
Abstract
Biological tissues exhibit complex spatial heterogeneity that directs the functions of multicellular organisms. Quantifying protein expression is essential for elucidating processes within complex biological assemblies. Imaging mass spectrometry (IMS) is a powerful emerging tool for mapping the spatial distribution of metabolites and lipids across tissue surfaces, but technical challenges have limited the application of IMS to the analysis of proteomes. Methods for probing the spatial distribution of the proteome have generally relied on the use of labels and/or antibodies, which limits multiplexing and requires a priori knowledge of protein targets. Past efforts to make spatially resolved proteome measurements across tissues have had limited spatial resolution and proteome coverage and have relied on manual workflows. Here, we demonstrate an automated approach to imaging that utilizes label-free nanoproteomics to analyze tissue voxels, generating quantitative cell-type-specific images for >2000 proteins with 100-µm spatial resolution across mouse uterine tissue sections preparing for blastocyst implantation.
Similar content being viewed by others


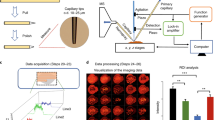
Introduction
Imaging mass spectrometry (IMS, also termed mass spectrometry imaging (MSI)) is a powerful tool for mapping the spatial distribution of biomolecules across a tissue of interest. In an IMS experiment, a probe, which may be a laser, ion beam, or liquid junction, is rastered across a surface to desorb or extract biomolecules that are then directly analyzed by mass spectrometry (MS). This allows for the creation of detailed spatial maps that reveal the native distribution of biomolecules at the surface without labels or pretreatment. However, there are limitations to these approaches, particularly for the analysis of proteins1,2. First, molecules are transmitted directly from the sample to the mass spectrometer without separation, limiting the dynamic range of observed analyte concentrations and restricting detection to the most abundant species. As a result, IMS experiments as applied to proteome profiling are limited to the most abundant 5% of proteins present in the tissue or cell model3,4,5. Second, the ionization efficiency for a given analyte is strongly impacted by the other constituents in the mixture, making quantitative comparisons challenging.
A single matrix-assisted laser deposition/ionization (MALDI) IMS experiment can produce thousands of ion images, providing molecular context to classical histological analysis, yet in order to identify the proteins, fragmentation data are often collected in separate experiments (reviewed in ref. 6) either directly from tissue or by liquid chromatography-tandem MS (LC-MS/MS) following extraction6,7,8. Protein coverage using IMS can be improved through on-tissue digestion, but confident in situ MS/MS peptide identification remains challenging due to low signal-to-noise ratios and high spectral complexity that impede database identifications. To increase the number of identified peptides, researchers have coupled IMS with LC-MS, where one tissue section is analyzed by IMS, while an adjacent section is homogenized and analyzed by LC-MS/MS9,10,11. However, linking these two MS modalities is challenging due to the high complexity of mammalian tissue sections, which has led to false-positive assignments12. As a result, there is currently no IMS technology capable of in-depth proteome imaging.
Proteomics methods based on LC-MS/MS analysis have become an indispensable tool for biological research13,14. Significant investment has been made in developing robust methodologies for quantitative proteomics to monitor changes in the proteome between different patients and/or treatment conditions15,16,17. This powerful approach offers a highly comprehensive and quantitative molecular profile of the specimen of interest. To achieve this in-depth coverage and measurement accuracy, proteins need to be extracted, digested into peptides, and separated by LC for effective MS analysis18. Analyte losses during this multistep processing due to surface adsorption can lead to larger sample requirements than would otherwise be necessary. Consequently, the requisite bulk extraction process blurs spatial information about differing cell types and tissue contexts, which are critical to obtaining a systems-level understanding of the specimen. The approach has been extended to proteome mapping using a “voxelation” approach, though the lateral resolution was limited to 1 mm due to sample handling and technical constraints19. To address this challenge, proteomic approaches have been combined with isolation techniques such as laser capture microdissection (LCM) and fluorescence-activated cell sorting (FACS); however, these applications are narrow to date, due to the limited amount of sample mass obtainable8,20,21,22,23,24.
To address the sensitivity limitations of existing proteomics workflows, we have developed a microfluidic sample preparation platform termed nanoPOTS (Nanodroplet Processing in One pot for Trace Samples), which dramatically increases proteome coverage for small samples, extending to single mammalian cells25. The combination of robotic nanopipetting, a microfabricated glass nanowell chip, and a one-pot processing workflow enable all sample preparation steps to take place in a ~200-nL volume, thereby reducing adsorptive losses to the surface of the reaction vessel and maintaining sufficient protein concentrations for efficient in-solution proteolytic digestion. In combination with FACS and ultrasensitive nanoLC-MS/MS, nanoPOTS has enabled nearly 700 proteins to be identified from single mammalian cells26. In addition, nanoPOTS has been combined with LCM to isolate and profile proteins within regions of interest in pancreas, brain and liver thin sections, as well as plant tissues8,25,27,28. Following sample processing of biological material into digested peptides, the samples were collected into capillary columns and concentrated onto solid-phase extraction (SPE) columns. These columns were manually inserted at the head of a nanoLC column and then injected for LC-MS/MS analysis. While in principle the previously described workflow could have been extended to high-resolution proteome imaging, the manual analysis and the lack of informatics tools precluded the generation and interpretation of in-depth proteome images.
In this work, we demonstrate high-resolution and in-depth proteome imaging using an automated workflow. First, we have coupled the nanoPOTS sample-processing platform with LCM, which provides automated sample collection and processing with unprecedented sensitivity25,29. Second, we paired these nanogram-quantity samples with a custom-designed LC system to achieve sensitive, reproducible analysis with robust, automated data capture, allowing confident analysis of the large sample sets required to create proteome maps.
