
Abstract
Organofluorine compounds have shown their great value in many aspects. Moreover, allenes are also a class of important compounds. Fluorinated or fluoroalkylated allenes might provide an option as candidates for drug and material developments, as allenes allow a great number of valuable transformations. Herein, we report a metal-free synthesis of difluoromethylated allenes via regioselective trifunctionalization of 1,3-enynes. This method proceeds through double C–F bond formation with concomitant introduction of an amino group to the allene. Synthetic applications are conducted and preliminary mechanistic studies suggest that a two-step pathway is involved. DFT calculations revealed an unusual dibenzenesulfonimide-assisted fluorination/fluoroamination with NFSI. In addition, kinetic reaction study revealed the induction period of both major and side products to support the proposed reaction mechanism. This work offers a convenient approach for the synthesis of a range of difluoromethylated allenes and is also a rare example of trifunctionalization of 1,3-enynes.
Similar content being viewed by others
Introduction
Organofluorine compounds have shown great importance over the years in the fields such as medicinal chemistry and agrochemistry, as the introduction of fluorine atom(s) onto molecules would modify considerably their chemical and physicochemical properties1,2,3,4,5,6. Thus, methods for the synthesis of organofluorine compounds have been flourished in both profundity and scope7,8,9,10,11,12,13. The difluoromethyl group (CF2H), as an analogue of the well-known trifluoromethyl group (CF3), has recently received increasing attentions and many powerful methods for its introduction into organic compounds have been developed14,15,16,17,18,19. On the other hand, allenes are key intermediates in organic synthesis and are important structural motifs that can also be found in natural products20,21,22,23,24. For the purpose of providing diverse fluorinated molecules to meet the increasing demand for drug discovery and new materials development, methods for efficient synthesis of various fluorinated or fluoroalkylated allenes are evidently of great value, as such allenes allow a great number of valuable skeletal and stereochemical transformations21,24.
Over the past few decades, many difluoromethylation reactions have been successfully developed, enabling facile synthesis of different types of difluoromethylated compounds. Most commonly, the CF2H group (or CF2R group) is accessed from a CF2-containing building block or agent14,15,16,17,18,19,25,26,27,28,29,30,31. For example, the syntheses of difluoromethylated allenes typically require the use of difluoromethyl metal compounds32,33,34,35. Recently, Wang et al. developed the nickel-catalyzed carbofluoroalkylation affording difluoroalkylated allenes using ethyl bromodifluoroacetate36. Notwithstanding these significant breakthroughs, the development of efficient and diversified approaches for the facile construction of difluoromethylated allenes is still challenging especially starts from non-CF2-containing building blocks and methods for the synthesis of difluoroalkylated allenes are still demanded.
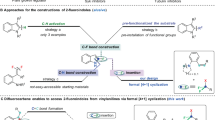
Inspired by earlier reports on gem-difluorination (Fig. 1a)37,38,39,40 and fluoroamination of styrenes (Fig. 1b)41,42,43,44,45, also as one part of our continuous efforts on functionalization of unsaturated compounds46,47,48,49, herein, we report a metal-free consecutive trifunctionalization of 1,3-enynes for the assembly of various difluoromethylated allenyl amines (Fig. 1c) using NFSI as the fluorination and amination source41,44,50,51,52,53,54,55,56,57,58,59. The synthetic potential of difluoromethylated allenes is demonstrated by transformations to a variety of difluoromethylated compounds21.

a Gem-difluorination of styrenes. b Fluoroamination of styrenes. c This work: facile synthesis of difluoromethylated allenes.
