig. 4). In addition, both unbound and surface-bound polymers were characterized by matrix-assisted laser desorption ionization imaging mass spectrometry (MALDI-IMS). First, unbound polymer was generated in the photochemical printer with irradiation times of 1, 2, 5, 10, and 40 min, then analyzed by MALDI-IMS to determine mass distributions and peak spacings (Supplementary Fig. 5a). A non-Gaussian distribution of peaks with spacings of 100 Da was observed, corresponding to the MMA monomer mass. Unfortunately, the non-Gaussian distribution, most likely a result of fragmentation of PMMA by MALDI-MS (ref. 52), prevented the quantification of polymer molecular weight distribution and dispersity. However, overall increases in signal intensity were observed for polymers generated under longer irradiation time (e.g., 10 vs. 40 min) up to the measured range of 20,000 Da, suggesting that higher molecular weight polymers were generated with longer irradiation and fragmenting during MALDI analysis. Next, patterned substrates with polymer covalently bound to the surface were analyzed (Supplementary Fig. 4b). Similar to the studies with the unbound PMMA polymer, uniform peak spacings were observed with a non-Gaussian distribution. We note that the peak spacings observed for the patterned substrate were 107 Da. It is currently unclear as to why the spacings of the polymer units are increased, but because the spectral peaks themselves are also broader, we believe these observations may be a result of a difference in ionization of the covalently bound polymer by the MALDI instrument, in comparison to the unbound polymer, despite identical sample preparation and ionizing conditions. Definitive polymer signal was detected at irradiation times as low as 1 min, with increasing mass signal intensity over time. This is consistent with changes in height measured by AFM and fluorescence signal intensity changes over time (Fig. 2c). Overall, MALDI-IMS definitively identified polymer generated within our photochemical printer, whether the polymer was unbound or covalently bound to a substrate surface.
A major challenge in polymer brush lithography is creating gradients, where brush density or brush height is varied across the surface. Here, we show how the kinetic data described above are used to create such a gradient pattern, where the h at each pixel was controlled independently to print an image of the Statue of Liberty. To do so, we chose a black and white picture of the Statue of Liberty (Fig. 2a) that was converted to a 1074 × 768 bitmap image, so each DMD mirror projects a single pixel with a corresponding width of 4.3 μm. The image was then inverted (Fig. 2b) and converted to five levels of gray (Fig. 2c), and finally, five black and white images based upon five threshold levels (Fig. 2d) were created. Each black pixel of each image is translated by the instrument into one mirror projecting light to the surface. To create the images, we start with all the black pixels ON at t0 = 0 (Fig. 2e), and we sequentially turn mirrors OFF to obtain polymers of different heights at the desired positions. Finally, all mirrors are turned OFF at t5. The last mirrors turning OFF, in t5, are the ones that have been ON during all the process and for a total time of t5, which corresponds to the longest exposure time (i.e., longest polymers). Using the kinetic fit obtained in Fig. 2c, we calculated the exposure times (t0 = 0; t1 = 1.37; t2 = 3.17; t3 = 5.55; t4 = 9.70; and t5 = 16.97 min) required at each pixel for obtaining the selected NF values (1.5, 3, 4, 5, and 6). We then sequentially printed the series of five images to produce a pattern, where the last image corresponds to the higher/brighter layer. Figure 2f shows a composite of nine fluorescence microscopy images of the polymer brush pattern obtained after rinsing the substrate with EtOH and sonicating in DMF for 10 min. AFM measurements of the marked areas in Fig. 2f (Fig. 2g, h) confirm that the different NF intensities correspond to polymer brushes of different h. The smallest isolated pixel size obtained, however, is 10 μm as a result of light dispersion, although with larger feature dimensions, light dispersion is not an issue as 100 mirrors project a pattern of 430 μm.
