
Abstract
The human gut microbiota (HGM), which is critical to human health, utilises complex glycans as its major carbon source. Glycosaminoglycans represent an important, high priority, nutrient source for the HGM. Pathways for the metabolism of various glycosaminoglycan substrates remain ill-defined. Here we perform a biochemical, genetic and structural dissection of the genetic loci that orchestrates glycosaminoglycan metabolism in the organism Bacteroides thetaiotaomicron. Here, we report: the discovery of two previously unknown surface glycan binding proteins which facilitate glycosaminoglycan import into the periplasm; distinct kinetic and genetic specificities of various periplasmic lyases which dictate glycosaminoglycan metabolic pathways; understanding of endo sulfatase activity questioning the paradigm of how the ‘sulfation problem’ is handled by the HGM; and 3D crystal structures of the polysaccharide utilisation loci encoded sulfatases. Together with comparative genomic studies, our study fills major gaps in our knowledge of glycosaminoglycan metabolism by the HGM.
Similar content being viewed by others

Introduction
The human gut microbiota (HGM) is a vast microbial community that is crucial to human health1. The maintenance of the HGM is dependent on its ability to utilise complex glycans as a nutrient source, in the form of dietary and host glycans2. The mechanisms by which this community metabolises these glycans is therefore critical to understanding the ecology of the system and how it could be manipulated to benefit human health. Bacteroidetes is one of the two major phyla that dominate the HGM3. These organisms dedicate up to 20% of their genome to complex glycan metabolism in the form of genes primarily encoding carbohydrate active enzymes such as, polysaccharide lyases (PLs), glycoside hydrolases, glycan transport and binding proteins, and carbohydrate sulfatases. Bacteroides arrange these genes into polysaccharide utilisation loci (PUL) which are co-localised, co-regulated genes, in response to a specific glycan4,5. The vast majority of our understanding of how Bacteroidetes metabolise complex glycans is based on dietary plant glycans6,7,8,9 yet host glycans represent a substantial carbon reservoir, which is also accessed by the HGM10,11. Glycosaminoglycans (GAGs) have been demonstrated to be a high-priority carbon source for the HGM organism Bacteroides thetaiotaomicron (B. theta)11,12, and although a pathway for Heparin/Heparan sulfate13 (H/HS) has been described, the metabolism of the complex, and distinct, GAGs chondroitin sulfate (CS-A and CS-C), dermatan sulfate (DS) and hyaluronic acid (HA) is poorly described. Enzymes of the HGM that metabolise CS have been described13,14,15, yet how they co-ordinate together, their mechanistic details, mode of action and sulfation tolerances remain largely opaque. In addition, the role and mechanism of action of the surface glycan binding proteins (SGBPs) in the degradation of CS, DS and HA is unknown. Here we report how a core PUL has evolved to metabolise the complexity and heterogeneity of CS-A, CS-C, DS and HA. The PULCS/DS/HA is co-regulated with distant genes, including a hyaluronidase belonging to the recently classified PL3316, to complement its core activities and thus fully enabling multiple GAG breakdown. Crystal structures of the two PUL-encoded carbohydrate sulfatases provide key insights into substrate recognition by these critical, and therapeutically relevant17, enzymes. The data enable a general model to be proposed for the metabolism of CS, DS and HA by the HGM and the Bacteroidetes phylum in a wider context.
Results
Structure of CS-A, CS-C, DS and HA
CS-A and CS-C are both composed of repeating disaccharide units composed of d-glucuronic acid (GlcA) and N-acetyl-D-galactosamine (GalNAc). GlcA is linked β(1,3) to GalNAc which is then linked β(1,4) to the next GlcA. CS-A and CS-C differ only in sulfation with CS-A highly enriched in O4 sulfation on GalNAc whilst CS-C is enriched in O6 sulfation on GalNAc. DS is related to CS-A except GlcA is predominantly replaced with l-iduronic acid which is connected to GalNAc via an α(1,4) linkage. Finally, HA is similar to CS-A but replaces GalNAc with N-acetyl-D-glucosamine (GlcNAc) and contains no sulfation (Fig. 1b).

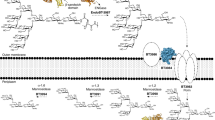
a Organisation of the PULCS/DS/HA locus in Bacteroides thetaiotaomicron. b Schematic depiction of the activity, location and specificity of PULCS/DS/HA encoded enzymes. Asterisk indicates structures were solved in this study.
PULCS/DS/HA structure
B. theta grows on CS-A, CS-C, DS and HA and transcriptomics, on CS-C and HA, demonstrate only PULCS/DS/HA and two additional genes, BT1596-S1_92S-sulf and BT4410PL33 are upregulated4,13. PULCS/DS/HA encodes three PLs (BT3324PL8, BT3328PL29 and BT3350PL8), one GH88 (BT3348GH88), 2-O-sulfatases (BT3333-S1_156S-sulf and BT3349-S1_274S-sulf), a single SusC/D-like transporter system (BT3331susD-BT3332susC), a hybrid two-component sensor (BT3334HTCS) and two proteins of unknown function (BT3329 and BT3330). The non-PUL encoded genes, BT1596-S1_92S-sulf and BT4410PL33, encode a 2-O sulfatase and a hyaluronidase, respectively (Fig. 1a, b). A paradigm for how PULs organise their protein apparatus has been established. A surface enzyme(s), with usually one SGBP, degrades and captures large polysaccharides/oligosaccharides at the cell surface. These partially degraded polysaccharides/oligosaccharides are transported into the periplasm through the action of the highly conserved, and critical, SusC/D-like transporter system. Small polysaccharides/large oligosaccharides transported into the periplasm are then metabolised to their constituent parts, usually monosaccharides, and transported into the cytoplasm to enter cytoplasmic metabolic pathways.
