
Abstract
Perfluorinated alkyl substances, such as perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), are toxic materials that are known to globally contaminate water, air, and soil resources. Strategies for the simultaneous detection and removal of these compounds are desired to address this emerging health and environmental issue. Herein, we develop a type of guanidinocalix[5]arene that can selectively and strongly bind to PFOS and PFOA, which we use to demonstrate the sensitive and quantitative detection of these compounds in contaminated water through a fluorescent indicator displacement assay. Moreover, by co-assembling iron oxide nanoparticle with the amphiphilic guanidinocalix[5]arene, we are able to use simple magnetic absorption and filtration to efficiently remove PFOS and PFOA from contaminated water. This supramolecular approach that uses both molecular recognition and self-assembly of macrocyclic amphiphiles is promising for the detection and remediation of water pollution.
Similar content being viewed by others
Introduction
Water pollution is a serious threat to the health of organisms worldwide and is widely regarded as a major environmental issue1. The contamination of surface and ground water2 by perfluorinated alkyl substances has particularly emerged as an environmental crisis impacting hundreds of millions of people3,4 due to the increasing use of these compounds in the production of fluoropolymers5, stain guard products6, and fire-fighting foams7. The most common perfluorinated alkyl pollutants are perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) (Fig. 1), which have been found in water worldwide, including the polar zones8. PFOS and PFOA can bind to proteins9 and then deposit within the body, resulting in undesirable effects10, including kidney11 and liver12 damage, thyroid disease13, immunotoxicity14, reproductive toxicity15, and cancer16. Moreover, these pollutants are highly stable as a result of the thermodynamic stability of the C–F bonds17 and demonstrate significant bioaccumulation, making them a persistent chemical threat to the environment18.

Chemical structures of the employed calixarene hosts (GC5A-6C and GC5A-12C), the Fl reporter dye, and the perfluorinated pollutants (PFOS and PFOA) in this work.
According to the U.S. Environmental Protection Agency, the health advisory level for the combined concentration of PFOA and PFOS in drinking water is 70 ng L−1 3. Nevertheless, the levels of them in drinking water typically exceed that threshold in communities near industrial areas, airports, and military facilities3,19,20. As a result, there is an increasing need for rapid and sensitive techniques for the detection of PFOS and PFOA in contaminated water, as well as efficient remediation methods.
Typical non-labeled detection methods of PFOS and PFOA, include gas chromatography with electron capture detection21 and chromatography-mass spectrometry3. However, the long analysis time and requirement of expensive instrumentation hinders the application of these techniques in high-throughput screening of environmental samples. There have been limited studies on the development of simple, inexpensive, and sensitive optical methods (e.g., via fluorescence or colorimetric changes) for the detection of PFOS22 and PFOA23,24. Furthermore, the lack of a specific receptor design can limit the sensitivity and selectivity of these optical detection methods25.
Degrading PFOA and PFOS is difficult and will produce new toxic byproducts26. Therefore, absorption may be the most suitable technique for purifying PFOS and PFOA from contaminated water27. Typical absorbents are made of activated carbon28, carbon nanotubes29, resins28, polymers30, mineral materials, biomaterials, and molecularly imprinted polymers31. However, there are few absorbents specifically designed for perfluorinated alkyl substances32, and no absorption system has been integrated with real-time detection through simple optical techniques.
Supramolecular chemistry represents an elegant approach to construct multifunctional materials for challenging applications33,34, with the virtues of molecular recognition and self-assembly. Macrocyclic hosts, such as crown ether35, cyclodextrin36, calixarene37,38,39,40, pillararene41,42, cucurbituril43,44, and others45,46, are families of well-studied artificial receptors with a cavity that can be selective for the recognition of particular guests. Efficient molecular recognition by macrocycles in aqueous media has been demonstrated for various applications, including the detection44,47 and absorption48,49 of pollutants. One representative example is β-cyclodextrin50, which displays a binding affinity of ~104 M−1 for PFOS and PFOA51, enabling a β-cyclodextrin polymer network to efficiently sequester PFOA27,52. Exploring artificial receptors with extraordinarily high affinities to PFOS and PFOA is crucial for both the detection and absorption of these compounds, as stronger binding will result in higher sensitivity and absorption efficiency.