Grafted-from polymer brush hypersurfaces
Another major challenge we overcome with this platform is the ability to simultaneously control both h and monomer composition at each pixel. That is, create hypersurfaces with potentially thousands of different polymer brushes in the array. Such patterns could be important for applications ranging from microarrays to modeling and understanding interactions of polymers with cells, bacteria, and viruses.
We produced a multicomponent pattern by sequentially introducing three differently colored fluorescent monomer solutions into the reaction cell. This required first calibrating printing conditions for copolymers containing red (methacryloxyethyl thiocarbamoyl rhodamine B (RMA)) and blue ((trifluoromethyl)coumarin]methacrylamide (CMA)). First, the relationship between NF and t was studied for CMA and RMA mixtures at 223:1 and 5000:1 molar ratios to MMA, respectively, following the same protocol as described above. The NFmax values were 6.5 ± 0.6 for RMA and 1.12 ± 0.02 for CMA (Supplementary Fig. 6). The low NF observed on the CMA-based brushes is likely because of the lower incorporation of CMA into the PMMA brush because of the slower polymerization of acrylates compared to methacrylates in free radical polymerizations53. To create the multicolor pattern, a painting of Barcelona was decomposed into three channels (red, green, and blue, Supplementary Fig. 7), each of which was subsequently assigned to a monomer (RMA:MMA, FMA:MMA, and CMA:MMA mixtures). These three images were then processed following the same procedure described for the printing of the Statue of Liberty, but with four threshold levels per channel (Supplementary Fig. 8). Three DMF solutions containing FMA, RMA, and CMA were prepared and sequentially flowed at 5 µL/min over the substrate, while each solution was exposed to the corresponding pattern, followed by 5 min of rinsing at 100 µL/min upon changing solutions. Figure 3 shows a composite of 75 fluorescent microscopy images of the pattern (25 images per channel, see Supplementary Fig. 9). The processing of the merged images is explained in Supplementary Methods and Supplementary Fig. 10. This print demonstrates that, in principle, an unlimited number of monomer compositions and mixtures can be sequentially introduced onto the printing platform to control the polymer composition and h at each pixel, thereby overcoming a major limitation in polymer brush lithography.

a Composite of 75 microscopy images (25 images per channel) showing all three different fluorophores utilized to print an image of the Barcelona skyline (courtesy of Ana Maria Edulescu). b and c magnification of the areas marked in a.
Block copolymer hypersurfaces
Multiplexed block copolymer patterns were printed, where the monomer composition at each x- and y-coordinate, as well as along the chain, was independently controlled, a challenge for which there exists no solution amongst micro- and nanolithography methods. In this pattern, each pixel contained brushes composed of either poly(ethylene glycol dimethacrylate) (pEGDMA), poly(tert-butyl methacrylate) (ptBMA), or block copolymers of the two. The blocks are printed by sequential introduction of the monomer solutions into the reaction cell, and the h of each block is a function of the t. In the first of these patterns, EGDMA and tBMA doped with FMA (300:1) and RMA (6000:1), respectively, were printed into homopolymer and block copolymer brushes that could be analyzed by fluorescence microscopy. The pattern consisted of four rows, in which first and third rows (from top to bottom) were printed forming nine features at nine different t (2–25 min). The second and fourth rows were printed to form a continuous feature at 18 different t (2–25 min) (Fig. 4a). Then, p(tBMA-co-RMA) was printed partially overlapped with the p(EGDMA-co-FMA) pattern (Fig. 4b). The composite fluorescence microscopy image (Fig. 4c) shows 18 different proportions of p(EGDMA-co-FMA)-block-p(tBMA-co-RMA) in rows 1 and 3 and two different gradients in rows 2 and 4. The colocalization of the two dyes is evidence of block copolymer formation.