Extracellular glycan binding and degradation
Two genes of unknown function BT3329 and BT3330 exist within PULCS/DS/HA. Both proteins had no enzymatic activity against any GAG substrates. However, they were shown to exhibit binding by isothermal titration calorimetry (ITC). Both proteins were able to bind all four GAGs and were thus classified as SGBPs (Supplementary Fig. 1 and Supplementary Table 1). BT3329SGBP bound to CS-A and HA with approximately ten and fourfold higher affinity than BT3330SGBP whilst against CS-C and DS BT3329SGBP bound with an ~100 and ~50-fold higher affinity. BT3329SGBP but not BT3330SGBP bound a CS oligosaccharide of DP10. BT3330SGBP was, however, able to bind a CS oligosaccharide of DP20. The SGBPs thus differ in their polysaccharide and oligosaccharide preferences. BT3329SGBP has the ability to bind more variably sulfated and smaller glycans than BT3330SGBP, and displays a greater tolerance to iduronic acid (Supplementary Fig. 1 and 2a). Both proteins were confirmed to be extracellular by immunofluorescence assays (Supplementary Fig. 2b). BT3331susD demonstrated a very strict substrate specificity binding only to CS-A with a measurable affinity, similar to that of BT3329SGBP (Supplementary Table 1). The surface lyase, BT3328PL29, showed its highest enzymatic activity and affinity against CS-A > CS-C > HA and little activity on DS, differing from previous data18. End point assays, the total number of glycosidic bonds cleaved, followed the relationship of HA > CS-C > CS-A (Fig. 2 and Supplementary Table 2). This demonstrates that although O4 sulfation increases BT3328PL29 activity on CS-A it also limits glycosidic bond access in some contexts. BT3328PL29 was only able to cleave HA oligosaccharides with a DP ≥ 8, indicating the enzyme has at least eight subsites (Supplementary Fig. 4a). Interestingly, BT3328PL29 displays very similar substrate specificities to BT3331susD which, could suggest that the proteins work closely together at the cell surface. When BT3328PL29 was incubated with HA oligosaccharides of d.p. 12, 10 and 8, labelled at their reducing end with a fluorescent tag, a labelled trisaccharide was produced from d.p. 12 substrate and a labelled disaccharide was produced from d.p. 10 and 8 substrates. These data indicate that BT3328PL29 cleaves glycans from the reducing end (Supplementary Fig. 2c, d and Supplementary discussion). We identified a conserved region of ~300 amino acids at the N-terminal of BT3328PL29 and BT3329SGBP. This region was also shared with SGBP-positioned genes from other PULs, which do not share a detectable sequence similarity downstream of this ~300 amino acid at the N-terminal region, like BT3328PL29 and BT3329SGBP. This might constitute a ‘spacer’ domain, not homologous but, functionally equivalent to the BACON domain found at the N-terminal region of the surface-anchored GH5 in the xyloglucan PUL7 and in many other SGBPs. Such domains would allow extracellular proteins to extend out of the capsular envelope and/or increase their mobility.

Peaks were identified by comparison with commercially purchased standards. The black, blue, green and red lines represent 0, early, mid-point and late stages of the reaction time course for each enzyme (CS-A chondroitin sulfate A, DS dermatan sulfate, CS-C chondroitin sulfate C and HA hyaluronic acid). All reactions were carried out in 50 mM MES, pH 6.5, with 150 mM NaCl at 37 °C.
Protein–protein interactions
The interaction of the cell surface proteins with one another was assessed by Native PAGE and co-immunoprecipitation (Co-IP). Native PAGE demonstrated that BT3328PL29, BT3329SGBP, BT3330SGBP and BT3331SusD displayed no observable interaction in vitro (Supplementary Fig. 3a, b). Co-IP data, however, demonstrated that at the cell surface BT3328PL29, BT3329SGBP and BT3330SGBP do indeed interact (Supplementary Fig. 3c–e). Co-IPs using anti-BT3328PL29 and anit-BT3329SGBP antibodies revealed the presence of both BT3329SGBP and BT3330SGBP in BT3328PL29-IP and the presence of both BT3328PL29 and BT3330SGBP in BT3329SGBP-IP. When the BT3330SGBP precipitant was analysed only BT3329SGBP could be observed and not BT3328PL29. Collectively these data suggest that BT3328PL29, BT3329SGBP and BT3330SGBP interact with each other at the bacterial cell surface in vivo. This interaction requires either membrane attachment or the presence of the integral membrane protein BT3332SusC.
Periplasmic glycan degradation apparatus
Biochemical analysis of the periplasmic lyases: BT3324PL8 and BT3350PL8 are both PL8s whilst BT4410 has recently been classified into family PL33. BT3350PL8 is a broad specificity lyase having similar activity on CS-A, CS-C and DS but had reduced activity on HA (Supplementary Table 2). BT3350PL8 displayed an endo mode of action but disaccharides were observed earlier in the reaction time course on the sulfated substrates (Fig. 2). End point assays revealed that BT3350PL8 could cleave similar amounts of glycosidic linkages in CS-A and DS but around half as much in the GAGs CS-C and HA (Supplementary Table 2). The end products of BT3350PL8 HA degradation are mainly of DP2, 4 and 6, with oligos of ≥DP4 making up the majority of the population (Fig. 2). Against CS-A the end products were mainly DP2 and DP4, whilst against CS-C and DS a greater quantity of products >DP6 were observed. Surprisingly, BT3350PL8 was able to cleave HA oligos down to disaccharides (Supplementary Fig. 4a). BT3324PL8 is an exo-acting lyase releasing exclusively disaccharides from all the GAGs tested (Fig. 2 and Supplementary Fig. 4a). BT3324PL8 displayed its highest activity against CS-A and DS with much reduced activities against CS-C and HA. Thus, O4 sulfation is required for maximal activity. BT4410PL33 displayed a highly endo-processive mode of action and displayed >100-fold preference for HA. End point assays showed the presence of sulfation limited substrate accessibility, consistent with BT4410PL33 being tailored towards HA (Fig. 2 and Supplementary Table 2).