In this first-of-its-kind application, we analyzed uterine cross sections from pregnant mice prior to the adhesion of early embryos. The luminal epithelial (LE) cells lining the uterine cavity are surrounded by stromal (S) cells and dispersed glandular epithelial (GE) cells. These cells show unique cell-type-specific protein expression in preparation for the attachment of early embryos to the LE, and subsequent invasion into the S. Distinct molecular signatures across the heterogeneous landscape of the mouse uterus during early pregnancy has made this an ideal model system for evaluating other imaging techniques such as matrix-assisted laser desorption ionization (MALDI) IMS30,31 and nanodesorption electrospray ionization (NanoDESI) IMS32,33,34,35 in previous studies. In this study, our proteomic imaging platform was capable of mapping >2000 proteins with 100-µm spatial resolution, thereby capturing the unique protein expression patterns of the LE, S, and GE cell types. Visualization of this large dataset was made possible through the development of a custom implementation of the powerful, open-source platform Trelliscope36, which also serves as an interactive, web-based interface for facile data dissemination. The application of this innovative platform to proteome mapping in a mouse uterus model system clearly demonstrates the exciting potential of proteome imaging to advance biomedical research.
Results
NanoPOTS imaging platform workflow
Our approach combines existing technology with a suite of technologies recently developed in our lab to achieve the robustness, sensitivity, and throughput that are essential for proteome-level imaging. A schematic of the workflow is shown in Fig. 1. Briefly, tissue voxels are created using LCM and captured directly into the nanoPOTS chip by pre-populating the nanowells with DMSO “capture solvent”8. Automated proteomic sample preparation is then carried out on-chip to minimize surface area exposure as was described previously25. The digested peptides are then transferred to a 96-well plate that has been prepopulated with 20 µL of LC buffer A (0.1% formic acid in water) in each well, which aids in reproducible droplet transfer while minimizing peptide losses. Samples are then transferred to a custom LC system equipped with a zero-dead-volume injection needle (Supplementary Fig. 1) to ensure full sample injection. Peptide separation is achieved using an in-house-packed capillary column with a 50-µm internal diameter and an integrated electrospray emitter tip to maximize sensitivity while maintaining the robust operation required for increased throughput. MS analysis was done using a QExactive Orbitrap in data-dependent mode with a 150-ms maximum ion time to allow for longer accumulation to accommodate lower ion fluxes. Datasets are then processed with MaxQuant utilizing the match-between-runs (MBR) option to reduce missing data. Sample datasets are then registered with coordinates from LCM dissection and protein and peptide data are visualized using a Trelliscope.

The authors thank PNNL Graphic Designer Nathan Johnson for preparing the figure.
Platform sensitivity and reproducibility
To demonstrate the reproducibility and sensitivity achievable with this platform, we used mouse liver tissue as a model system. Liver was chosen due to its relative homogeneity on the size scale used in this study. First, square tissue voxels of decreasing area were cut from the liver tissue, with four replicate voxels analyzed at each size. Figure 2a, b shows the peptide and protein coverage, respectively, as a function of lateral resolution. As expected, protein coverage decreases as voxel area is decreased due to the resulting reduction in protein-loading mass. However, when MaxQuant MBR is employed, >800 proteins can still be identified with two unique peptides at 50-µm lateral resolution. Second, reproducibility is critical to producing quantitative protein maps. To establish the reproducibility of our imaging platform, 20 replicate voxels were dissected from a homogeneous region of liver tissue and analyzed using the nanoPOTS imaging platform. Figure 2c shows coefficients of variation (CVs) for protein quantification across the 20 datasets. Using normalized MaxQuant LFQ intensity gives a median CV of 14.4%, which indicates that robust quantification is achievable with this platform.

a Mean number of peptide identifications from four replicate analyses of liver tissue voxels at different lateral resolutions, with and without MBR enabled. b Mean number of protein identifications from four replicate analyses of liver tissue voxels at different lateral resolutions, with and without MBR enabled. c Histogram of protein LFQ intensity coefficient of variation (CV) for 20 replicate voxels from homogeneous tissue sections. Error bars, standard deviation.
Proteome analysis of cell types in Wnt5a-null uterine tissue
To validate the findings in our proteomic images, we performed a complementary study in which we used LCM, nanoPOTS, and LC-MS/MS analyses to isolate, characterize, and statistically compare LE, S, and GE cells across multiple Wnt5a-null mouse uterine tissue sections. This dominant cell population study contained 15 LC-MS/MS instrument runs associated with 15 unique biological samples, 5 S samples, 5 LE samples, and 5 GE samples, in which 100–200 ng of these unique cell populations were captured from three to five sections for each of the 15 samples. From the MaxQuant MBR search, 19,952 peptides had at least two observations across the 15 analyses. The algorithm RMD-PAV was used to identify any outlier biological samples37. Samples were also examined via Pearson correlation and no samples were identified as outliers. Peptides found to have inadequate data for either qualitative or quantitative statistical tests were also removed from the dataset, resulting in a final dataset for normalization that included 15 unique biological samples and 17,387 measured unique peptides corresponding to 2940 unique proteins. Median centering based on rank-invariant peptides (with p value threshold for rank invariance of 0.2) was used for normalization38. Protein quantification was performed using R-rollup, which scales the peptides associated with each protein by a reference peptide and then sets their median as the protein abundance39. The peptide having the least missing data is selected as the reference peptide. Pairwise-univariate statistical comparisons were carried out between each of the three cell types using a Tukey-adjusted ANOVA or a Holm-adjusted g test to compare each pair of dominant cell types for each of the 2940 proteins38. The three statistical comparisons of interest were (1) LE vs. GE, (2) S vs. GE, and (3) S vs. LE. The number of significant proteins (adjusted p value <0.05) for each of the three comparisons based on the ANOVA-adjusted p values were (1) 1220 proteins increasing in the LE and 46 proteins increasing in the GE, (2) 1673 proteins increasing in the S and 42 proteins increasing in the GE, and (3) 777 proteins increasing in the S and 196 proteins increasing in the LE.