In this work, we report the nanomolar binding of two different guanidinocalix[5]arenes (GC5A-6C and GC5A-12C, which feature 6 and 12 carbon atoms in each alkyl chains at the lower rim of the macrocycles, respectively) (Fig. 1) towards PFOS and PFOA. Based on molecular recognition, we achieve sensitive fluorescence detection of PFOA and PFOS using an indicator displacement assay (IDA) with fluorescein (Fl) as the reporter dye (Fig. 1). Moreover, efficient magnetic absorption of PFOA and PFOS is achieved by loading magnetic iron oxide nanoparticle (MNP) into the GC5A-12C assembly.
Results
Complexation studied by theoretical calculations and NMR
Both PFOS and PFOA possess two potential binding sites: a head group composed of sulfonate (PFOS) or carboxylic acid (PFOA) and a C–F chain tail. Based on these structural features, we explored GC5A-6C as a candidate receptor mainly due to its complementary size and shape compared to the chain structures of PFOS and PFOA, as well as its salt-bridge interactions. GC5A-6C was synthesized and purified according to our previous method53. Geometry optimizations of the GC5A-6C•PFOA and GC5A-6C•PFOS complexes were performed using the B3LYP-D3/6–31G(d)/SMD(water) method (Supplementary Note 1)54,55,56,57, in which we found that both complexes feature a threading geometry (Fig. 2a and Supplementary Fig. 1).

a The optimized binding geometry of the GC5A-6C•PFOS complex (right) at the B3LYP-D3/6–31G(d)/SMD(water) level of theory and the MEP-mapped molecular van der Waals surfaces of GC5A-6C (left) and PFOS (middle). b δginter = 0.01 a.u. isosurfaces colored by the sign of (λ2)ρ for the GC5A-6C•PFOS complex (the meaning of δginter and sign(λ2)ρ were in ref. 58). Blue indicates strong attraction, while red indicates strong repulsion. c The atoms of GC5A-6C colored according to their contributions to the binding with PFOS. White indicates no contribution to the complexation, and blue indicates the largest relative contribution.
To gain further understanding of the molecular recognition behavior, we calculated and mapped the molecular electrostatic potential (MEP)59 on the molecular van der Waals surfaces of GC5A-6C and PFOS (Fig. 2a). GC5A-6C is electron-deficient, especially at its upper rim, and PFOS is electron-rich, particularly at its sulfonate head group. The optimized binding geometry is reasonable since molecules tend to approach each other in a complementary manner of the MEP. Furthermore, independent gradient model analysis58 (Fig. 2b) reveals the strong N–H···O hydrogen bonds (blue areas in the isosurfaces) between the guanidinium groups of GC5A-6C and the head group of PFOS. The green areas in the isosurfaces indicate the existence of (1) weak C–H···F–C hydrogen bonds between alkyl chains in the GC5A-6C and the fluorocarbon chain in PFOS60, and (2) van der Waals interactions of the aromatic rings and oxygen atoms in GC5A-6C with the fluorocarbon chain in PFOS. Coloring the atoms of GC5A-6C according to their contribution to the host-guest complexation clearly shows that the main contributions derive from the guanidinium groups and aromatic rings, though the alkyl chains also provide weak interactions (Fig. 2c). As a result, the synergistic effect of these different interactions contributes to the strong binding between GC5A-6C and PFOS or PFOA as desired. We verified the complexation of PFOA with GC5A-6C by 19F nuclear magnetic resonance (NMR) experiments in CD3OD. The significant upfield shift in the fluorine nuclei link to Cα was observed (Supplementary Fig. 2), which may be caused by the intermolecular N–H···F hydrogen bonds between guanidinium groups of GC5A-6C and fluorine atoms link to Cα of PFOA when PFOA was encapsulated into the cavity of GC5A-6C (Supplementary Fig. 1).