Genetic analyses of the PULCS/DS/HA lyases: The contribution of each lyase in GAG metabolism was further assessed through growth experiments with single, double and triple genetic mutants. Using CS-A and CS-C as the substrate, we observed that individual PL mutants exhibited very little evidence of growth defects; Δbt3324 exhibited a slight defect in growth rate on CS-A (Fig. 3a). The Δbt3350/Δbt4410 showed a stronger phenotype than Δbt3324/Δbt3350 on CS-A, whilst on CS-C both mutants exhibited the same level of defect. This is due to O6 and/or O2 sulfation in CS-C being inhibitory to BT4410PL33. Whilst for CS-A the endo 4S sulfatase, BT3349-S1_274S-sulf (see ‘O-sulfatases’ section), is able to remove 4S sulfation thus allowing enhanced activity of BT4410PL33 on CS-A (Further detail in Supplementary discussion). The double-mutant Δbt3324/Δbt4410 showed extremely limited or no growth on all substrates, comparable with the triple-mutant Δbt3324/Δbt3350/Δbt4410 (Fig. 3a). The Δbt3324/Δbt4410 mutant was previously shown to have drastically reduced BT3333-S1_156S-sulf mRNA levels in the presence of CS13, thus, BT3350PL8 alone cannot generate disaccharides at a significant rate as to upregulate the PUL. BT3350PL8 must, therefore, work in conjunction with BT3324PL8 and BT4410PL33 to complete GAG catabolism to their disaccharide repeating units. These data are consistent with BT3350PL8 generating new non-reducing ends for BT3324PL8 and BT4410PL33 to act on, demonstrating an endo–exo synergy as commonly observed in other glycan-degrading systems. When HA is the substrate, all mutants that show a defect involved the deletion of BT4410PL33. The Δbt4410 mutant by itself was sufficient to cause a severe growth defect, whilst the Δbt3324/Δbt3350 mutant showed only a minor defect, reinforcing the critical role of BT4410PL33 as a hyaluronan specific lyase (Fig. 3a). When DS is the growth substrate, loss of any periplasmic lyase alone is sufficient to cause a significant defect, but loss of either BT4410PL33 or BT3350PL8 is particularly severe (Fig. 3a). Both enzymes have endo character indicating that the ability to cleave linkages internally in DS is critical to its breakdown by B. theta. All double mutants failed to grow at all on DS (Fig. 3a).

a Growth of genetic knockouts on glycosaminoglycan substrates (CS-A chondroitin sulfate A, DS dermatan sulfate, CS-C chondroitin sulfate C and HA hyaluronic acid); b TLC analysis of purified oligosaccharide (X), left behind in media from Δbt3324 growths, against PULCS/DS/HA encoded lyases; c HPLC analysis of X treated with BT3324PL8. Data represent technical triplicates and source data are provided in the source data file labelled as mutant growths.
Analyses of spent supernatants from genetic mutants: Analysis of spent supernatants from mutant growths revealed that the Δbt3324 mutant, produced a unique product. This substrate was purified and treated with the different lyases; only BT3324PL8 showed activity (Fig. 3b). The products were analysed by HPLC and two peaks were identified as UA2S-GalNAc6S disaccharide and either UA-GalNAc4S or UA-GalNAc6S in a ratio of 3:1 (Fig. 3c). The accumulation of this substrate reveals that BT3324PL8 is the only enzyme in B. theta capable of cleaving regions within CS-C enriched in 2S sulfation.
O-Sulfatases
PULCS/DS/HA encodes two O sulfatases belonging to the sulfatase subfamilies S1_27 and S1_15, the endo 4S sulfatase BT3349-S1_274S-sulf and the exo-acting 6S sulfatase BT3333-S1_156S-sulf, as classified by the sulfatlas database19. BT1596-S1_92S-sulf is located outside the PUL and belongs to subfamily S1_9. BT3349-S1_274S-sulf and BT1596-S1_92S-sulf are active on all O4 and O2 sulfated CS disaccharides, respectively. This is consistent with previous work on these subfamilies20. Whilst BT3333-S1_156S-sulf was only active on GalNAc6S and is the first member to be functionally and structurally characterised within the S1_15 subfamily (Supplementary Fig. 4b, c). BT3349-S1_274S-sulf contains an SpII signal peptide (Supplementary Table 3) and, due to its endo mode of action, it was hypothesised that the enzyme may be attached to the extracellular surface. Measures of the enzymatic activity at the cell surface by whole-cell assays (see ‘Methods’) did not result in any degradation of Δ4,5UA-GalNAc4S, a substrate for BT3349-S1_274S-sulf, even after an incubation time of 16 h. However, when the cells were sonicated and the disaccharide incubated with the CFE, Δ4,5UA-GalNAc4S was completely degraded within 1 h (Supplementary Fig. 4d). This confirms BT3349-S1_274S-sulf is a periplasmic enzyme. BT3333-S1_156S-sulf also contains an SpII signal peptide but whole-cell assays, using GalNAc6S, indicate that BT3333-S1_156S-sulf is periplasmic (Supplementary Fig. 4d). This is consistent with the requirement for the periplasmic BT3348GH88 to produce its GalNAc6S substrate (Supplementary Fig. 4d).
Crystal structures of BT3349-S1_274S-sulf and BT3333-S1_156S-sulf: Both enzymes are α/β proteins, composed of two domains, a larger N-terminal domain and a smaller C-terminal ‘sub-domain’ as is typical of formylglycine sulfatases21. An inactive form of BT3349-S1_274S-sulf was crystallised with a CS-A tetrasaccharide, but only a trisaccharide of Δ4,5UA-GalNAc4S-GlcA could be modelled (Fig. 4a, b, Supplementary Fig. 5a). Subsite nomenclature for carbohydrate sulfatases is such that an S site denotes the position of the labile sulfate, which is attached to a 0 subsite sugar. Additional subsites then increase in number toward the reducing end of the glycan, i.e. +1, +2, etc., and decrease in number toward the non-reducing end of the glycan, i.e. −1, −2, etc22. Electron density for the −1, 0 and S subsites was well ordered whilst the +1 GlcA was partially disordered and makes no interactions with the protein. A detailed description of the mutants is provided in the supplementary discussion (Supplementary Table 5 and Supplementary Fig. 5e). The endo activity of BT3349-S1_274S-sulf is conferred by an ‘opening up’ of either side of the 0 subsite, allowing glycan extension in both the +1 and −1 direction. Structural comparison of HGM, marine and human carbohydrate sulfatases reveals that the linker between β-strand 4 and α-helix 5 is extended, and/or alterations of the C-terminal subdomain (Fig. 4c–h) commonly occur in the exo sulfatases, precluding endo activity. In BT3333-S1_156S-sulf, however, occlusion of potential positive subsites is achieved by an insertion between β-strand 3 and α-helix 4. These features would also prevent extension of the sugar chain beyond the 0 subsite in the endo sulfatases.