Proteome imaging of Wnt5a-null uterine tissue
The nanoPOTS proteomic imaging platform was then used to create 2D protein images of tissue sections comprising the three cell types of interest. Pseudocolor optical images of the imaging area and voxel boundaries are shown in Fig. 3a, b. To display these 2D protein images, we developed a Trelliscope software platform that allowed us to explore the images and correlate them to the statistically significant results from the dominant cell population study. This software platform enabled us to share all results from this study in a searchable and customizable approach (see Supplementary Trelliscope Video Tutorial).

a Stromal-dominant image and b luminal epithelium-dominant image. Scale bar, 100 µm. The authors thank PNNL Graphic Designer Nathan Johnson for preparing the figure.
Imaged areas were taken from the center of uterine sections, enabling visualization of the proteomic landscape of the uterus orchestrating embryo implantation. The S-dominant tissue section (depicted in Figs. 4–6) comprises 24 LC-MS/MS instrument runs associated with 24 unique voxels, 4 containing GE and S, 8 containing LE, and 12 containing S (Fig. 3a). The LE-dominant tissue section (depicted in Supplementary Figs. 2 and 3) also contains 24 LC-MS/MS instrument runs associated with 24 voxels, in this case with 2 containing GE and S, 14 containing LE, and 8 containing S (Fig. 3b). MaxQuant analysis of our S-dominant section characterized 8065 unique peptides corresponding to 1658 unique proteins that had at least two observations across the 24 analyses. Employing the MBR feature characterized 9411 unique peptides corresponding to 1764 unique proteins that had at least two observations across the 24 runs. MaxQuant analysis of the LE-dominant section characterized 11,803 unique peptides corresponding to 2212 unique proteins that had at least two observations across the 24 runs. Employing MBR characterized 13,797 unique peptides corresponding to 2357 unique proteins that had at least two observations across the 24 runs. Median centering based on rank-invariant peptides (0.2) was used for normalization. Our searchable Trelliscope software platform ([http://msc-viz.emsl.pnnl.gov/nanoPOTS_PI_MS/]) contains images of all 2298 and 2447 quantifiable MaxQuant and MaxQuant MBR proteins, respectively, and 12,495 and 14,673 quantifiable MaxQuant and MaxQuant MBR peptides, respectively, from the S- and LE-dominant sections.

(1) Armadillo repeat protein deleted in velo-cardio-facial syndrome homolog (ARVC), reticulon-4 (RTN4), and CD166 antigen (CD166); (2) junctional adhesion molecule A (JAM1); (3) voltage-dependent anion-selective channel protein 2 (VDAC2); (4) coronin-2A (COR2A); (5) annexin A1 (ANXA1), keratin type I cytoskeletal 19 (K1C19), and catenin beta-1 (CTNB1); (6) erlin-2 (ERLN2), a neutral cholesterol ester hydrolase 1 (NCEH1). Scale bars, 100 µm. The authors thank PNNL Graphic Designer Nathan Johnson for preparing the figure.

(1) Serine protease inhibitor A3K (SPA3K), pregnancy zone protein (PZP); (2) apolipoprotein A-I (APOA1); (3) collagen alpha-1(I) chain (CO1A1), collagen alpha-4(VI) chain (CO6A4); (4) basement membrane-specific heparan sulfate proteoglycan core protein (PGBM), EMILIN-1 (EMIL1), Decorin (PGS2); (5) serum albumin (ALBU), complement C3 (CO3), and immunoglobulin heavy constant mu (IGHM). Scale bars, 100 µm. The authors thank PNNL Graphic Designer Nathan Johnson for preparing the figure.

Prostaglandin H2 (PGH2), prostaglandin E2 (PGE2), 12(S)-hydroperoxyeicosatetraenoic acid (12(S)-HpETE), 15(S)-hydroperoxyeicosatetraenoic acid (15(S)-HpETE). The authors thank PNNL Graphic Designer Nathan Johnson for preparing the figure.
Functional analysis of tissue-type differences
Proteins of interest discussed in the paper were statistically significant (<0.05 adjusted p value) in our dominant cell-type data and had complementary spatial distributions in our proteome-imaging data. Of these proteins of interest, 149 are enriched in the LE and 175 are enriched in the S (Supplementary Tables 1 and 2). Supplementary Data 1 and 2 contain the images, from both MaxQuant and MaxQuant MBR, for these proteins in addition to the associated box plots from the dominant cell population study. Although we provide both MaxQuant and MaxQuant MBR data in Supplementary Data 1 and 2, the images in the paper are from MaxQuant unless otherwise specified.
Proteins detected in the GE exhibit a high degree of overlap with the LE and S-expression patterns since 50 of the 149 proteins enriched in the LE were also enriched in the GE and 27 of the 175 proteins enriched in the S were also enriched in the GE (Supplementary Tables 1 and 2). An important goal of this study was to characterize the unique proteomic landscapes of LE cells, which are the first cells to attract and make contact with the early embryo (blastocyst), and the S cells, which support embryo growth during early pregnancy. Our 100-µm voxel size was sufficient to capture the LE cells lining on both sides of the uterine cavity. The LE-localized protein images in Fig. 4 and S-localized protein images in Fig. 5 were selected by correlating gene ontology (GO) categories between our images and our statistically significant results. Negative log10 p values from these tests are indicated by white-to-blue color intensity enrichment scores in Figs. 4 and 5.