Results
Reaction optimization
We commenced reaction condition optimization using CuTC as the catalyst and 1,10-phenanthroline as the ligand. As shown in Table 1, the reaction between 1,3-enyne 1a and NFSI was originally thought to involve intermolecular 1,4-fluoroamination, providing the monofluoromethylated allene 3a as the desired product49. However, only a trace amount of 3a was detected, and a new compound, the difluoromethylated allene 4a, was obtained as major product albeit in a low yield (Table 1, entry 1). Formation of the product 4a alluded to a previously unknown reaction, and we therefore decided to optimize the conditions towards formation of 4a. With the Pd(OAc)2/BC catalyst system45, product 4a was obtained in 34% yield (Table 1, entry 2). Use of a CoCl2 catalyst failed to provide any targeted 4a (Table 1, entry 4), while Pt(COD)Cl2 and NiCl2 showed a better performance than the Pd(OAc)2/BC catalyst system (Table 1, entry 5 v. entry 6 v. entry 2). After extensive screening experiments, it was found that the reaction of the 1,3-enyne 1a with NFSI afforded 4a in 28% yield, even in the absence of catalyst and ligand (Table 1, entry 7). The yield of 4a was further improved to 44% upon running the reaction at an elevated temperature (Table 1, entry 8). Other solvents were screened and it was found that toluene and chloroform provide the product 4a in 66 and 61% yield (Table 1, entries 9 and 10), while acetonitrile, a polar solvent, afforded the targeted product in only 12% yield (Table 1, entry 11). Furthermore, the yield of 4a dropped when the reaction was carried out in toluene at 100 oC (Table 1, entry 12). Notably, a small amount of the monofluoromethylated allene 3a was observed in all these reactions.
Substrate scope
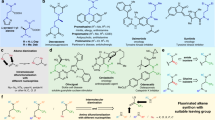
With the optimal reaction conditions in hand, the substrate scope was studied and various difluoromethylated allenes were successfully prepared in moderate to good yields. As shown in Fig. 2, the phenyl group at position 2 can be replaced by a substituted phenyl group (4b-4v). Electron-donating groups on the benzene ring, such as a methyl group, an iso-butyl group, or a tert-butyl group, and electron-withdrawing substituents such as Br, F, and CN on the benzene ring are tolerated. Enynes with primary, secondary, or tertiary alkyl groups linked with the C–C triple bond could also be diversified. As examples, 1,3-enynes with a n-hexyl group (1b-1i), a cyclopropyl group (1k, 1l, and 1n), a chloroalkyl group (1m), or a tert-butyl group (1o) are suitable substrates for the direct difluoroamination reaction. 1,3-Enynes bearing a silyl group (1r), a silyl ether moiety (1s), or an ester group (1t) also engage in this reaction. Moreover, 2,4-diaryl substituted 1,3-enynes can be used as the substrates to afford the corresponding products in moderate yields (see 4u and 4v). However, (3-methylbut-3-en-1-yn-1-yl)benzene (1w), as an example for non-styrene systems, only provided the desired product 4w in 24% yield, which might be due to the less stability of the intermediate compared to that of 1-alkynyl styrenes. The structures of the difluoromethylated allenes 4k and 4l were unambiguously confirmed by single crystal X-ray diffraction.

aWith CHCl3 (1 mL) as the solvent. bThe reaction was performed in cyclohexane and the monofluoromethylated allene 3x was obtained in 46% yield.
Encouraged by the results using terminal 1,3-enynes, we next investigated difluoroamination of internal 1,3-enynes aiming at the preparation of difluoroalkylated allenes. With 1,3-enyne 1x, bearing a primary alkyl group linked with the C–C triple bond, only a 28% yield of the product 4x was obtained, and the accompanying monofluoroamination product 3x was produced in 46% yield as major compound. However, it was found that the substrates bearing a bulky group at the triple bond deliver the corresponding products 4y-4ae in higher yields and functionalities such as the ester group (4ac) and silyl groups (4ad and 4ae) are tolerated.