a Fluorescence microscopy image (λex = 530–550 nm, barrier filter λem = 575 nm) of a p(tBMA-FMA) (300:1) pattern composed of four rows printed at 1–25 minutes (left to right). b Fluorescence microscopy image (λex = 530–550 nm, barrier filter λem = 575 nm) of the same area as a showing a p(EGDMA-RMA) (6000:1) pattern composed of four rows printed at 1–25 min from right to left (top rows) and left to right (bottom rows). c Composite of a and b. d AFM height image of an array composed of p(tBMA) brush features (triangles and middle row) printed at 5, 10, and 20 min t from right to left. Bottom row is composed of p(EGMA) brushes which were also printed over the p(tBMA) on middle row at 5, 10, and 20 min from right to left. e Average height profiles of bands marked by colored squares in d.
Another pattern was printed for analysis by AFM, where the changes in h confirmed the formation of block copolymer brushes. To this end, 432 repeats of a 3 × 3 pattern composed of rows of p(tBMA), p(EGDMA), and p(tBMA)-block-p(EGDMA) were printed (Fig. 4). First, in the top and middle rows of this pattern, features of p(tBMA) were grown with t of 5, 10, and 20 min. Then, the reaction cell was rinsed with an EGDMA solution in DMF at 100 µL/min for 2 min. The EGDMA was then patterned at same t in the middle and bottom rows, where in the middle row the p(tBMA) blocks grow from the living p(EGMA) block. Figure 4d shows AFM analysis of one of these 3 × 3 patterns. In the top and bottom row, which are the p(tBMA) and p(EGDMA) homopolymers, respectively, h increases with increasing t. The middle row, where the blocks were grown on top of each other, h is greater than either the top or bottom row, indicating that the chain ends remain living, and that the polymers continue to grow upon the introduction of the tBMA.
Discussion
We have developed a platform for the photochemical patterning of block copolymer arrays, where the position and composition of each of the > 750,000 pixels can be independently controlled, and with micrometer-scale feature resolution. Because the surface is irradiated by a computer-modulated DMD, arbitrary patterns can be printed without necessitating an expensive series of photomasks. The integration of microfluidics and an air-free reaction chamber with the DMD is the key innovation that allows the spatiotemporally controlled grafting of different materials onto the substrate, and could, in principle, be used to make polymer patterns composed of a practically unlimited number of unique brush compositions. Although living SI-ATRP offers broad monomer scope and narrow molecular weight distribution, it is also challenging since it requires oxygen-free conditions, but the environmental control enabled by the air-free reaction cell means that this platform is compatible with even the most demanding photochemical reactions. We also demonstrate the ability to quantify polymer brush kinetics in a single print, and have printed random- and block copolymer microarrays, where in the latter, monomer composition along the chain was carefully regulated. While the SI-ATRP polymerization was studied here, this printer is a general tool for combinatorial surface photochemistry, and the facility with which hundreds or even thousands of different reactions conditions can be attempted in each print promises to rapidly accelerate progress in research disciplines where interfacial organic composition has a critical role. In the future, we will explore different chemistries, automate the microfluidics to coordinate them with the DMD, and integrate beam pen arrays54 to improve throughput, resolution, and versatility. Ultimately, we envision a new era of soft lithography where the fabrication of synthetic surfaces with complexity comparable to what is found in biological interfaces will soon become a reality.
Methods
Substrate functionalization
All materials were purchased from VWR or Fisher unless otherwise noted. Four-inch <100> silicon wafers with 500 nm thermal oxide layer (Nova Electronic Materials, USA) were cleaned in piranha solution (3:1 H2SO4:H2O2) for 15 min, and rinsed with Milli-Q water (18 MΩ). The wafer pieces were then functionalized with (3-aminopropyl)triethoxysilane (Gelest, USA) following a previously reported method55. These amine-terminated substrates were immersed in 120 mL of CH2Cl2 with 1.67 mL (12 mmol) Et3N for 30 min. A total of 1.48 mL (12 mmol) of α-bromoisobutyryl bromide was then added, and the solutions were kept for 18 h in the dark. Substrates were then rinsed with CH2Cl2, EtOH, and Milli-Q H2O, dried with an air gun, and stored in an Ar atmosphere in the dark until used.