The top six LE GO categories and the corresponding images depicted in Fig. 4 include (1) cadherin binding and cell adhesion molecule binding with protein images armadillo repeat protein deleted in velo-cardio-facial syndrome homolog (“ARVC [https://www.uniprot.org/uniprot/P98203]”), reticulon-4 (“RTN4 [https://www.uniprot.org/uniprot/Q99P72]”), and CD166 antigen (“CD166 [https://www.uniprot.org/uniprot/Q61490]”); (2) cell junction with protein image junctional adhesion molecule A (“JAM1 [https://www.uniprot.org/uniprot/O88792]”); (3) ion transport with protein image voltage-dependent anion-selective channel protein 2 (“VDAC2 [https://www.uniprot.org/uniprot/Q60930]” with MBR); (4) brush border with protein image coronin-2A (“COR2A [https://www.uniprot.org/uniprot/Q8C0P5]” with MBR); (5) apical, lateral, and basolateral plasma membrane with protein images annexin A1 (“ANXA1 [https://www.uniprot.org/uniprot/P10107]”), keratin type I cytoskeletal 19 (“K1C19 [https://www.uniprot.org/uniprot/P19001]”), and catenin beta-1 (“CTNB1 [https://www.uniprot.org/uniprot/Q02248]”); (6) lipid metabolic process with protein images erlin-2 (“ERLN2 [https://www.uniprot.org/uniprot/Q8BFZ9]” with MBR) and neutral cholesterol ester hydrolase 1 (“NCEH1 [https://www.uniprot.org/uniprot/Q8BLF1]” with MBR). The corresponding protein images for the LE-dominant tissue section can be found in Supplementary Fig. 2. These proteins have functional roles molecularly linked to epithelial cell crypt formation such as actin cytoskeleton remodeling, cell polarization, and cell migration. In addition, proteins such as “CTNB1 [https://www.uniprot.org/uniprot/Q02248]” are molecularly linked to our Wnt5a-null phenotype40.
The top 5 S GO categories and the corresponding images depicted in Fig. 5 include (1) peptidase inhibitor activity with protein images serine protease inhibitor A3K (“SPA3K [https://www.uniprot.org/uniprot/P07759]”) and pregnancy zone protein (“PZP [https://www.uniprot.org/uniprot/Q61838]”); (2) high-density lipoprotein particle with protein image apolipoprotein A-I (“APOA1 [https://www.uniprot.org/uniprot/Q00623]”); (3) collagen organization with protein images collagen alpha-1(I) chain (“CO1A1 [https://www.uniprot.org/uniprot/P11087]”) and collagen alpha-4(VI) chain (“CO6A4 [https://www.uniprot.org/uniprot/A2AX52]” with MBR); (4) extracellular matrix and basement membrane with protein images basement membrane-specific heparan sulfate proteoglycan core protein (“PGBM [https://www.uniprot.org/uniprot/Q05793]” with MBR), EMILIN-1 (“EMIL1 [https://www.uniprot.org/uniprot/Q99K41]” with MBR), Decorin (“PGS2 [https://www.uniprot.org/uniprot/P28654]” with MBR); (5) blood microparticle with protein images serum albumin (“ALBU [https://www.uniprot.org/uniprot/P07724]”), complement C3 (“CO3 [https://www.uniprot.org/uniprot/P01027]”), and immunoglobulin heavy constant mu (“IGHM [https://www.uniprot.org/uniprot/P01872]” with MBR). The corresponding protein images for the LE-dominant tissue section can be found in Supplementary Fig. 3. Molecular epithelial–stromal cell crosstalk is essential for successful embryo implantation, and many of these significantly changing S proteins have functional roles molecularly linked to remodeling of the extracellular matrix. In addition, blood-associated proteins, including immune proteins IGHM and CO3, significantly increased in S cells compared with LE cells where they were not detected or detected at low levels; the avascular LE cells aid the blastocyst in escaping the maternal immune surveillance at the time of implantation.
Comparison to known lipid-mediated metabolic processes
Corroborating our LE-specific increase in lipid metabolic processes (Fig. 4), arachidonic acid-derived lipid mediators are known to play an essential role in embryo implantation41,42,43. We have recently shown, using in situ metabolome-imaging techniques, that prostaglandins (PG) including PGE2 localize to the LE in uterine sections obtained from the same Wnt5a-null mouse analyzed in this study32. As illustrated in Fig. 6, our nanoPOTS proteomic images mapped cytosolic phospholipase A2 (“PA24A [https://www.uniprot.org/uniprot/P47713]”), prostaglandin G/H synthase 1 (“PGH1 [https://www.uniprot.org/uniprot/P22437]”, also named cyclooxygenase-1), and prostaglandin E synthase 2 (“PGES2 [https://www.uniprot.org/uniprot/Q8BWM0]” with MBR) expression to LE cells. PA24A hydrolyzes arachidonic acid from the sn-2 position of phospholipids; free arachidonic acid is then metabolized into prostaglandins such as PGH2 and PGE2. In addition, our nanoPOTS proteomic images also captured the LE localization of arachidonate 12-lipoxygenase (“LX12E [https://www.uniprot.org/uniprot/P55249]”) and arachidonate 15-lipoxygenase (“LOX15 [https://www.uniprot.org/uniprot/P39654]”), which metabolize arachidonic acid into bioactive lipid mediators, 12(S)-hydroperoxyeicosatetraenoic acid (12(S)-HpETE) and 15(S)-hydroperoxyeicosatetraenoic acid (15(S)-HpETE). Progesterone-induced synthesis of the 12/15-LOX-derived lipid mediators in LE cells activates a critical regulator of embryo implantation, the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) and its downstream gene networks44. In addition to an LE-specific increase in lipid metabolic processes, our nanoPOTS proteomic images mapped oxysterol-binding protein-related protein 8 (“OSBL8 [https://www.uniprot.org/uniprot/B9EJ86]”) expression to voxels containing GE (Fig. 6). OSBL8 plays an essential role in lipid transfer between cell membrane bilayers at contacts between the endoplasmic reticulum and other membranes to aid in maintaining membrane lipid homeostasis45.
Discussion
Herein, we establish the potential for in-depth, high-lateral-resolution imaging of the proteome across tissues using an automated nanoPOTS workflow. The approach combines the high sensitivity of the nanoPOTS approach with LCM and a custom, automated sample transfer and analysis platform that enables complete sample utilization and robust operation. When taken together, this platform enables quantitative mapping of >2000 proteins with 100-µm spatial resolution. The sensitivity of this approach was demonstrated through replicate analysis of liver tissue voxels of decreasing size. Further, analyzing 20 replicate tissue voxels from this relatively homogeneous tissue produced CVs similar to bulk analysis, indicating platform reproducibility and stability. We then verified the ability of our imaging platform to find meaningful differences using a mouse embryo implantation model system. Many of the proteins quantified in the proteome images showed differential expression across different tissue features. These differences were then confirmed by adjusted p values from multiple comparison testing of highly enriched cell-type pools from the same tissues using LCM. The high level of agreement between these analyses indicates the great potential of this approach as a discovery tool. The depth of proteome coverage and robust quantitation achievable with this platform greatly exceed those of existing IMS platforms.