Synthetic applications
Synthetic applications of this consecutive difluoroamination of 1,3-enynes have been demonstrated and are shown in Fig. 3. In the presence of N-iodosuccinimide (NIS), the difluoromethylated allene 4m was regioselectively transformed into the (Z,Z)-diene 5 in 54% yield. Note that during iodination one of the sulfonyl groups of the -N(SO2Ph)2 functionality was removed. Interestingly, when the allene 4u bearing two phenyl groups was subjected to the reaction with NIS, the difluoromethylated vinyl imine 6 was formed. Such vinyl iodides, 5 and 6, are also pivotal building blocks for further transformations60. Moreover, the silyl group of the allene 4s could be readily removed to afford the allene 7 with a free alcohol group (81%), which could be further cyclized in 57% yield to the multisubstituted 3,6-dihydro-2H-pyran 8.

a Derivatizations of difluoromethylated allene 4m. b Derivatizations of difluoromethylated allene 4u. c Synthesis of multisubstituted 3,6-dihydro-2H-pyran 8 from difluoromethylated allene 4s.
Mechanistic studies
Finally, preliminary experiments to probe the mechanism of the difluoromethylated allene formation reaction were performed. Since the fluoroamination product 3a was found as a side product (see Table 1), 3a was supposed to be the reaction intermediate and was therefore subjected to the standard conditions. However, the desired product 4a was not detected (Fig. 4a). Notably, during reaction optimization the fluorinated 1,3-enyne61,62,63,64,65,66,67,68 9a was observed. In addition, treatment of the 1,3-enyne 1a with a reduced amount of NFSI afforded compound 9a along with targeted 4a. Therefore, we suspected that 9a might be a key intermediate in this cascade. Indeed, when compound 9a was subjected to the reaction with NFSI, product 4a was isolated in 74% yield (Fig. 4b), suggesting that 9a, rather than 3a, is a possible intermediate for the difluoroamination and this reaction probably relies on a two-step process.

a Test of 3a as the possible reactive intermediate. b Test of 9a as the possible reactive intermediate.
DFT study
On the other hand, DFT study was also performed according to the experiments of mechanism studies. Initially, three kinds of 1,3-enyne radical cation species oxidized by NFSI were considered, but the enthalpies were too high (ΔH > 57 kcal/mol) to support the cation radical process (Supplementary Fig. 1)44.
We then focus on the allenyl cation pathways. Two-component reactions of an 1,3-enyne with NFSI were investigated in different orientations of NFSI to transfer the fluoride to 1,3-enyne, 1. Two transition states, TS1 and TS1’, were located leading to the reactive intermediate 9 with a dibenzenesulfonimide (DBSI) moiety synchronously and the 1,2-adduct N-(1-fluoro-2-phenylhept-3-yn-2-yl)-N-(phenylsulfonyl)benzenesulfonamide (10), with activation free energy of 25.1 and 31.1 kcal/mol, respectively (Supplementary Fig. 2), in which producing DBSI via TS1 is a concerted process after fluorine transfer. Intrinsic reaction coordinate (IRC) calculations have been conducted for the validity of concerted proton transfer and 1,2-addition from TS1 and TS1’, respectively (Supplementary Figs. 3 and 4). In addition, TS6, the transition state corresponding to the bisphenylsulfonyl imide moiety of NFSI directly addition to the 4 C atom of 1, was calculated as well. However, the activation energy of TS6 is so much higher (59.4 kcal/mol), and it results in a 3,4-addition compound N-(3-fluoro-2-phenylhepta-1,3-dien-4-yl)-N-(phenylsulfonyl)benzenesulfonamide (11) (Supplementary Fig. 2) which was confirmed by IRC calculation as well as the calculations from TS1 and TS1’. It is worth noting that no transition state corresponding to 1,4-addition to form desired product 4 can be located. This is reasonable because the bond distance of N-F of the optimized NFSI molecule is 1.40 Å much less than the distance between 1 C and 4 C in the optimized structure of 1 (d(1C-4C) = 3.54 Å).