Printing solutions
FMA (Millipore-Sigma, USA) was purified through a silica column (SiO2 CH2Cl2:MeOH 97:3). Methacryloxyethyl thiocarbamoyl rhodamine B (RMA) (Polysciences Inc., USA) was purified through a silica column (SiO2 CH2Cl2:MeOH 98.5:1.5). Inhibitor was removed from MMA, EGDMA, and tBMA (Alfa-Aesar, USA) by flowing each monomer through a short plug of basic alumina. After the alumina plug, monomers were degassed in a vial wrapped with aluminum by bubbling Ar for 1 hour. 7-[4-(Trifluoromethyl)coumarin]methacrylamide (CMA) (Millipore-Sigma, USA), and Tris[2-phenylpyridinato-C2,N]iridium(III) (Ir(ppy)3) (Millipore-Sigma, USA) were used without further purification. Printing solutions were prepared as follows: Ir(ppy)3 (1.82 mM in distilled DMF) stock solution was first prepared and kept in the glovebox. FMA solution (24.9 mM FMA, 7.5 M of MMA, and 0.36 mM of Ir(ppy)3 in DMF), RMA (1.25 mM RMA, 7.5 M MMA, and 0.61 mM of Ir(ppy)3 in DMF), CMA (33.6 mM CMA, 7.5 M MMA, and 0.36 mM Ir(ppy)3 in DMF), EGDMA (4.24 M EGDMA, 0.364 mM of Ir(ppy)3 in DMF), and tBMA (4.92 M tBMA and 0.364 mM of Ir(ppy)3 in DMF) stock solutions were prepared for printing.
Photochemical patterning
The printer uses a TERA-Fab E Series instrument (TERA-print, USA) equipped with DMD containing 1024 × 768 micromirrors, a 405 nm LED light source (32 mW cm−2), and a piezoactuated x, y, and z platform. An air-free chamber was made with two polystyrene dishes, equipped with an Ar inlet with a valve, and two apertures for the microfluidic tubing connection. After setting the reactive substrate inside the glovebox, the valve is closed and the chamber is sealed with Parafilm, including inlet/outlet apertures for the microfluidics. Once out of the glovebox, the Ar inlet is plugged to a T connection with two more valves for purging of the air from the tubing. After purging the gas lines, the Ar valve is opened, the tubing seals are removed, and the PEEK 1/16˝ tubing introduced. The solutions are then flowed into the reactive droplet using NE-1000 Programmable Syringe Pumps (Pump Systems Inc., USA). All prints were performed at 8 mW cm−2. The t used to print Fig. 2a were: 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 16, 18, 20, and 22 min.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Klein, J., Kumacheva, E., Mahalu, D., Perahia, D. & Fetters, L. J. Reduction of frictional forces between solid surfaces bearing polymer brushes. Nature 370, 634–636 (1994).
Wei, Q. et al. Protein Interactions with polymer coatings and biomaterials. Angew. Chem. Int. Ed. 53, 8004–8031 (2014).
Zhai, L. Stimuli-responsive polymer films. Chem. Soc. Rev. 42, 7148–7160 (2013).
Hucknall, A. et al. Simple fabrication of antibody microarrays on nonfouling polymer brushes with femtomolar sensitivity for protein analytes in serum and blood. Adv. Mater. 21, 1968–1971 (2009).
Barbey, R., Kauffmann, E., Ehrat, M. & Klok, H.-A. Protein microarrays based on polymer brushes prepared via surface-initiated atom transfer radical polymerization. Biomacromolecules 11, 3467–3479 (2010).
Godula, K. & Bertozzi, C. R. Density variant glycan microarray for evaluating cross-linking of mucin-like glycoconjugates by lectins. J. Am. Chem. Soc. 134, 15732–15742 (2012).