This nanoPOTS proteomic imaging analytical platform enabled us to visualize proteome-level cell-type-specific alterations across mouse uterine tissue sections preparing for blastocyst implantation. A reciprocal interaction between a blastocyst and the receptive uterus is critical to successful pregnancy. In mice, LE cells undergo extensive remodeling to create implantation chambers (crypts) formed by the evagination of these epithelial cells; during the initial apposition, adhesion, and attachment steps of embryo implantation, blastocysts are positioned within these crypts46. Mice with uterine-specific deletion of Wnt5a, which exhibit haphazard crypt formation and enhanced molecular transformation across all LE cells, were imaged in this study. Thus, proteins detected represent samples of deleted uteri and may vary in normal uteri. The nanoPOTS imaging platform generated quantitative images for >2000 proteins across cellular regions in the Wnt5a-null uterus with 100-µm spatial resolution. An important goal of this study was to characterize the unique proteomic landscapes of LE cells, the first cells to attract and make direct contact with the blastocyst, and the S cells, which support embryo growth during early pregnancy. Images of proteins localizing to the LE had functional roles molecularly linked to epithelial cell crypt formation, such as actin cytoskeleton remodeling, cell polarization, and cell migration. Images of proteins localizing to the S had functional roles molecularly linked to remodeling of the extracellular matrix, a primary component of the S. In addition to visualizing tissue-type-specific expression with our protein images, we were also able to visualize region-specific bioactivity. We have recently shown that prostaglandins, including prostaglandin E2, localize to the LE in uterine sections obtained from the same Wnt5a-null mouse used in this study32. Our protein images were able to visualize the metabolism of arachidonic acid into bioactive lipid mediators where prostaglandin G/H synthase 1 and prostaglandin E synthase 2 localized to the LE. In addition, our protein images were able to characterize unique tissue microenvironments within the same cell populations by visualizing the gradient expression increase of stroma proteins along the mesometrial (top)–antimesometrial (bottom) axis (Fig. 5).
The in-depth proteome-mapping results from the innovative nanoPOTS imaging platform clearly demonstrate the exciting potential of spatially resolved proteomics to provide previously unobtainable insights into tissue proteomes. Further, the utilization of Trelliscope for data visualization makes these powerful datasets quickly and easily accessible to the broader research community, greatly increasing their impact.
Methods
Mouse liver tissue
All mice used in this study were housed at PNNL according to NIH and institutional guidelines for the use of laboratory animals. All protocols for this study were reviewed and approved by the Institutional Animal Care and Use Committee of Battelle, Pacific Northwest Division. C57BL/6J mice were obtained from Jackson Labs and tissues were prepared for LCM as previously described8. Briefly, harvested mouse livers were washed with phosphate-buffered saline (PBS) before being snap frozen and stored at –80 °C until analysis. Samples were placed in the cryostat (NX-70; Thermo Fisher Scientific) and allowed to warm to cutting temperature, approximately –15 °C. At that point, a lobe was separated from the rest of the liver with a sterile scalpel and mounted with water onto a chuck. Sections were obtained at 10 µm, thaw-mounted onto PEN membrane slides (Carl Zeiss Microscopy, Germany), and stored at −80 °C until use.
Mouse uterine tissue
All mice used in this study were housed in the Cincinnati Children’s Animal Care Facility according to NIH and institutional guidelines for the use of laboratory animals. All protocols for this study were reviewed and approved by the Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee. Utilizing the LoxP-Cre system, mice with uterine-specific inactivation of Wnt5ad/d (Wnt5aloxP/loxP) were generated as previously described46. Uterine tissues were collected on day 4 of pregnancy. This transgenic mouse model of impaired embryo implantation contains cellular and molecular changes in the uterus, including disrupted luminal epithelial evaginations (crypts) at the antimesometrial and mesometrial domains46. These crypts are an essential step in the receptive uterus prior to embryo attachment; in wild-type mice these luminal epithelial projections localize only to the antimesometrial pole. Wnt5ad/d mice were chosen for these foundational nanoPOTS proteomic imaging experiments because of these exaggerated morphological changes. Uterine tissue from one Wnt5ad/d mouse was sectioned with a thickness of 12 µm using a cryostat (NX-70). The temperatures of chuck and blade were set at −16 and −20 °C for liver tissues and −16 and −20 °C for uterus tissues. The tissue sections were deposited on Zeiss PEN membrane slides and stored at −80 °C.
Tissue fixative solution (70% ethanol) was precooled to 4 °C before use. Tissue sections were immediately immersed into 70% ethanol for 15 s after removal from the −80 °C freezer or dry-ice box. Rehydration was performed for 30 s in deionized water. Next, the tissue sections were immersed in Mayer’s hematoxylin solution (Sigma-Aldrich, St. Louis, USA) for 1 min, dipped twice in deionized water to remove excess dye solution, and immersed in Scott’s Tap Water Substitute (Sigma-Aldrich) for 15 s to dye the tissues. Finally, tissue dehydration was performed by sequentially immersing the tissue sections in 70% ethanol for 1 min, 95% ethanol for 1 min, 100% ethanol for 1 min, and xylene for 2 min. The sections were dried in a fume hood for 10 min; subsequently fixed tissue sections can be directly used or stored at −80 °C for future use.