Since the product (N-(1,1-difluoro-2-phenyldeca-2,3-dien-4-yl)-4-methyl-N-tosylbenzenesulfonamide, 12) adducted by N(Ts)2 can be observed in presence of the HN(Ts)2 (see crossover reaction in Supplementary Methods), three-component reactions of 1,3-enyne with NFSI and DBSI were further considered and discussed. Figure 5 shows the overall potential energy surface from 1 to produce the 1,4-addition major product 4 and minor product 3. As described above, 1 reacts with NFSI first to form the reactive intermediate 9 with the DBSI through the lowest energetic transition state TS1. Then, the reactive intermediate 9 reacts with a second NFSI to form an ion pair of difluoromethylallenyl cation and bisphenylsulfonyl imide anion (int1) through TS2 with the energy barrier of 25.5 kcal/mol. Subsequently, an adjacent DBSI, generated from the first step, participates in the reaction to suspend the 2-addition of bisphenylsulfonyl imide anion to difluoromethylallenyl cation. A low barrier transition state TS3 (13.9 kcal/mol) corresponding to simultaneous imide addition‒proton transfer was then located to form the product 4 and to regenerate a DBSI.

The free energy profile of formations of fluoroamination and difluoroamination products, 3 and 4.
On the other hand, the pathway of three-component reaction combining 1 and NFSI with DBSI to deliver the minor product 3 was also calculated. A three-component transition state of fluorine transfer, TS4, was located with the energy barrier of 29.0 kcal/mol which is higher than that of TS1 but lower than that of TS1’. As expectation, an intermediate int3 was then conducted similar to int2 in which the DBSI suspends the proton abstraction and the 2-addition of bisphenylsulfonyl imide anion. Subsequently, transition state TS5 corresponding to simultaneous imide addition‒proton transfer can also be located with 11.7 kcal/mol of barrier resulting in the monofluoroamination product 3 and regenerating a DBSI. It is noteworthy that the barrier of TS4, 3.9 kcal/mol higher than that of TS1, may be somewhat too high to compete with the path of 9 generation due to the artificial overestimation of unfavorable DBSI association entropy, as seen in comparison with the energy difference between int1 and int2. Nevertheless, according to the crossover reaction, the proposed trimolecular pathway with higher barrier to form the side product 3 is believed to take place in the reactions.
Kinetic studies
For further supporting this hypothesis, kinetic studies of the reaction was therefore conducted to track the formations of side product 3, major product 4 and active intermediate 9 (Supplementary Fig. 5). Because of lacking 9 and DBSI, the induction period of formation of 3 and 4 should thus be seen if the trimolecular pathway is the favorable one. Figure 6 presents the kinetic profiles for the formations of 3a, 4a and 9a.

Kinetic profiles for the formation of 3a, 4a and active intermediate 9a.
As expected, the reaction rate of 9a is faster than that of 3a, and the induction period of 3a and 4a can be observed. Initially, even no trace amount of 3a and 4a can be detected in the first 6 minutes. After that, trace amounts of 3a can be observed, but the rate of generation is very slow; nevertheless, the yield of 9a has been over 10% at the 20 minute. Although the yields of 3a and 4a are almost the same in the first one hour, the generation rate of 4a seems to accelerate probably due to the increase of concentration of 9a. Finally, the active intermediate 9a is consumed to deliver the major product 4a at the end of reaction, and the amounts of 3a should stop to grow after expending all of the reactant 1a. The fast generation rate of 9 and the induction periods of formations of 3 and 4 are well consistent with the proposed reaction mechanism.
Proposed mechanism
Based on our mechanistic experiments, theoretical studies and published work41,44,45,69,70, the proposed mechanism for the synthesis of difluoromethylated allenes from trifunctionalization of 1,3-enynes is depicted in Fig. 7. Initially, electrophilic fluorination of the 1,3-enyne 1 by NFSI synchronously generates the dibenzenesulfonimide and the fluorinated enyne 9 as the reactive intermediate. The second NFSI then reacts with 9 to afford the major product 4 assisted by the DBSI, adjacent to 9. On the other hand, accumulation of DBSI will aid the formation of fluorination product 3, which is observed as a side product cannot be converted into product 4 under the standard reaction conditions. The surplus DBSI can also assists the NFSI to react with the substrate 1 to form the side product 3.