Sensitive and selective detection of PFOS and PFOA
We further determined the binding affinities of GC5A-6C to PFOS and PFOA using an IDA (Fig. 3a), in which a fluorescent indicator first reversibly binds to the receptor (Supplementary Fig. 3). Then, an analyte is added into the solution, displacing the indicator from the cavity of the receptor, which changes the optical signal61. By employing GC5A-6C•Fl as the reporter pair (Ka = 5.0 × 106 M−1)53, we obtained binding affinities of (3.5 ± 1.0) × 107 M−1 for PFOS (Fig. 3b) and (1.7 ± 0.3) × 107 M−1 for PFOA (Fig. 3c). The binding affinities are about three orders of magnitude higher than those of previously reported supramolecular hosts toward PFOS, which are around 104 M−1 (Supplementary Table 3).

a Illustration of the IDA principle. Competitive titrations of GC5A-6C•Fl (0.4/0.5 μM) with b PFOS (up to 2.4 μM) and c PFOA (up to 7.6 μM). (Inset) The associated titration curves of b PFOS and c PFOA fit according to a 1:1 competitive binding model. All experiments were performed in HEPES buffer at 25 °C, λex = 500 nm, and λem = 513 nm. Data represent mean ± s.d. (n = 3 independent experiments).
The principle of the IDA allows for fluorescence “switch-on” sensing of PFOS and PFOA using the GC5A-6C•Fl reporter pair. The fluorescence increase linearly with increasing PFOS (Fig. 4a) and PFOA (Fig. 4b) concentrations, respectively. Based on these results, we calculated the limit of detection (LOD) values as 21.4 ± 0.4 nM (11.3 ± 0.2 μg L−1) for PFOS and 26.4 ± 0.2 nM (10.9 ± 0.1 μg L−1) for PFOA by a 3σ/slope method62. The sensitivity of this assay to PFOS and PFOA is superior to or comparable with those of gas chromatography and high-performance liquid chromatography techniques63, indicating that this facile and sensitive fluorescence assay has potential for rapid detection of heavy contamination by PFOS and PFOA21,63. For example, the PFOA concentration in drinking water near a fluorochemical facility in Washington, West Virginia, USA, is as high as 13.3 μg L−1, which is 190-fold greater than the health advisory level (70 ng L−1) recommended by the U.S. Environmental Protection Agency3,64. Given the sensitivity of our technique, it should be possible to quickly detect PFOS and PFOA in the drinking water of such contaminated regions.

Plots of I/I0 against PFOS (left) and PFOA (right) concentrations in a, b HEPES buffer, c, d tap water, and e, f water of Mati Lake. I and I0 are the intensities of fluorescence of the GC5A-6C•Fl (0.8/1.0 μM for a, b; 3.0/1.0 μM for c, d; and 10.0/1.0 μM for (e, f) reporter pair with and without the guest molecules, respectively. All experiments were performed at 25 °C, λex = 500 nm, and λem = 513 nm. Error bars represent mean ± s.d. (n = 3 independent experiments).
We also evaluated the detection selectivity of GC5A-6C for PFOA and PFOS. For comparison, we determined that octanesulfonic acid and octanoic acid (C–H chain surfactants) exhibit affinities of (6.0 ± 1.1) × 104 M−1 (Supplementary Fig. 4) and (7.6 ± 0.8) × 104 M−1 (Supplementary Fig. 5) with GC5A-6C, respectively. For these C–H chain surfactants, the hydrogen atoms feature a 23% smaller van der Waals radius compared to fluorine44, which leads to smaller molecular volumes. As a result, their binding affinities are almost three orders of magnitude lower than those of PFOS and PFOA. Furthermore, the addition of common anionic/cationic surfactants, perfluorohexane and anions caused no significant enhancement in the fluorescence intensity of the GC5A-6C•Fl complex (Fig. 5). Salt concentrations are considered to be orders of magnitude higher than PFOS and PFOA in contaminated water. Even 1000-fold excess salts (NaCl, KCl, and MgCl2) resulted in much smaller fluorescence recovery of the GC5A-6C•Fl reporter pair than PFOS and PFOA (Supplementary Fig. 6). Considering that other unknown pollutants in real highly contaminated water might interfere with the detection selectivity of GC5A-6C, we collected the waste water samples from the manufacturing facility (Cangzhou, Hebei Province, China), and freeze-dried these highly contaminated water samples. The addition of the obtained powder with mass concentration more than ten times higher than PFOS and PFOA also caused no significant enhancement in the fluorescence intensity, which validated GC5A-6C could bind preferentially to PFOS and PFOA over other pollutants in the real world. Moreover, the selectivity to various interferences in Fig. 5 were established in the real highly contaminated water (Supplementary Fig. 7).