Nanowell chip fabrication
Nanowell chips were fabricated on glass slides with precoated chromium and photoresist layers (Telic company, Valencia, USA) using standard photolithography and wet chemical-etching procedures. An array of 3 × 9 nanowells with a diameter of 1.2 mm and a center-to-center spacing of 4.5 mm was designed in AutoCAD and printed with a Direct-Write Lithography System (SF-100; Intelligent Micro Patterning LLC, St. Petersburg, USA). After exposure, development, and chromium etching, the slides were etched in a solution of 2:4:4 (v:v:v) buffered hydrofluoric acid, hydrochloric acid, and water at an etch rate of 1 µm/min for 10 min. After drying at 120 °C for 2 h, the slides were treated with 2% (v/v) heptadecafluoro-1,1,2,2-tetrahydrodecyldimethylchlorosilane in 2,2,4-trimethylpentane. After removing the remaining chromium layer, an array of hydrophilic spots was formed on a hydrophobic background. A glass frame (machined by Coherent Inc., Santa Clara, CA) with a thickness of 1 mm and a width of 5 mm was affixed to the nanowell slide using silicone adhesive. Finally, a sealing cover plate was fabricated by spin-coating a layer of polydimethylsiloxane (30 µm in thickness). The sealing cover slide was used to reversibly seal the nanowell chip during reaction incubation.
Laser capture microdissection of tissue sections
Before experiments, nanowells were prepopulated with 200-nL DMSO droplets that served as a low-vapor-pressure capture medium. Laser capture microdissection (LCM) was performed on a PALM MicroBeam system (Carl Zeiss MicroImaging, Munich, Germany). A slide adapter (SlideCollector 48, Carl Zeiss MicroImaging) was used to mount a nanowell chip on the LCM microscope. Voxelation of the tissue section was achieved by first drawing a grid on the tissue using PalmRobo software, followed by tissue cutting and catapulting. Both liver and uterine tissues were cut at an energy level of 42 and with an iteration cycle of 2 to completely separate 100 × 100 µm tissue voxels. The “CenterRoboLPC” function with an energy level of delta 10 and a focus level of delta 5 was used to catapult tissue voxels into DMSO droplets. The “CapCheck” function was activated to confirm successful sample collection from tissue sections to DMSO droplets. The collected samples can be processed directly or stored at −20 °C for weeks until use.
Proteomic sample processing
The nanowell chip was heated to 70 °C for 10 min to evaporate the DMSO droplet. A nanoliter-resolution robotic liquid-handling platform was employed to dispense reagents into nanowells. First, a cell lysis buffer containing 0.2% (w/v) n-dodecyl-β-d-maltoside (DDM; Sigma-Aldrich), 5 mM dithiothreitol (DTT), and 1× PBS was applied into each nanowell with a volume of 100 nL. The chip was incubated at 70 °C for 1 h for cell lysis, protein extraction, and denaturation. Next, 50 nL of 30 mM iodoacetamide in 50 mM ammonium bicarbonate (ABC) buffer (pH 8.0) was added to each well and incubated in the dark for 30 min. Protein digestion was performed by dispensing 50 nL of 0.01 ng/nL Lys-C (MS grade, Promega, Madison, USA) and trypsin (Promega) in ABC buffer, and incubated for 4 and 8 h, respectively. Finally, the enzymatic reaction was terminated by adding 50 nL of 0.5% trifluoroacetic acid in aqueous buffer and incubated for 30 min.
The processed samples were transferred into 96-well PCR plates (twin.tec PCR Plates; Eppendorf, Hauppauge, USA) for LC-MS analysis. The 96-well plate was prefilled with 25 µL of 0.1% FA and 0.02% DDM aqueous buffer. The robotic platform was used to aspirate nanoliter samples from nanowells and dispense into the 25-µL buffer. Each nanowell was washed twice with 200 nL of the same buffer to maximize sample recovery. Finally, the 96-well plates were sealed with sealing tape (Nunc; Thermo Scientific) and stored at −20 °C.
Sample analysis with SPE-LC-MS/MS
A homebuilt LC system was employed to automatically perform sample injection, sample cleanup, and LC separation. The platform consisted of a PAL autosampler (CTC Analytics AG, Zwingen, Switzerland), two Cheminert six-port injection valves (Valco Instruments, Houston, USA), a binary nanoUPLC pump (Dionex UltiMate NCP-3200RS; Thermo Scientific), and a HPLC sample loading pump (1200 Series; Agilent, Santa Clara, USA). Both SPE precolumn (150 µm i.d., 4-cm length) and LC column (50 µm i.d., 70-cm Self-Pack PicoFrit column, New Objective, Woburn, USA) were slurry-packed with Jupiter C18 packing material (300-Å pore size, trapping column 5 µm, and analytical column 3-µm particle sizes; Phenomenex, Terrence, USA). The sample was injected into a 20-µL loop and loaded onto the trapping column using Buffer A (0.1% formic acid in water) at 3 µL/min for 20 min. After trapping, the sample was reverse-flow eluted onto the analytical column at 150 nL/min and separated by a gradient of 5–8% (0–2 min), 8–12% (2–20 min), 12–35% (20–75 min), and 35–60% (75–97 min) of Buffer B (0.1% formic acid in acetonitrile). The LC column was then washed using 75% Buffer B for 10 min and re-equilibrated using 5% Buffer B for 50 min.
A QExactive Plus Orbitrap MS (Thermo Scientific) was used to analyze the separated peptides. A 2.2-kV high voltage was applied at the ionization source to generate electrospray and ionize peptides. The ion transfer capillary was heated to 250 °C to desolvate droplets. The S-lens RF level was set at 70. Data-dependent mode was employed to automatically trigger precursor scan and MS/MS scans. Precursors were scanned at a resolution of 35,000, an AGC target of 3E6, a maximum ion trap time of 50 ms, and mass range of 375–1800. Top-12 precursors were isolated with an isolation window of 2, an AGC target of 1E5, and a maximum ion trap time of 150 ms, and then fragmented by high-energy collision with an energy level of 32%. A dynamic exclusion of 30 s was used to minimize repeated sequencing. MS/MS spectra were scanned at a resolution of 17,500.