a Cartoon representation of BT3349-S1_274S-sulf ramped blue to red from its N- to C-terminus; b stick representation of binding site interactions with the protein in green and the bound trisaccharide yellow; c surface representation of BT3349-S1_274S-sulf showing how the +1 and −1 ends of the trisaccharide are free to be extended; d overlays of surface representations of BT3349-S1_274S-sulf and a cartoon of the marine endo 4S ι-carrageenan sulfatase (PsS1_19A). CS and ι-carrageenan oligosaccharides are shown in yellow and cyan, respectively. e Overlay of a surface of BT3349-S1_274S-sulf with a cartoon of the exo-sulfatase BT1596-S1_92S-sulf. f Overlay of a surface of BT3349-S1_274S-sulf with a cartoon of the exo-sulfatase BT3333-S1_156S-sulf. g Overlay of a surface of BT3349-S1_274S-sulf with a cartoon of the human exo-sulfatase 1FSU4S-sulf. h Overlay of a surface of BT3349-S1_274S-sulf with a cartoon of the human exo-sulfatase 5FQL2S-sulf. All cartoon representations are ramped blue to red from their N- to C-terminus.
The structure of BT3333-S1_156S-sulf was solved in complex with GalNAc6S and displays a pocket topology (Fig. 5a, Supplementary Fig. 5b). The 0 subsite is made up primarily of polar interactions (Fig. 5b). The axial O4 of the GalNAc (the unique difference of Gal versus Glc-configured substrates, the latter having an equatorial O4) coordinates with Oδ2 of D173 and NH1 of R174 (for further interactions see Supplementary discussion). These interactions probably drive the specificity for GalNAc versus GlcNAc substrates. R174 also coordinates with O1 and the endocyclic ring oxygen through NH2 whilst O3 interacts with the Nε2 of H225A (Fig. 5b). Comparing overlays of BT3333-S1_156S-sulf with BT4656-S1_116S-sulf, the GlcNAc6S sulfatase encoded by PULHep, reveals that the two substrates bind perpendicular to one another and thus no interactions are spatially conserved (Fig. 5c). Despite this both enzymes use an Asp and Arg to bind O4 and a His to co-ordinate O3. In BT3333-S1_156S-sulf these residues are from the N-terminal side of the protein (D173, R174 and H225) whilst in BT4656-S1_116S-sulf they are from the C-terminal side of the protein (D361, R363 and H447). In addition, when compared with GALNS (4FDJ), the human 6S GalNAc sulfatase, there is no conservation of the sugar-binding residues between the two proteins (Supplementary Fig. 5c–e). Currently no substrate complex of GALNS exists and the specificity determinants remain undescribed.

a A close up of a surface representation showing the pocket topography of BT3333-S1_156S-sulf; b stick representations of the binding site interactions with the protein in green and GalNAc6S in yellow; c overlay of BT3333-S1_156S-sulf in complex with GalNAc6S (green) and BT4656-s1-116S-sulf in complex GlcNAc6S (cyan).
GH88 enzyme
The BT3348GH88 is an essential enzyme for CS metabolism13. BT3348GH88 showed a similar catalytic efficiency against Δ4,5UA-GalNAc, Δ4,5UA-GalNAc6S and Δ4,5UA-GlcNAc but was inactive against Δ4,5UA2S-GalNAc, Δ4,5UA-GalNAc4S, Δ4,5UA-GalNAc4S6S and Δ4,5UA2S-GalNAc4S6S (all β1,3 linked). BT3348GH88 was also inactive on the β1,4 linked heparin disaccharide Δ4,5UA-GlcNAc (Supplementary Table 5). BT3348GH88 can tolerate O6 sulfation but not O2 or O4 sulfation, and is specific for the β1,3 linkage but has no preference for GlcNAc or GalNAc at its +1 subsite.
Growth and bioinformatic analyses of gut Bacteroides species
Eleven Bacteroides species were grown on each GAG as sole carbon source and classified as either strong, intermediate or non-utilisers. For CS-A and CS-C B. theta, B. ovatus, B. eggerthii, B. xylanisolvens and B. plebeius were all strong utilisers whilst B. caccae, B. cellulosilyticus, and B. intestinalis demonstrated intermediate growth. When DS was the substrate there was a shift in classifications compared with CS-A and CS-C. B. eggerthii and B. plebeius switched to being non-utilisers whilst B. intestinalis was classified as a strong utiliser. When HA was the substrate B. ovatus and B. eggerthii out performed all other strains. Only B. theta grew as fast but was classified as intermediate due to a ~20% lower final OD600 nm (Fig. 6).

The different species were divided into strong, intermediate and non-utilisers based on analysis of growth curve rates and final optical densities. Data represent technical triplicates and source data are provided in the source data file labelled as strain growths.
Orthologous proteins were searched to identify PULCS/DS/HA relatives in the eleven species. The three strains B. vulgatus, B. dorei and B. fragilis lack PULCS/DS/HA and only harbour a BT3348GH88 orthologue (and a separated BT4410PL33 in B. fragilis). Consistently, all three showed no growth on any GAGs. We observed BT3328PL29-BT3330SGBP were more rapidly diverging and completely lost in B. cellulosilyticus, B. intestinalis and B. eggerthii. However, no impact on growth was observed, consistent with Δbt3328 mutant growth data. The only genetic differences with potential growth impacts were the loss of BT4410PL33 orthologues in both B. eggerthii and B. plebeius (which additionally lacks a BT3350PL8 orthologue). The importance of BT4410PL33 for B. theta growth on DS and HA (i) explains the switch of B. eggerthii and B. plebeius from strong utilisers on CS-A and CS-C to non-utilisers on DS; (ii) suggests that alternative pathways for HA catabolism exist in these organisms compared with B. theta. B. eggerthii notably outcompeted most other strains on CS-A, CS-C and HA. This could mean members of the PL35 (BACPLE_02279, BACEGG_01448) or PL37 (BACEGG_00231) which are active on GAGs16,23 and not present in B. theta, are contributing to this enhanced growth.
Comparative genomic analyses on a thousand Bacteroidetes species revealed that PULCS/DS/HA is largely associated with the Bacteroides genus but not ubiquitous. Different sections are variably conserved and may have accessory interest, like the aforementioned BT3328PL29-BT3330SGBP region (Supplementary Fig. 6 and Supplementary Table 5). Also, many insertions are observed which separate the PUL elements into several islands, without impairing the PUL activity, according to growth data. Examples span from the three-gene insertion specific to B. theta strains (BT3344-BT3347 that includes an unrelated susCD pair) to hundreds of genes separating B. plebeius orthologues into three genomic regions (Supplementary Fig. 6). Thus, orthologue conservation is more relevant over a tight organisation when predicting growth. Searches beyond the human gut Bacteroides genus reveal species having all required orthologues for growth in the Chitinophaga, Cytophaga and Sphingobacteria classes (some from human skin, chicken gut and soil environments). Notably, a tightly packed PUL was found in Propebela bergensis that even incorporates BT4410PL33 within the core PUL (Supplementary Fig. 6).