Luzinov, I., Minko, S. & Tsukruk, V. V. Adaptive and responsive surfaces through controlled reorganization of interfacial polymer layers. Prog. Polym. Sci. 29, 635–698 (2004).
Stuart, M. A. C. et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 9, 101–113 (2010).
Leslie, D. C. et al. A bioinspired omniphobic surface coating on medical devices prevents thrombosis and biofouling. Nat. Biotechnol. 32, 1134–1140 (2014).
Anseth, K. S. et al. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control. Release 78, 199–209 (2002).
Khademhosseini, A., Langer, R., Borenstein, J. & Vacanti, J. P. Microscale technologies for tissue engineering and biology. Proc. Natl Acad. Sci. USA 103, 2480–2487 (2006).
Krishnamoorthy, M., Hakobyan, S., Ramstedt, M. & Gautrot, J. E. Surface-initiated polymer brushes in the biomedical field: applications in membrane science, biosensing, cell culture, regenerative medicine and antibacterial coatings. Chem. Rev. 114, 10976–11026 (2014).
Gunnewiek, M. K. et al. Creeping proteins in microporous structures: polymer brush-assisted fabrication of 3D gradients for tissue engineering. Adv. Healthc. Mater. 4, 1169–1174 (2015).
Zhao, B. & Brittain, W. J. Polymer brushes: surface-immobilized macromolecules. Prog. Polym. Sci. 25, 677–710 (2000).
Edmondson, S., Osborne, V. L. & Huck, W. T. S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 33, 14–22 (2004).
Chen, M., Zhong, M. & Johnson, J. A. Light-controlled radical polymerization: mechanisms, methods, and applications. Chem. Rev. 116, 10167–10211 (2016).
Percec, V. et al. Aqueous room temperature metal-catalyzed living radical polymerization of vinyl chloride. J. Am. Chem. Soc. 124, 4940–4941 (2002).
von Werne, T. A. et al. A versatile method for tuning the chemistry and size of nanoscopic features by living free radical polymerization. J. Am. Chem. Soc. 125, 3831–3838 (2003).
Zoppe, J. O. et al. Surface-initiated controlled radical polymerization: state-of-the-art, opportunities, and challenges in surface and interface engineering with polymer brushes. Chem. Rev. 117, 1105–1318 (2017).
Pan, X., Fantin, M., Yuan, F. & Matyjaszewski, K. Externally controlled atom transfer radical polymerization. Chem. Soc. Rev. 47, 5457–5490 (2018).
Sumerlin, B. S., Neugebauer, D. & Matyjaszewski, K. Initiation efficiency in the synthesis of molecular brushes by grafting from via atom transfer radical polymerization. Macromolecules 38, 702–708 (2005).
Fors, B. P. & Hawker, C. J. Control of a living radical polymerization of methacrylates by light. Angew. Chem. 124, 8980–8983 (2012).
Poelma, J. E., Fors, B. P., Meyers, G. F., Kramer, J. W. & Hawker, C. J. Fabrication of complex three-dimensional polymer brush nanostructures through light-mediated living radical polymerization. Angew. Chem. Int. Ed. 52, 6844–6848 (2013).
Wei, W., Balamurugan, A., Dwyer, J. H. & Gopalan, P. Substrate-independent approach to dense cleavable polymer brushes by nitroxide-mediated polymerization. ACS Macro Lett. 7, 100–104 (2018).
Shah, R. R. et al. Using atom transfer radical polymerization to amplify monolayers of initiators patterned by microcontact printing into polymer brushes for pattern transfer. Macromolecules 33, 597–605 (2000).
Hyun, J. & Chilkoti, A. Surface-initiated free radical polymerization of polystyrene micropatterns on a self-assembled monolayer on Gold. Macromolecules 34, 5644–5652 (2001).