Data analysis
All raw files were processed using MaxQuant47,48 (version 1.5.3.30) for feature detection, database searching, and protein/peptide quantification. Mass spectra were searched against the Uniprot Mus Musculus database downloaded in October 2016, containing 16,825 sequence entries. Carbamidomethylation of cysteine was set as a fixed modification and N-terminal acetylation and oxidation of methionine were allowed as variable modifications. A peptide length > 6 was required with a maximum of two missed cleavages allowed, and a false discovery rate of 0.01. The searches were completed twice with these settings, first with the match-between-runs (MBR) feature enabled and then without, for comparison. Contaminants and reverse sequences were removed from the peptides.txt file prior to use for downstream statistical analysis and image display.
Statistical analysis
In the quantified data, values that were not observed were indicated by NA and data were then log2 transformed. Peptides not observed in at least two samples across all instrument runs within a study were removed. Biological outliers were identified via RMD-PAV37 (p value threshold 0.001) and Pearson correlation. Data were normalized by median centering based on rank-invariant peptides38, where rank invariance was determined by a p value threshold of 0.2. Protein quantification was performed using R-rollup49, which scaled the peptides associated with each protein by a reference peptide (the peptide with the least missing data) and then set the median of the scaled peptides as the protein abundance. Pairwise-univariate statistical comparisons were carried out between each of the three cell types using a Tukey-adjusted ANOVA or a Holm-adjusted g test to compare each pair of dominant cell types50.
Gene Ontology Enrichment Analysis
Uniprot IDs were converted to gene symbols using the ID conversion service at www.uniprot.org. Using the R statistical programming environment, lists of proteins identified as differentially abundant in each experiment were subjected to the EASE-adjusted Fisher exact test51 using the complete human GO categories available at [http://geneontology.org/].
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data supporting this study are available from the corresponding authors upon reasonable request. Mass Spectrometry Interactive Virtual Environment (MassIVE; [https://massive.ucsd.edu]) accession number MSV000084421 contains raw proteomics datasets and their corresponding MaxQuant searching results. Visualization of these large datasets is possible through our searchable Trelliscope software platform ([http://msc-viz.emsl.pnnl.gov/nanoPOTS_PI_MS/]).
References
Amstalden van Hove, E. R., Smith, D. F. & Heeren, R. M. A concise review of mass spectrometry imaging. J. Chromatogr. A 1217, 3946–3954 (2010).
Vaysse, P. M., Heeren, R. M. A., Porta, T. & Balluff, B. Mass spectrometry imaging for clinical research—latest developments, applications, and current limitations. Analyst 142, 2690–2712 (2017).
Spraggins, J. M. et al. MALDI FTICR IMS of intact proteins: using mass accuracy to link protein images with proteomics data. J. Am. Soc. Mass Spectrom. 26, 974–985 (2015).
van de Ven, S. M. et al. Protein biomarkers on tissue as imaged via MALDI mass spectrometry: a systematic approach to study the limits of detection. Proteomics 16, 1660–1669 (2016).
Hsu, C. C., Chou, P. T. & Zare, R. N. Imaging of proteins in tissue samples using nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 87, 11171–11175 (2015).
Ryan, D. J., Spraggins, J. M. & Caprioli, R. M. Protein identification strategies in MALDI imaging mass spectrometry: a brief review. Curr. Opin. Chem. Biol. 48, 64–72 (2019).
Stauber, J. et al. On-tissue protein identification and imaging by MALDI-ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 338–347 (2010).
Zhu, Y. et al. Spatially resolved proteome mapping of laser capture microdissected tissue with automated sample transfer to nanodroplets. Mol. Cell Proteomics 17, 1864–1874 (2018).
Meding, S. et al. Tryptic peptide reference data sets for MALDI imaging mass spectrometry on formalin-fixed ovarian cancer tissues. J. Proteome Res. 12, 308–315 (2013).
Stauber, J. et al. MALDI imaging of formalin-fixed paraffin-embedded tissues: application to model animals of Parkinson disease for biomarker hunting. J. Proteome Res. 7, 969–978 (2008).
Rauser, S. et al. Classification of HER2 receptor status in breast cancer tissues by MALDI imaging mass spectrometry. J. Proteome Res. 9, 1854–1863 (2010).
Schober, Y., Schramm, T., Spengler, B. & Rompp, A. Protein identification by accurate mass matrix-assisted laser desorption/ionization imaging of tryptic peptides. Rapid Commun. Mass Spectrom. 25, 2475–2483 (2011).
Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
Nilsson, T. et al. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 7, 681–685 (2010).
Bantscheff, M., Lemeer, S., Savitski, M. M. & Kuster, B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965 (2012).
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J. & Kuster, B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 (2007).
Xie, F., Liu, T., Qian, W. J., Petyuk, V. A. & Smith, R. D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 286, 25443–25449 (2011).
Piehowski, P. D. et al. Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J. Proteome Res. 12, 2128–2137 (2013).
Petyuk, V. A. et al. Spatial mapping of protein abundances in the mouse brain by voxelation integrated with high-throughput liquid chromatography–mass spectrometry. Genome Res. 17, 328–336 (2007).
Braakman, R. B. et al. Optimized nLC-MS workflow for laser capture microdissected breast cancer tissue. J. Proteomics 75, 2844–2854 (2012).
Clair, G. et al. Spatially-resolved proteomics: rapid quantitative analysis of laser capture microdissected alveolar tissue samples. Sci. Rep. 6, 39223 (2016).
Wisniewski, J. R., Ostasiewicz, P. & Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 10, 3040–3049 (2011).
Hughes, C. S. et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014).
Lombard-Banek, C., Moody, S. A. & Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angew. Chem. Int. Ed. Engl. 55, 2454–2458 (2016).
Zhu, Y. et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat. Commun. 9, 882 (2018).
Zhu, Y. et al. Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive nanoLC-MS. Angew. Chem. Int. Ed. Engl. 57, 12370–12374 (2018).
Xu, K. et al. Benchtop-compatible sample processing workflow for proteome profiling of <100 mammalian cells. Anal. Bioanal. Chem. 411, 4587–4596 (2019).