Discussion
PULCS/DS/HA performs a highly co-ordinated degradation of GAGs but in a contrasting way to the previously described PULHep, which degraded H/HS. PULHep encodes four PLs all of which displayed significant processivity with variable sulfation tolerances24. PULCS/DS/HA, however, utilises endo–exo synergy in the periplasm between BT3350PL8 and BT3324PL8 for the degradation of CS-A, CS-C and DS. The PUL also deploys an endo-acting O4 sulfatase, BT3349-S1_274S-sulf, the rationale behind its endo mode of action, however, is not obvious. BT3349-S1_274S-sulf may enable BT4410PL33 to contribute to the degradation of CS-A and DS, where O4 sulfation would be inhibitory. A surprising finding was the important role BT4410PL33 plays in DS metabolism within the Bacteroides genus, a result that could not be predicted. The anchoring of BT3349-S1_274S-sulf to the periplasmic side of the membrane may localise it close to the SusC and where it could remove O4 sulfation as glycans enter the periplasm. Alternatively, it could be localised close to BT3334HTCS and BT3348GH88, neither of which can utilise O4 sulfated disaccharides13 and are also membrane associated. This may allow O4 sulfated disaccharides to be desulfated close to the HTCS before BT3348GH88 can degrade them. BT1596-S1_92S-sulf must remove any 2S sulfation present for BT3348GH88 to act. BT3333-S1_156S-sulf then removes the O6 sulfation of GalNAc leaving the final degradation products as GalNac and 4-deoxy-L-threo-5-hexosulose-uronate. For HA metabolism BT4410PL33 is the only lyase required due to its endo-processive mode of action, and after BT3348GH88 has acted the final products would be GlcNAc and 4-deoxy-L-threo-5-hexosulose-uronate. 4-deoxy-L-threo-5-hexosulose-uronate is able to feed directly into metabolism in the cytoplasm25 whilst both GlcNAc and GalNAc require phosphorylation before further metabolism. In contrast to PULHep, which encodes the GlcNAc kinase BT4654ROK, PULCS/DS/HA does not encode for any kinases. Therefore, phosphorylation of GalNAc and GlcNAc, must be completed by constitutively active kinases. Support for a constitutively active GlcNAc kinase is that deletion of BT4654ROK in PULHep has no growth defect on H/HS24. In addition, no differentially expressed kinases are observed during CS growth.
Although the extracellular glycan apparatus appears to confer no advantage in mono-cultured systems, it may be important in a multi-organism, competitive, environment. When B. theta, B. caccae and B. cellulosilyticus were co-cultured on CS for 5 days it was observed that B. cellulosilyticus CFUs were ≤100-fold lower than B. theta and B. caccae and ≤100 than B. cellulosilyticus when mono-cultured26. B. cellulosilyticus lacks any surface glycan apparatus whilst it is present in both B. theta and B. caccae.
Goodman et al.27 identified, through transposon mutagenesis, genes in B. theta which were important for colonisation of the mouse gut. Strains with insertion in bt3348 showed a marked decrease in output compared with input. In addition, when mice were colonised with an artificial microbiome of multiple Bacteroidetes species (including transposon mutagenised B. theta strains), B. theta strains with insertions in bt3330 and BT3349-S1_27 were much reduced in bacterial number compared with monocolonized mice. Thus, CS, DS and HA metabolism are under increased selection pressure in a competitive environment with other Bacteroidetes species. Conversely, bt3328 mutants showed increased bacterial numbers, compared with monocolonized mice, when colonised with other Bacteroidetes, Firmicutes and Actinobacteria.
Indomethacin release, in the gut, from cross-linked CS is dependent upon biodegradation by the caecal contents28. The detailed biochemical analysis of the extracellular glycan apparatus in this study may facilitate strategies to more tightly control drug release to the digestive tract using cross-linked CS. The structural data of BT3349-S1_274S-sulf shows how endo-sulfatase activity has evolved and could be used to predict/guide engineering of sulfatase function. The active site of BT3333-S1_156S-sulf reveals the specificity determinants for Gal over Glc-configured substrates when compared with the GlcNAc6S sulfatase BT4656-S1-116S-sulf. These data highlight how much more highly variable the active sites of related sulfatase are than could be predicted from the nature of their respective substrates. The knowledge of the highly variable nature of the glycone-binding site of sulfatases may help design future glycosulfate-specific inhibitors. This is of timely relevance because of the recently revealed role of sulfatase action in eliciting colitis in a colitis sensitive mouse model17.
Conclusions
The data presented here reveal how the HGM has evolved a core GAG utilisation machinery (PULCS/DS/HA) to target diverse GAGs in the human gut. PULCS/DS/HA components included orphan, physically connected and constitutively active genes with complementary and in some cases unique activities which together enable the complete metabolism of diverse GAGs. PULCS/DS/HA components are widely conserved in Bacteroides and thus presents a general model of CS, DS and HA metabolism, which can be applied to other members of the HGM.
Methods
Sources of carbohydrates. Carbohydrates were from Sigma, carbosynth or Dextra Laboratories. All other chemical reagents were purchased from Sigma.
Bacteroides culture and genetic manipulation: Various Bacteroides strains and B. theta VPI-5482 were cultured anaerobically in a Whitley A35 Workstation (Don Whitley) at 37 °C in either tryptone yeast extract glucose medium or minimal medium (MM) containing GAGs (described below). B. theta strains containing specific gene deletions or inactivated versions of enzymes were made by counter selectable allelic exchange as described below18. All genetic mutants, except Δbt3328, were provided by Dr Guy E. Townsend and Prof. Eduardo Groisman13. The in frame mutants were generated using counter selectable allelic exchange29. Briefly, 1-kb fragments either side of the target gene were cloned, ‘stitched together’ by PCR, and ligated into the pExchange plasmid using SalI/XbaI sites. This recombinant plasmid was then conjugated into a tdk− version of B. theta (referred to as wildtype (WT) in this study) using the S17-1 λ pir Escherichia coli strain. Initial homologous recombination, plasmid insertion, is selected for using erythromycin resistance. Then a second low-frequency recombination event, exclusion of the pExchange plasmid from the genome, is selected for using FUdR containing plates. In frame deletion mutants were detected and checked using PCR and sequencing. Insertional mutations were constructed by cloning a 750-bp sequence from the gene to be disrupted into the pKNOCK tet vector, followed by its introduction into the B. theta chromosome30. Growth of the WT and mutants was measured on glycans by mixing the autoclave-sterilised polysaccharides (0.5% final) with minimal media (per 100 ml was: 0.1 g of Ammonium sulfate and sodium carbonate, 0.05 g cysteine, 10 ml 1 M potassium phosphate pH 7.2, 0.1 ml of 1 mg ml−1 vitamin K, 1 ml of 0.4 mg ml−1 iron sulfate, 0.4 ml of 0.25 mg ml resazurin, 0.05 ml of 0.01 mg ml−1 vitaminB12 and 5 ml of mineral salts for defined media) and monitoring growth continuously in 96-well plates using a Biotek Epoch plate reader. Growth curves presented are averages of three technical replicates.