Fluorescence responses of GC5A-6C•Fl (0.8/1.0 μM) after adding PFOS, PFOA, and interfering species (0.8 μM for small species and 5 μg mL−1 for waste water samples). I and I0 are the intensities of fluorescence of the GC5A-6C•Fl reporter pair with and without the guest molecules, respectively. All experiments were performed in HEPES buffer at 25 °C, λex = 500 nm, and λem = 513 nm. The CTAB is hexadecyltrimethylammonium bromide. Error bars represent mean ± s.d. (n = 3 independent experiments).
To evaluate the applicability and reliability of the proposed method and considering interfering species commonly found in environmental water samples from different sources, we applied the assay to detect PFOS and PFOA in tap water and lake water samples. The water samples were obtained from tap water and Mati Lake (Nankai University, Tianjin, China), which were immediately filtered through 0.45 μm micropore membranes to remove insoluble particles and stored in brown glass bottles as blank samples. Then, we performed the IDA of PFOS and PFOA in the tap and lake water samples containing variable concentrations of the compounds. Despite the existence of various interfering species in these water samples, linear increases in the fluorescence of the GC5A-6C•Fl complex were observed as we increased the concentrations of PFOS and PFOA (Fig. 4c–f). These linear relationships allowed us to establish calibration curves of the intensity of fluorescence to determine the unknown concentrations of PFOS and PFOA in the tap and lake water samples. The LOD values for PFOS were 30.9 ± 0.1 nM (16.6 ± 0.1 μg L−1) in the tap water and 120.8 ± 0.5 nM (65.0 ± 0.3 μg L−1) in Mati Lake. Meanwhile, the LOD values for PFOA were 39.1 ± 2.8 nM (16.2 ± 1.2 μg L−1) in the tap water and 120.3 ± 2.0 nM (49.8 ± 0.8 μg L−1) in Mati Lake.
According to the LOD values of the water samples, our method can be directly applied for the detection of PFOS and PFOA in heavily contaminated sources, such as communities near industrial areas64, airports3, and military facilities3, in which the total concentrations of PFOS and PFOA may reach 1174 μg L−165. For measuring contamination in regular drinking water with lower PFOS and PFOA concentrations, we can employ our assay after preconcentrating the water sample with a solid-phase extraction. Aliquots of blank tap water samples from different regions and Haihe River water samples and those spiked with 50 ng L−1 of PFOS or PFOA were extracted with HLB cartridges (Supplementary Note 2). We then performed the displacement assay using the GC5A-6C•Fl reporter pair and obtained the concentrations of PFOS and PFOA from the established calibration curves. As shown in Supplementary Table 4, the experimental results showed that no PFOS or PFOA was detected in any of the blank tap water samples and Haihe River water samples. The standard addition recoveries of those water samples are in the range of 90.3–102.7%. These values validated our assay, demonstrating its precision and accuracy for the detection of PFOS and PFOA in contaminated drinking water.
Real-time/on-site scanometric monitoring of PFOS and PFOA
To make the assay more available to daily use, the obvious fluorescence changes of GC5A-6C•Fl reporter pair with various concentrations of PFOS and PFOA were further applied as a real-time/on-site visual detection mode by using a smartphone with easy-to-access color scanning application (app). The green color intensities (G values) of the fluorescent images can be directly scanned from the app (Fig. 6 and Supplementary Note 3). According to the G values, calibration curves can be set up with increasing PFOS and PFOA concentrations, respectively.

a The images of the GC5A-6C•Fl reporter pair (8.0/10.0 μM) with various concentrations of PFOS and PFOA (up to 6.0 μM) taken by an iPhone 7. b The images recorded by iPhone 7 with a color-scanning app. Plots of G values against PFOS (c) and PFOA (d) concentrations in HEPES buffer at 25 °C. G values are green color intensities directly scanned from the color scanning app.