Dibenzenesulfonimide assisted three-component trifunctionalization of 1,3-enynes.
In conclusion, a metal-free synthesis of various difluoromethylated allenes through difluoroamination of 1,3-enynes has been developed. NFSI was used as the reactant and a broad substrate scope was obtained. The synthetic potential of difluoromethylated allenes has been demonstrated by transformations of them to a variety of useful difluoromethylated compounds. Moreover, this reaction is also a rare example of trifunctionalization of 1,3-enynes. Preliminary mechanistic studies suggest that a two-step pathway is involved and DFT studies revealed a dibenzenesulfonimide-assisted fluorination/fluoroamination with NFSI.
Methods
General procedure
In a flame-dried Schlenk tube, NFSI (1.5 mmol, 3.0 equiv.) was dissolved in toluene (1 mL) under a nitrogen atmosphere. Then, 1,3-enyne (0.5 mmol, 1.0 equiv.) was added. The reaction mixture was stirred at 80 oC for 12 h. After the reaction completion as detected by TLC, the solvent was evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel (PE/EA or PE/DCM) to afford the allene product.
Data availability
Detailed experimental procedures and characterization of compounds can be found in the Supplementary Information. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (4k: CCDC 1891507; 4l: CCDC 1891508; 5: CCDC 1891506). These data could be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All data are available from the authors upon request.
References
Hiyama, T. Organofluorine Compounds: Chemistry and Applications. (Springer, 2000).
Ojima, I. (ed) Fluorine in Medicinal Chemistry and Chemical Biology. (Wiley-Blackwell, 2009).
Kirsch, P. Modern Fluoroorganic Chemistry. (Wiley-VCH, 2013).
Muller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Zhou, Y. et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II-III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 116, 422–518 (2016).
Besset, T., Poisson, T. & Pannecoucke, X. Recent progress in direct introduction of fluorinated groups on alkenes and alkynes by means of C-H bond functionalization. Chem. Eur. J. 20, 16830–16845 (2014).
Charpentier, J., Fruh, N. & Togni, A. Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 115, 650–682 (2015).
Liang, T., Neumann, C. N. & Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 52, 8214–8264 (2013).
Gao, P., Song, X.-R., Liu, X.-Y. & Liang, Y.-M. Recent developments in the trifluoromethylation of alkynes. Chem. Eur. J. 21, 7648–7661 (2015).
Merino, E. & Nevado, C. Addition of CF3 across unsaturated moieties: a powerful functionalization tool. Chem. Soc. Rev. 43, 6598–6608 (2014).
Ni, C. & Hu, J. The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations. Chem. Soc. Rev. 45, 5441–5454 (2016).
Tomashenko, O. A. & Grushin, V. V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 111, 4475–4521 (2011).
Belhomme, M.-C., Besset, T., Poisson, T. & Pannecoucke, X. Recent progress toward the introduction of functionalized difluoromethylated building blocks onto C(sp2) and C(sp) centers. Chem. Eur. J. 21, 12836–12865 (2015).
Feng, Z., Xiao, Y.-L. & Zhang, X. Transition-metal (Cu, Pd, Ni)-catalyzed difluoroalkylation via cross-coupling with difluoroalkyl halides. Acc. Chem. Res. 51, 2264–2278 (2018).
Hu, J., Zhang, W. & Wang, F. Selective difluoromethylation and monofluoromethylation reactions. Chem. Commun. 45, 7465–7478 (2009).