Co-immunoprecipitation and western blotting: Bacterial cells were collected in the mid-exponential phase and bacterial pellets were resuspended in the lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.2 mm Na3VO4, 1% Nonidet P-40 alternative, 1 mm PMSF, 1 mm DTT, and 1× protease inhibitors (Roche Applied Science)). Lysates were sonicated followed by incubation with 1 μg of antibodies as indicated for 16 h at 4 °C, antibodies were pulled down using protein G-Sepharose beads. Samples were then run on 12.5% SDS-PAGE gels, then transferred to a nitrocellulose membrane using standard buffers. Both primary and secondary antibodies were used as 1/1000 dilutions in 5% milk powder. The secondary antibody was mouse anti-rabbit IgG-HRP (sc-2357). Normal mouse IgG (sc-2025) was used as a negative control. Both antibodies were purchased from santa-cruz.
Microscopy: GAG grown cells were harvested at mid-exponential phase and fixed for 90 min in formalin (10% formaldehyde in PBS buffer). Primary Ab was incubated 1/500 for 2 h at room temperature (RT), followed by incubation with the secondary Ab (Santa Cruz goat anti-rabbit FITC) at 1/500 at RT for 1 h. Cells were immobilised for the microscopy by transfer on 1.2% agarose/H2O pads. Fluorescence microscopy was carried out with Nikon Eclipse Ti equipped with Nikon Plan Apo 100×/1.40 NA phase contrast objective, Prime sCMOS camera (Teledyne Photometrics) and Lambda LS Xenon-arc lamp (Sutter Instruments), and Chroma 49002 filter set (Chroma Technology). Images were acquired with Metamorph 7.7 (Molecular Devices) and analysed with Fiji31.
Whole-cell assays. Whole-cell assays are a direct readout of cell surface activity. After washing with PBS there is no longer any sugar transport by the TonB dependent SuSC transporter. Therefore, product profiles and activities of surface enzymes can be assessed. B. theta was grown in 5 mL MM with 1% (wt/vol) appropriate GAG as the sole carbon source to mid-exponential phase in glass test tubes. Cells were harvested by centrifugation at 5000 × g for 10 min at RT and washed in 5 mL PBS (pH 7.2) before being resuspended in 1 mL PBS. Washed cells were assayed against 10 mg mL−1 of the appropriate substrate at 37 °C for up to 24 h. Assays were analysed by thin layer chromatography (TLC), and 2 μL each sample was spotted onto silica plates and resolved in butanol:acetic acid:water (2:1:1) running buffer. The plates were dried, and the sugars were visualised using diphenylamine stain.
Recombinant protein production. Genes were amplified by PCR using the appropriate primers and the amplified DNA cloned in pET28a using BamHI/XhoI or NheI/XhoI restriction sites generating constructs with either N- or C-terminal His6 tags (Supplementary Table 7). Recombinant genes were expressed in E. coli strains BL21 (DE3) or TUNER (Novagen), containing the appropriate recombinant plasmid, and cultured to mid-exponential phase in LB supplemented with 50 μg/mL kanamycin at 37 °C and 180 rpm. Cells were then cooled to 16 °C, and recombinant gene expression was induced by the addition of 0.1 mM isopropyl β-D-1-thiogalactopyranoside; cells were cultured for another 16 h at 16 °C and 180 rpm. The cells were then centrifuged at 5000 × g and resuspended in 20 mM Hepes, pH 7.4, with 500 mM NaCl before being sonicated on ice. Recombinant protein was then purified by immobilised metal ion affinity chromatography using a cobalt-based matrix (Talon, Clontech) and eluted with 100 mM imidazole. For the proteins selected for structural studies (BT3333-S1_156S-sulf and BT3349-S1_274S-sulf), another step of size-exclusion chromatography was performed using a Superdex 16/60 S200 column (GE Healthcare), with 10 mM Hepes, pH 7.5, and 150 mM NaCl as the eluent, and they were judged to be ≥95% pure by SDS-PAGE. Protein concentrations were determined by measuring absorbance at 280 nm using the molar extinction coefficient calculated by ProtParam on the ExPasy server (web.expasy.org/protparam/).
Isothermal Calorimetry (ITC). The affinity of BT3329SGBP, BT3330SGBP and BT3331SusD for oligo- and polysaccharides was quantified by ITC using a Microcal VP calorimeter. The protein sample (50 μM), stirred at 394 rpm in a 1.4-mL reaction cell, was injected with 26 × 10-μL aliquots of ligand. Titrations were carried out in 50 mM Tris-HCl buffer, pH 8.0, at 25 °C. Integrated binding heats minus dilution heat controls were fit to a single set of sites binding model to derive KA, ΔH and n (number of binding sites on each molecule of protein) using Microcal Origin v7.0. The molar concentration of binding sites present in polysaccharides for BT3329SGBP was determined by altering the concentration of ligand used for regression of the isotherm until the fit yielded a value of 1 for n. For BT3330SGBP and BT3331SusD n was fixed to 1 and a ligand concentration of 2 mM was used.