The absorption and magnetic separation of PFOS and PFOA
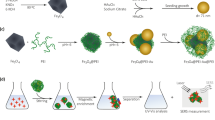
To add absorption capability to this sensing system, we constructed a hybrid material composed of amphiphilic calixarene nanoparticle (Fig. 7). The pegylated GC5A-12C nanoparticle66 was generated by co-assembling GC5A-12C and 4-(dodecyloxy)benzamido-terminated methoxy poly(ethyleneglycol) (PEG-12C) (Supplementary Fig. 8) at a 2:1 molar ratio (Supplementary Note 4). PEG-doping was implemented to enhance the water-solubility and mechanical stability of the material to prevent coagulation and settling67. We determined the binding constants for the pegylated GC5A-12C nanoparticle for PFOS and PFOA to be (1.3 ± 0.3) × 107 M−1 and (4.8 ± 0.4) × 106 M−1, respectively (Supplementary Figs. 9–11). The binding strength of the pegylated GC5A-12C nanoparticle is comparable with that of GC5A-6C and should be suitable for absorption applications. Furthermore, we obtained hybrid nanoparticle (MNP@GC5A-12C) by encapsulating hydrophobic MNP into the hydrophobic domain of the GC5A-12C nanoparticle during preparation (Supplementary Note 5). Dynamic light scattering measurements revealed the MNP@GC5A-12C have a hydrated diameter of 213 ± 3 nm (Supplementary Fig. 12).

Illustration of the absorption and magnetic separation procedure of PFOS and PFOA by the MNP@GC5A-12C nanoparticle.
Al (III) phthalocyanine chloride tetrasulfonic acid (AlPcS4, Supplementary Fig. 8) was chosen as a model dye to explore the absorption ability of MNP@GC5A-12C. MNP@GC5A-12C was dispersed into the solution of AlPcS4 and then isolated with an external magnetic field for 1 h. Subsequently, the supernatant was filtered through a mixed cellulose esters film (Millipore, 0.025 μm) and collected for ultraviolet–visual (UV–vis) experiments. Negligible absorbance of AlPcS4 was observed after absorption (Fig. 8a), indicating the complete removal of AlPcS4 by MNP@GC5A-12C. As a control experiment, filtration without MNP@GC5A-12C resulted in barely any effect on the concentration of the AlPcS4 in solution (Supplementary Fig. 13).

a Absorption spectra of AlPcS4 (10 μM) without any treatment and after magnetic separation by MNP@GC5A-12C and filtration. (Inset) Photographs of MNP@GC5A-12C ([GC5A-12C] = 100 μM) (left), MNP@GC5A-12C ([GC5A-12C] = 100 μM) with AlPcS4 (10 μM) (middle), and the filtrate after magnetic separation and filtration (right). b Concentrations of PFOS and PFOA with only filtration, and with both magnetic separation and filtration are quantified by the UPLC–ESI-MS/MS system. Concentrations of PFOS and PFOA without magnetic separation are shown as a control. Error bars in (b) represent mean ± s.d. (n = 3 independent experiments).
We further applied MNP@GC5A-12C to absorb PFOS and PFOA. The quantifications of PFOS and PFOA were performed by means of ultra-performance liquid chromatography–electrospray ionization–tandem mass spectrometry (UPLC–ESI–MS/MS). The calibration curves were set up and quantitative parameters were evaluated (Supplementary Note 6). The PFOS and PFOA absorption efficiencies of each samples were characterized at [PFOS]0 = [PFOA]0 = 1000 ng mL−1. After the removal procedure, the solutions were pre-concentrated to accurately determine PFOS and PFOA concentrations by the UPLC–ESI–MS/MS. There were only (0.43 ± 0.07)% residual PFOS and (1.53 ± 0.04)% residual PFOA, respectively (Fig. 8b). The regeneration of the present supramolecular material is considered to be feasible owing to its response to specific organic solvent68. In DMSO, GC5A-12C neither formed amphiphilic aggregates indicated by very low scattering intensity (Supplementary Fig. 14), nor complexed with PFOS revealed by no change in chemical shifts of 19F NMR spectra of PFOS (Supplementary Fig. 15). Therefore, we envisage that regeneration of the supramolecular material could be achieved by using routine purification methods in organic synthesis such as column chromatography.