Landelle, G. et al. Recent advances in transition metal-catalyzed Csp(2)-monofluoro-, difluoro-, perfluoromethylation and trifluoromethylthiolation. Beilstein J. Org. Chem. 9, 2476–2536 (2013).
Rong, J., Ni, C. & Hu, J. Metal-catalyzed direct difluoromethylation reactions. Asian J. Org. Chem. 6, 139–152 (2017).
Yerien, D. E., Barata-Vallejo, S. & Postigo, A. Difluoromethylation Reactions of Organic compounds. Chem. Eur. J. 23, 14676–14701 (2017).
Hoffmann-Röder, A. & Krause, N. Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed. 43, 1196–1216 (2004).
Ma, S. Some typical advances in the synthetic applications of allenes. Chem. Rev. 105, 2829–2872 (2005).
Brummond, K. M. & DeForrest, J. E. Synthesizing allenes today (1982-2006). Synthesis 2007, 795–818 (2007).
Aubert, C. et al. Transition metal catalyzed cycloisomerizations of 1,n-Allenynes and -Allenenes. Chem. Rev. 111, 1954–1993 (2011).
Ma, S. Electrophilic addition and cyclization reactions of allenes. Acc. Chem. Res. 42, 1679–1688 (2009).
Cao, P., Duan, J.-X. & Chen, Q.-Y. Difluoroiodomethane: practical synthesis and reaction with alkenes. J. Chem. Soc., Chem. Commun. 737–738 (1994).
Xia, J.-B., Zhu, C. & Chen, C. Visible light-promoted metal-free C-H activation: diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc. 135, 17494–17500 (2013).
Tang, X.-J. & Dolbier, W. R. Jr. Efficient Cu-catalyzed atom transfer radical addition reactions of fluoroalkylsulfonyl chlorides with electron-deficient alkenes induced by visible light. Angew. Chem. Int. Ed. 54, 4246–4249 (2015).
Lin, Q.-Y., Ran, Y., Xu, X.-H. & Qing, F.-L. Photoredox-catalyzed bromodifluoromethylation of alkenes with (difluoromethyl)triphenylphosphonium bromide. Org. Lett. 18, 2419–2422 (2016).
Arai, Y. et al. Oxydifluoromethylation of alkenes by photoredox catalysis: simple synthesis of CF2H-containing alcohols. Chem. Eur. J. 22, 1262–1265 (2016).
Zhang, M., Lin, J. H. & Xiao, J. C. Photocatalyzed cyanodifluoromethylation of alkenes. Angew. Chem. Int. Ed. 58, 6079–6083 (2019).
Testa, C. et al. Palladium-catalysed C–H bond electrophilic fluorination of highly substituted arylpyrazoles: experimental and DFT mechanistic insights. Adv. Synth. Catal. 357, 2913–2923 (2015).
Burton, D. J. & Hartgraves, G. A. Regioselective preparation of difluoromethyl allenes. J. Fluor. Chem. 49, 155–158 (1990).
Burton, D. J., Hartgraves, G. A. & Hsu, J. A facile, general route to perfluoroalkyl allenes. Tetrahedron Lett. 31, 3699–3702 (1990).
Burton, D. J. & Hartgraves, G. A. The preparation of HCF2CdX and HCF2ZnX via direct insertion into the carbon halogen bond of CF2HY (Y=Br, I). J. Fluor. Chem. 128, 1198–1215 (2007).
Gu, Y., Lu, C., Gu, Y. & Shen, Q. Ligand-controlled copper-catalyzed highly regioselective difluoromethylation of allylic chlorides/bromides and propargyl bromides. Chin. J. Chem. 36, 55–58 (2018).
Zhang, K.-F. et al. Nickel-catalyzed carbofluoroalkylation of 1,3-enynes to access structurally diverse fluoroalkylated allenes. Angew. Chem. Int. Ed. 58, 5069–5074 (2019).
Banik, S. M., Medley, J. W. & Jacobsen, E. N. Catalytic, asymmetric difluorination of alkenes to generate difluoromethylated stereocenters. Science 353, 51–54 (2016).