Enzyme assays. All assays, unless stated, were carried out in 50 mM MES, pH 6.0, or Bis-Tris propane, pH 6.5, with 150 mM NaCl at 37 °C. PL activity was monitored continuously at A235 nm by the formation of the carbon double bond generated through the enzymes beta elimination mechanism of action. BT3334GH88 glucuronyl hydrolase activity was determined by monitoring loss of signal at A235nm. The kinetic parameters of BT3349-S1_274S were monitored via a continuous linked assay using BT3334GH88 and UA-GalNAc4S as the substrate. BT3334GH88 will only hydrolyse UA-GalNAc4S after it has been converted to UA-GalNAc by BT3349-S1_274S-sulf and thus loss of sulfation can be monitored indirectly by loss of signal at A235nm caused by BT3334GH88 hydrolysing any UA-GalNAc produced. The ability of BT3333-S1_156S-sulf, BT3349-S1_274S-sulf and BT1596-S1_92S-sulf to remove O2, O4 and O6 sulfation, respectively, from unsaturated CS disaccharides was also monitored qualitatively by TLC. Reaction profiles of PLs and sulfatase digestion of GAGs were monitored by HPAEC using an AD25 or a VWD, Thermofisher, absorbance detector at A235nm to detect the carbon–carbon double bond products, with H2O, pH 3.5, as the eluent and a second eluent of H2O with 3 M NaCl used to generate a linear NaCl gradient to 100% over 90 min. All enzymatic assays were performed in technical triplicate.
Labelling of glycans: Labelling with 2-aminobenzamide acid (2-AB) was performed using the ludgerTag 2-AB glycan labelling kit. Briefly, DMSO was added to acetic acid in a 0.75:1 ratio and 100 μl added to 5 mg of 2-AB. This mixture is then added to the reductant sodium cyanoborohydride and when fully dissolved 5 μl was added to 50 nmol of dried HA dodecasaccharide. The reaction was left at 65 °C for ~3 h. After labelling samples were cleaned up using the provided spin columns. Labelling of HA octasaccharide and decasaccharide was carried out by creating a saturated solution of 7-aminonapthalene-1,3-disulfonic acid (ANDSA) in formamide and mixing this with 500 μg of dried glycan. The mixture was left overnight at RT and the labelled glycans used the next day in a 1/5 dilution versus enzyme buffer. Saccharides labelled using the UV fluorescent tag, ANDSA were run on 33% acrylamide tris-acetate gels with tris-MES running buffer. Gels were viewed after removal from the glass plates on a UV transilluminator32.
Crystallisation of BT3333-S1_156S-sulf and BT3349-S1_274S-sulf. After purification, BT3333-S1_156S-sulf and BT3349-S1_274S-sulf were carried forward in the same eluent as used for the size-exclusion chromatography. All proteins were then concentrated in centrifugal concentrators with a molecular mass cutoff of 30 kDa. Sparse matrix screens were set up in 96-well sitting drop TTP Labtech plates (400-nL drops). Initial BT3333-S1_156S-sulf apo crystals were obtained at 20 mg/mL in 20% PEG 3350 and KCl. For BT3349-S1_274S-sulf ligand-bound initial hits were obtained at 20 mg mL−1 in 20% PEG 3350 and 0.2 M K acetate. BT3349-S1_274S-sulf crystals were soaked overnight with 10 mM of a CS tetrasaccharide prior to fishing. BT3333-S1_156S-sulf ligand-bound forms were crystallised at 20 mg mL−1 with 10 mM GlaNAc6S in 30% PEG 4000, 200 mM MgCl2, and 0.1 M Tris, pH 8.5. All crystals were cryo-cooled with the addition of 20% PEG 400. Data were collected at Diamond Light Source (Oxford) on beamlines I24 and I04-1 (0.98 Å) at 100 K. The data were integrated with XDS33 and scaled with Aimless34,35. Five percent of observations were randomly selected for the Rfree set. The phase problem was solved by molecular replacement using the program automated molecular replacement server Balbes36 for BT3349-S1_274S-sulf. The phase problem for BT3333-S1_156S-sulf was initially solved using Molrep37 and BT1624 (unpublished structure) as the search model. This gave a partial solution, which could not be fully solved due to tNCS and/or twinning. A good enough model of BT3333-S1_156S-sulf was constructed to be used to solve the phase problem for subsequent collected data, which showed no tNCS and/or twinning defects. Models underwent recursive cycles of model building in Coot38 and refinement cycles in Refmac539. The models were validated using Coot38 and MolProbity40. Structural Figures were made using Pymol (the PyMOL Molecular graphics system, Version 2.0 Schrodinger, LLC) and all other programmes used were from the CCP4 suite41. The data processing and refinement statistics are reported in Supplementary Table 6.
Site-directed mutagenesis. Site-directed mutagenesis was conducted using the PCR-based QuikChange kit (Stratagene) according to the manufacturer’s instructions using the appropriate plasmid as the template and appropriate primer pairs.
Comparative genomics analysis. Similar PULs were searched in >900 Bacteroidetes genomes as follows. First, we identified orthologous proteins to PULCS/DS/HA based on reciprocal best BlastP results between B. theta and each species. Orthologous proteins were mapped to their PUL predictions in PULDB to identify loci gathering several orthologues. For the 11 species tested for growth, PUL predictions were manually refined (enlarged) for boundaries to cover the whole PULCS/DS/HA region which included variable insertions between the core operon and BT3324 or BT3349-S1_27-BT3350 orthologues.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Protein structure data concerned with the structures solved in this work, 6S21 and 6S20, have been deposited with the protein data bank (PDB). Data underlying Figs. 3, 6, Supplementary Figs. 2–4 and Supplementary Tables 2 and 4 have been provided in a source data folder.
Change history
15 February 2020
The original version of this Article was updated shortly after publication. The error has now been fixed and the Supplementary Information PDF is available to download from the HTML version of the Article.
28 August 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Mohajeri, M. H. et al. The role of the microbiome for human health: from basic science to clinical applications. Eur. J. Nutr. 57, 1–14 (2018).
Koropatkin, N. M., Cameron, E. A. & Martens, E. C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335 (2012).
Sweeney, T. E. & Morton, J. M. The human gut microbiome: a review of the effect of obesity and surgically induced weight loss. JAMA Surg. 148, 563–569 (2013).
Martens, E. C. et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9, e1001221 (2011).
Lapebie, P., Lombard, V., Drula, E., Terrapon, N. & Henrissat, B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 2043 (2019).
Cartmell, A. et al. A surface endogalactanase in Bacteroides thetaiotaomicron confers keystone status for arabinogalactan degradation. Nat. Microbiol. 3, 1314–1326 (2018).
Larsbrink, J. et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 (2014).
Luis, A. S. et al. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nat. Microbiol. 3, 210–219 (2018).