Tu, H., Heitzman, C. E. & Braun, P. V. Patterned poly(N-isopropylacrylamide) brushes on silica surfaces by microcontact printing followed by surface-initiated polymerization. Langmuir 20, 8313–8320 (2004).
Xu, F. J., Li, Y. L., Kang, E. T. & Neoh, K. G. Heparin-coupled poly(poly(ethylene glycol) monomethacrylate)-Si(111) hybrids and their blood compatible surfaces. Biomacromolecules 6, 1759–1768 (2005).
Matsuura, K., Utoh, R., Nagase, K. & Okano, T. Cell sheet approach for tissue engineering and regenerative medicine. J. Control. Release 190, 228–239 (2014).
Bhat, R. R., Chaney, B. N., Rowley, J., Liebmann-Vinson, A. & Genzer, J. Tailoring cell adhesion using surface-grafted polymer gradient assemblies. Adv. Mater. 17, 2802–2807 (2005).
Wu, J., Mao, Z. & Gao, C. Controlling the migration behaviors of vascular smooth muscle cells by methoxy poly(ethylene glycol) brushes of different molecular weight and density. Biomaterials 33, 810–820 (2012).
Ren, T., Mao, Z., Guo, J. & Gao, C. Directional migration of vascular smooth muscle cells guided by a molecule weight gradient of poly(2-hydroxyethyl methacrylate) brushes. Langmuir 29, 6386–6395 (2013).
Pester, C. W. et al. Engineering surfaces through sequential stop-flow photopatterning. Adv. Mater. 28, 9292–9300 (2016).
Giam, L. R. & Mirkin, C. A. Cantilever-free scanning probe molecular printing. Angew. Chem. Int. Ed. 50, 7482–7485 (2011).
Garcia, R., Knoll, A. W. & Riedo, E. Advanced scanning probe lithography. Nat. Nano. 9, 577–587 (2014).
Kaholek, M., Lee, W.-K., LaMattina, B., Caster, K. C. & Zauscher, S. Fabrication of stimulus-responsive nanopatterned polymer brushes by scanning-probe lithography. Nano Lett. 4, 373–376 (2004).
Chen, L. et al. Biomimicking nano-micro binary polymer brushes for smart cell orientation and adhesion control. Small 12, 3400–3406 (2016).
Paik, M. Y. et al. Patterning of polymer brushes. A direct approach to complex, sub-surface structures. Nano Lett. 10, 3873–3879 (2010).
Sweat, D. P. et al. A dual functional layer for block copolymer self-assembly and the growth of nanopatterned polymer brushes. Langmuir 29, 12858–12865 (2013).
Bian, S. et al. Beam pen lithography as a new tool for spatially controlled photochemistry, and its utilization in the synthesis of multivalent glycan arrays. Chem. Sci. 5, 2023–2030 (2014).
Liu, X., Zheng, Y., Peurifoy, S. R., Kothari, E. A. & Braunschweig, A. B. Optimization of 4D polymer printing within a massively parallel flow-through photochemical microreactor. Polym. Chem. 7, 3229–3235 (2016).
Ge, Q., Qi, H. J. & Dunn, M. L. Active materials by four-dimension printing. Appl. Phys. Lett. 103, 131901 (2013).
Singh-Gasson, S. et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nat. Biotechnol. 17, 974 (1999).
Pellois, J. P. et al. Individually addressable parallel peptide synthesis on microchips. Nat. Biotechnol. 20, 922–926 (2002).
Choi, J.-W. et al. Fabrication of 3D biocompatible/biodegradable micro-scaffolds using dynamic mask projection microstereolithography. J. Mater. Process. Technol. 209, 5494–5503 (2009).
Nielson, R., Kaehr, B. & Shear, J. B. Microreplication and design of biological architectures using dynamic-mask multiphoton lithography. Small 5, 120–125 (2009).
Miri, A. K. et al. Microfluidics-enabled multimaterial maskless stereolithographic bioprinting. Adv. Mater. 30, 1800242 (2018).