Ilchenko, N. O., Tasch, B. O. A. & Szabo, K. J. Mild silver-mediated geminal difluorination of styrenes using an air- and moisture-stable fluoroiodane reagent. Angew. Chem. Int. Ed. 53, 12897–12901 (2014).
Kitamura, T., Muta, K. & Oyamada, J. Hypervalent iodine-mediated fluorination of styrene derivatives: stoichiometric and catalytic transformation to 2,2-difluoroethylarenes. J. Org. Chem. 80, 10431–10436 (2015).
Scheidt, F. et al. Catalytic geminal difluorination of styrenes for the construction of fluorine-rich bioisosteres. Org. Lett. 20, 8073–8076 (2018).
Zhang, H. et al. Regioselective radical aminofluorination of styrenes. Angew. Chem. Int. Ed. 53, 11079–11083 (2014).
Zhang, Q. & Li, Y. N-Fluorobenzenesulfonimide: an efficient nitrogen source for C–N bond formation. Synthesis 47, 159–174 (2014).
Xiong, T. & Zhang, Q. New amination strategies based on nitrogen-centered radical chemistry. Chem. Soc. Rev. 45, 3069–3087 (2016).
Zhang, Q. et al. Metal-free three-component regioselective aminofluorination of styrene derivatives. J. Org. Chem. 82, 8258–8266 (2017).
Qiu, S. et al. Palladium-catalyzed intermolecular aminofluorination of styrenes. J. Am. Chem. Soc. 132, 2856–2857 (2010).
Jian, W. et al. Iron-catalyzed decarboxylative alkyl etherification of vinylarenes with aliphatic acids as the alkyl source. Angew. Chem. Int. Ed. 56, 3650–3654 (2017).
Qian, B. et al. Iron-catalyzed carboamination of olefins: synthesis of amines and disubstituted beta-amino acids. J. Am. Chem. Soc. 139, 13076–13082 (2017).
Ye, C. et al. Copper-catalyzed 1,4-alkylarylation of 1,3-enynes with masked alkyl electrophiles. Chem. Sci. 10, 3632–3636 (2019).
Zhu, X. et al. Copper-catalyzed radical 1,4-difunctionalization of 1,3-enynes with alkyl diacyl peroxides and N-fluorobenzenesulfonimide. J. Am. Chem. Soc. 141, 548–559 (2019).
Sibbald, P. A. & Michael, F. E. Palladium-catalyzed diamination of unactivated alkenes using N-fluorobenzenesulfonimide as source of electrophilic nitrogen. Org. Lett. 11, 1147–1149 (2009).
Ingalls, E. L., Sibbald, P. A., Kaminsky, W. & Michael, F. E. Enantioselective palladium-catalyzed diamination of alkenes using N-fluorobenzenesulfonimide. J. Am. Chem. Soc. 135, 8854–8856 (2013).
Xie, J., Wang, Y. W., Qi, L. W. & Zhang, B. Access to aminated saturated oxygen heterocycles via copper-catalyzed aminooxygenation of alkenes. Org. Lett. 19, 1148–1151 (2017).
Xiong, T. et al. Palladium-catalyzed allylic C-H amination of alkenes with N-fluorodibenzenesulfonimide: water plays an important role. Chem. Commun. 48, 2246–2248 (2012).
Zhang, B. & Studer, A. Copper-catalyzed intermolecular aminoazidation of alkenes. Org. Lett. 16, 1790–1793 (2014).
Zhang, G. et al. Highly regioselective radical amination of allenes: direct synthesis of allenamides and tetrasubstituted alkenes. Angew. Chem. Int. Ed. 54, 12649–12653 (2015).
Zhang, H. et al. Copper-catalyzed intermolecular aminocyanation and diamination of alkenes. Angew. Chem. Int. Ed. 52, 2529–2533 (2013).