Ndeh, D. et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 544, 65–70 (2017).
Pudlo, N. A. et al. Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. mBio 6, e01282–01215 (2015).
Rogers, T. E. et al. Dynamic responses of Bacteroides thetaiotaomicron during growth on glycan mixtures. Mol. Microbiol. 88, 876–890 (2013).
Tuncil, Y. E. et al. Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. mBio 8, e01068–17 (2017).
Raghavan, V., Lowe, E. C., Townsend, G. E. 2nd, Bolam, D. N. & Groisman, E. A. Tuning transcription of nutrient utilization genes to catabolic rate promotes growth in a gut bacterium. Mol. Microbiol. 93, 1010–1025 (2014).
Salyers, A. A. & O’Brien, M. Cellular location of enzymes involved in chondroitin sulfate breakdown by Bacteroides thetaiotaomicron. J. Bacteriol. 143, 772–780 (1980).
Shaya, D. et al. Composite active site of chondroitin lyase ABC accepting both epimers of uronic acid. Glycobiology 18, 270–277 (2008).
Helbert, W. et al. Discovery of novel carbohydrate-active enzymes through the rational exploration of the protein sequences space. Proc. Natl Acad. Sci. USA 116, 6063–6068 (2019).
Hickey, C. A. et al. Colitogenic Bacteroides thetaiotaomicron antigens access host immune cells in a sulfatase-dependent manner via outer membrane vesicles. Cell Host Microbe 17, 672–680 (2015).
Ndeh, D. et al. The human gut microbe Bacteroides thetaiotaomicron encodes the founding member of a novel glycosaminoglycan-degrading polysaccharide lyase family PL29. J. Biol. Chem. 293, 17906–17916 (2018).
Barbeyron, T. et al. Matching the diversity of sulfated biomolecules: creation of a classification database for sulfatases reflecting their substrate specificity. PloS ONE 11, e0164846 (2016).
Ulmer, J. E. et al. Characterization of glycosaminoglycan (GAG) sulfatases from the human gut symbiont Bacteroides thetaiotaomicron reveals the first GAG-specific bacterial endosulfatase. J. Biol. Chem. 289, 24289–24303 (2014).
Hanson, S. R., Best, M. D. & Wong, C. H. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. 43, 5736–5763 (2004).
Hettle, A. G. et al. The molecular basis of polysaccharide sulfatase activity and a nomenclature for catalytic subsites in this class of enzyme. Structure 26, 747–758 e744 (2018).
Konasani, V. R., Jin, C., Karlsson, N. G. & Albers, E. A novel ulvan lyase family with broad-spectrum activity from the ulvan utilisation loci of Formosa agariphila KMM 3901. Sci. Rep. 8, 14713 (2018).
Cartmell, A. et al. How members of the human gut microbiota overcome the sulfation problem posed by glycosaminoglycans. Proc. Natl Acad. Sci. USA 114, 7037–7042 (2017).
Maruyama, Y. et al. Metabolic fate of unsaturated glucuronic/iduronic acids from glycosaminoglycans: molecular identification and structure determination of streptococcal isomerase and dehydrogenase. J. Biol. Chem. 290, 6281–6292 (2015).
Raghavan, V. & Groisman, E. A. Species-specific dynamic responses of gut bacteria to a mammalian glycan. J. Bacteriol. 197, 1538–1548 (2015).
Goodman, A. L. et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289 (2009).
Rubinstein, A., Nakar, D. & Sintov, A. Colonic drug delivery: enhanced release of indomethacin from cross-linked chondroitin matrix in rat cecal content. Pharm. Res. 9, 276–278 (1992).
Martens, E. C., Chiang, H. C. & Gordon, J. I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457 (2008).
Koropatkin, N. M., Martens, E. C., Gordon, J. I. & Smith, T. J. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115 (2008).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Drummond, K. J., Yates, E. A. & Turnbull, J. E. Electrophoretic sequencing of heparin/heparan sulfate oligosaccharides using a highly sensitive fluorescent end label. Proteomics 1, 304–310 (2001).
Kabsch, W. XDS. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D. Biol. Crystallogr. 62, 72–82 (2006).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67, 282–292 (2011).
Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. Sect. D. Biol. Crystallogr. 64, 125–132 (2008).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 22–25 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67, 355–367 (2011).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 12–21 (2010).
Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 50, 760–763 (1994).
Acknowledgements
The authors would like to thank Diamond Light Source for beamtime and the staff of beamlines I04 and I24 for assistance with crystal testing and collection. We would also like to thank Prof. Eduardo Groisman and Dr Guy Townsend for providing B. theta strains. We would also like to thank Carl Morland for his expert technical assistance. This work was supported by start up funds provided by Liverpool University to A.C. The work was also supported by the MRC CiC award MC_PC_17166 held by E.A.Y and a Wellcome ISSF grant 204822/Z/16/Z awarded to U.L.M. A.B. was supported on an ERC advanced award (Grant 322820).
Author information
Authors and Affiliations
Contributions
D.N. and A.C. performed enzymology and binding kinetics. D.N. and A.C. performed protein localisation. D.N. performed bacterial growths. H.S. carried out microscopy. N.T and B.H. performed bioinformatic analysis. A.C and A.B. carried out X-ray crystallography experiments. A.C and U.L.M. carried out Co-IP experiments. A.C. and E.A.Y. carried out sugar-labelling experiments. A.C. conceptualised the work and wrote first draft. A.C and N.T prepared Figures. All authors contributed to revision and editing of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Mirjam Czjzek and the other, anonymous, reviewer for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ndeh, D., Baslé, A., Strahl, H. et al. Metabolism of multiple glycosaminoglycans by Bacteroides thetaiotaomicron is orchestrated by a versatile core genetic locus. Nat Commun 11, 646 (2020). https://doi.org/10.1038/s41467-020-14509-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14509-4
This article is cited by
-
Bacteroides salyersiae is a potent chondroitin sulfate-degrading species in the human gut microbiota
Microbiome (2024)
-
The gut microbiota and its biogeography
Nature Reviews Microbiology (2024)
-
Host–microbiome orchestration of the sulfated metabolome
Nature Chemical Biology (2024)
-
Biochemical Characterization of Hyaluronate Lyase CpHly8 from an Intestinal Microorganism Clostridium perfringens G1121
Applied Biochemistry and Biotechnology (2024)
-
Harnessing gut microbes for glycan detection and quantification
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