Carbonell, C. et al. Massively multiplexed tip-based photochemical lithography under continuous capillary flow. Chem 4, 857–867 (2018).
Matyjaszewski, K. & Tsarevsky, N. V. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 1, 276–288 (2009).
Chmielarz, P., Krys, P., Wang, Z., Wang, Y. & Matyjaszewski, K. Synthesis of well-defined polymer brushes from silicon wafers via surface-initiated seATRP. Macromol. Chem. Phys. 218, 1700106 (2017).
Pyun, J., Kowalewski, T. & Matyjaszewski, K. Synthesis of polymer brushes using atom transfer radical polymerization. Macromol. Rapid Commun. 24, 1043–1059 (2003).
Iura, T. & Ohtani, H. Fragmentation behavior of poly(methyl methacrylate) during matrix-assisted laser desorption/ionization. Rapid Commun. Mass Spectrom. 29, 155–162 (2015).
Roos, S. G., Müller, A. H. E. & Matyjaszewski, K. Copolymerization of n-butyl acrylate with methyl methacrylate and PMMA macromonomers: comparison of reactivity ratios in conventional and atom transfer radical copolymerization. Macromolecules 32, 8331–8335 (1999).
Liao, X. et al. Desktop nanofabrication with massively multiplexed beam pen lithography. Nat. Commun. 4, 2103 (2013).
Nicholas, M. P., Rao, L. & Gennerich, A. Covalent immobilization of microtubules on glass surfaces for molecular motor force measurements and other single-molecule assays. Methods Mol. Biol. 1136, 137–169 (2014).
Acknowledgements
This work was carried out under government support from the National Science Foundation (DBI-1661702), a MURI award from the Department of Defense (MURI FA9550-16-1-0150), an Air Force Office of Science and Research (FA9550-17-1-0356), an AFOSR National Defense Science and Engineering Graduate Fellowship, 32 CFR 168a, to M.A.T., and an NSF Graduate Research Fellowship (DGE-1144086) to A.S.C. The authors would also like to acknowledge the Surface Science Facility of CUNY Advanced Science Research Center in Graduate Center for the training and use of XPS and AFM. We kindly thank S. Shafaie for technical support on MALDI-IMS. This work was performed in part at the Advanced Science Research Center Nanofabrication facility of the Graduate Center at the City University of New York, which receives support from the State of New York. This work made use of the IMSERC at Northwestern University, which received support from the State of Illinois. We are grateful to Oliver Kuehl and Ana Maria Edulescu, who provided the original images that were used to prepare Figs. 2f and 3, respectively.
Author information
Authors and Affiliations
Contributions
A.B.B. and N.C.G. conceived the research. A.B.B., N.C.G., and C.C. coordinated the research. A.B. and C.C. designed the printer and the experiments. C.C., D.V., A.M.W., A.S.C., M.A.T., and J. K. performed the experiments. All authors contributed to analysis and discussion of the data. The manuscript was written by C.C. and A.B.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carbonell, C., Valles, D., Wong, A.M. et al. Polymer brush hypersurface photolithography. Nat Commun 11, 1244 (2020). https://doi.org/10.1038/s41467-020-14990-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14990-x
This article is cited by
-
Research progress of quantum dot photolithography patterning and direct photolithography application
Nano Research (2024)
-
Electric field-induced morphological changes on polymer surface using phase-field model
Journal of Materials Science (2023)
-
Coarse-grained Dynamics Simulation in Polymer Systems: from Structures to Material Properties
Chemical Research in Chinese Universities (2022)
-
Dip-Pen Nanolithography(DPN): from Micro/Nano-patterns to Biosensing
Chemical Research in Chinese Universities (2021)
-
Design and Fabrication of a Disposable Nanoactuator Using 3D Printing
International Journal of Precision Engineering and Manufacturing (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