Zheng, G. et al. Radical cascade reaction of alkynes with N-fluoroarylsulfonimides and alcohols. Nat. Commun. 6, 7011 (2015).
Zheng, G. et al. Highly regio- and stereoselective intermolecular seleno- and thioamination of alkynes. Chem. Eur. J. 22, 3513–3518 (2016).
Knight, L. K. et al. β-Diketiminato scandium chemistry: synthesis, characterization, and thermal behavior of primary amido alkyl derivatives. Organometallics 23, 2087–2094 (2004).
Doucet, H. & Hierso, J.-C. Palladium-based catalytic systems for the synthesis of conjugated enynes by sonogashira reactions and related alkynylations. Angew. Chem. Int. Ed. 46, 834–871 (2007).
Prakash, G. K. S. et al. Synthesis of monofluoroalkenes via Julia–Kocienski reaction. J. Fluor. Chem. 131, 1192–1197 (2010).
Zhang, H. et al. Monofluorovinyl tosylate: a useful building block for the synthesis of terminal vinyl monofluorides via Suzuki-Miyaura coupling. Org. Lett. 13, 560–563 (2011).
Shao, Q. & Huang, Y. Direct fluorination of styrenes. Chem. Commun. 51, 6584–6586 (2015).
Wu, J., Xiao, J., Dai, W. & Cao, S. Synthesis of monofluoroalkenes through selective hydrodefluorination of gem-difluoroalkenes with Red-Al®. RSC Adv. 5, 34498–34501 (2015).
Hamel, J. D., Cloutier, M. & Paquin, J. F. Ability: an approach to beta-substituted monofluoroalkenes using gem-chlorofluoropropenes. Org. Lett. 18, 1852–1855 (2016).
Liu, Q., Shen, X., Ni, C. & Hu, J. Stereoselective carbonyl olefination with fluorosulfoximines: facile access to Z or E terminal monofluoroalkenes. Angew. Chem. Int. Ed. 56, 619–623 (2017).
Chen, X.-W. et al. Hydrogen-transfer-mediated alpha-functionalization of 1,8-naphthyridines by a strategy overcoming the over-hydrogenation barrier. Angew. Chem. Int. Ed. 56, 14232–14236 (2017).
Kojima, R., Kubota, K. & Ito, H. Stereodivergent hydrodefluorination of gem-difluoroalkenes: selective synthesis of (Z)- and (E)-monofluoroalkenes. Chem. Commun. 53, 10688–10691 (2017).
DesMarteau, D. D., Xu, Z. Q. & Witz, M. N-Fluorobis[(perfluoroalkyl)sulfonyl]imides: reactions with some olefins via.alpha.-fluoro carbocationic intermediates. J. Org. Chem. 57, 629–635 (1992).
Zhang, X. et al. Investigation of radical cation in electrophilic fluorination by ESI-MS. Org. Lett. 7, 3877–3880 (2005).
Acknowledgements
We thank the National Key R&D Program of China (2017YFA0700103), the NSFC (Grant Nos 21672213, 21871258, and 21922112), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), and the Haixi Institute of CAS (Grant No. CXZX-2017-P01) for financial support.
Author information
Authors and Affiliations
Contributions
M.T.M. and H.B. conceived the idea. M.T.M., Y.J., and C.Y. conducted the experiments and they contribute equally to this paper. M.-F.C. conducted the DFT calculation. M.I. and X.Z. synthesized the substrates and repeated the reactions. M.T.M., Y.J., C.Y., Y.L. and H.B. analyzed the data. Y.L., Z.W., A.S. and H.B. co-wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Paul Fleurat‐Lessard and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taj Muhammad, M., Jiao, Y., Ye, C. et al. Synthesis of difluoromethylated allenes through trifunctionalization of 1,3-enynes. Nat Commun 11, 416 (2020). https://doi.org/10.1038/s41467-019-14254-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-14254-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