Liang, Y. et al. Spatially resolved proteome profiling of <200 cells from tomato fruit pericarp by integrating laser-capture microdissection with nanodroplet sample preparation. Anal. Chem. 90, 11106–11114 (2018).
Zhu, Y. et al. Subnanogram proteomics: impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int J. Mass Spectrom. 427, 4–10 (2018).
Burnum, K. E. et al. Spatial and temporal alterations of phospholipids determined by mass spectrometry during mouse embryo implantation. J. Lipid Res. 50, 2290–2298 (2009).
Burnum, K. E. et al. Imaging mass spectrometry reveals unique protein profiles during embryo implantation. Endocrinology 149, 3274–3278 (2008).
Duncan, K. D. et al. Quantitative mass spectrometry imaging of prostaglandins as silver ion adducts with nanospray desorption electrospray ionization. Anal. Chem. 90, 7246–7252 (2018).
Lanekoff, I. et al. Three-dimensional imaging of lipids and metabolites in tissues by nanospray desorption electrospray ionization mass spectrometry. Anal. Bioanal. Chem. 407, 2063–2071 (2015).
Lanekoff, I. et al. Trp53 deficient mice predisposed to preterm birth display region-specific lipid alterations at the embryo implantation site. Sci. Rep. 6, 33023 (2016).
Burnum-Johnson, K. E., Baker, E. S. & Metz, T. O. Characterizing the lipid and metabolite changes associated with placental function and pregnancy complications using ion mobility spectrometry-mass spectrometry and mass spectrometry imaging. Placenta 60(Suppl. 1), S67–S72 (2017).
Hafen, R., et al. Trelliscope: a system for detailed visualization in the deep analysis of large complex data. In: 2013 IEEE Symposium on Large-Scale Data Analysis and Visualization (LDAV) (Atlanta, GA, 2013).
Matzke, M. M. et al. Improved quality control processing of peptide-centric LC-MS proteomics data. Bioinformatics 27, 2866–2872 (2011).
Webb-Robertson, B. J., Matzke, M. M., Jacobs, J. M., Pounds, J. G. & Waters, K. M. A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics 11, 4736–4741 (2011).
Matzke, M. M. et al. A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. Proteomics 13, 493–503 (2013).
Mikels, A. J. & Nusse, R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 4, e115 (2006).
Reese, J. et al. Coordinated regulation of fetal and maternal prostaglandins directs successful birth and postnatal adaptation in the mouse. Proc. Natl Acad. Sci. USA 97, 9759–9764 (2000).
Hirota, Y., et al. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J. Clin. Invest. 120, 803–815 (2010).
Cha, J. et al. Combinatory approaches prevent preterm birth profoundly exacerbated by gene-environment interactions. J. Clin. Invest. 123, 4063–4075 (2013).
Li, Q. et al. A novel pathway involving progesterone receptor, 12/15-lipoxygenase-derived eicosanoids, and peroxisome proliferator-activated receptor gamma regulates implantation in mice. J. Biol. Chem. 279, 11570–11581 (2004).
Chung, J. et al. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432 (2015).
Cha, J. et al. Appropriate crypt formation in the uterus for embryo homing and implantation requires Wnt5a-ROR signaling. Cell Rep. 8, 382–392 (2014).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Polpitiya, A. D. et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558 (2008).
Webb-Robertson, B. J. et al. Combined statistical analyses of peptide intensities and peptide occurrences improves identification of significant peptides from MS-based proteomics data. J. Proteome Res. 9, 5748–5756 (2010).
Hosack, D. A., Dennis, G. Jr, Sherman, B. T., Lane, H. C. & Lempicki, R. A. Identifying biological themes within lists of genes with EASE. Genome Biol. 4, R70 (2003).
Acknowledgements
The authors would like to thank PNNL Graphic Designer Nathan Johnson for preparing the figures. Wnt5a floxed mice were originally obtained from Terry P. Yamaguchi (NCI, Frederick, MD). This work was supported by the National Institutes of Health (NIH) Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R21 HD084788 and NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under award UG3HL145593 (HuBMAP Program) to K.E.B.-J., Laboratory Directed Research and Development award from Pacific Northwest National Laboratory (PNNL) to P.D.P., R33 CA225248 (PNNL) to R.T.K., NIH grants (R01HD068524, DA006668 to S.K.D.), and grant from the March of Dimes (22-FY17-889). PNNL is a multiprogram national laboratory operated by Battelle for the Department of Energy (DOE) under Contract DE-AC05-76RLO 1830. A portion of this research was performed using EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and loacted at PNNL.
Author information
Authors and Affiliations
Contributions
P.D.P., Y.Z., R.T.K. and K.E.B.-J. conceptualized and designed experiments. J.Y. and S.K.D. provided mouse models and dissected uterine tissues. Y.Z. and P.D.P. prepared samples for LC-MS/MS analysis. P.D.P., Y.Z., R.J.M. and D.J.O. designed and built automated LC system. R.Z. and Y.Z. ran LC-MS/MS analysis. P.D.P. carried out LC-MS/MS data processing. L.M.B., K.G.S., and B.-J.M.W.-R. performed statistical analysis and built the Trelliscope data visualization platform. K.E.B.-J., Y.G. and H.D.M. carried out biological data interpretation. P.D.P., Y.Z. and K.E.B.-J. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piehowski, P.D., Zhu, Y., Bramer, L.M. et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-μm spatial resolution. Nat Commun 11, 8 (2020). https://doi.org/10.1038/s41467-019-13858-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13858-z
This article is cited by
-
Multiscale biochemical mapping of the brain through deep-learning-enhanced high-throughput mass spectrometry
Nature Methods (2024)
-
Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications
Bone Research (2024)
-
A Review of the Application of Spatial Transcriptomics in Neuroscience
Interdisciplinary Sciences: Computational Life Sciences (2024)
-
Mapping microhabitats of lignocellulose decomposition by a microbial consortium
Nature Chemical Biology (2024)
-
Exploration of cell state heterogeneity using single-cell proteomics through sensitivity-tailored data-independent acquisition
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