Discussion
In conclusion, our artificial GC5A-6C receptor successfully encapsulated PFOA and PFOS with nanomolar affinity in aqueous media. Taking advantage of the strong recognition and supramolecular assembly, we achieved not only sensitive and quantitative detection of PFOA and PFOS in tap and lake water through the fluorescent IDA strategy, but also the efficient removal of them by the hybrid MNP@GC5A-12C nanoparticle via a simple magnetic absorption and filtration procedure. These results will facilitate the development of detection and absorption methods for PFOA and PFOS. Although the present LOD and removal efficiency cannot reach the health advisory level in drinking water, the proposed supramolecular approach can be practically operated in heavily polluted areas, such as industrial regions, airports, and military facilities. For daily drinking water detection, we can use the solid-phase extraction for sample preconcentration. Higher removal efficiency may be achieved by applying the GC5A-12C nanoparticle as solid-phase extraction absorbents. This work made full use of the molecular recognition and self-assembly of artificial receptors, offering a promising strategy for the detection and remediation of water pollution.
Methods
Chemicals
All the reagents and solvents were commercially available and used as received unless otherwise specified purification. Ammonium acetate and 2,2,2-trifluoroethanol were purchased from Sigma-Aldrich. Fl was purchased from Tokyo Chemical Industry. Al (III) phthalocyanine chloride tetrasulfonic acid (AlPcS4) was purchased from Frontier Scientific. PFOA, PFOS, hexadecyltrimethylammonium bromide, octanoic acid, octanesulfonic acid, and perfluorohexane were purchased from Energy Chemical. Iron oxide nanoparticle stabilized by oleic acid (MNP) was purchased from Ji Cang Nano Company. The waste water samples were provided by the manufacturing facility located in Cangzhou, Hebei province, China. 5,11,17,23,29-Pentaguanidinium-31,32,33,34,35-penta(4-methylpentloxy)calix[5]arene (GC5A-6C), 5,11,17,23,29-pentaguanidinium-31,32,33,34,35-penta dodecyloxy-calix[5]arene (GC5A-12C) and 4-(dodecyloxy)benzamido-terminated methoxy poly(ethylene glycol) (PEG-12C) were synthesized according to the previous literature53,66.
Samples
The 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) buffer solution of pH 7.4 was prepared by dissolving 2.38 g of HEPES in approximate 900 mL double-distilled water. Titrate to pH 7.4 at the lab temperature of 25 °C with NaOH and make up volume to 1000 mL with double-distilled water. The pH value of the buffer solution was then verified on a pH-meter calibrated with three standard buffer solutions.
Apparatus
19F NMR data were recorded on a Bruker AV400 spectrometer. Steady-state fluorescence spectra were recorded in a conventional quartz cell (light path 10 mm) on a Cary Eclipse equipped with a Cary single-cell peltier accessory. UV–vis spectra were recorded in a quartz cuvette (light path 10 mm) on a Cary 100 UV–vis spectrophotometer equipped with a Cary dual cuvette peltier accessory. The sample solutions for dynamic light scattering measurements were examined on a laser light scattering spectrometer (NanoBrook 173plus) equipped with a digital correlator at 659 nm at a scattering angle of 90°. Quantification of PFOS and PFOA from the absorption studies were performed by means of UPLC–ESI–MS/MS (Waters, Milford, MA, USA).
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information, and from the corresponding author upon reasonable request. The source data underlying Figs. 3b, c, 4a–f, 5, 6, 8a, b, Supplementary Figs. 4, 5–7, 9–14, and Supplementary Table 4 are provided as a Source Data file.
References
Schwarzenbach, R. P., Egli, T., Hofstetter, T. B., von Gunten, U. & Wehrli, B. Global water pollution and human health. Annu. Rev. Environ. Resour. 35, 109–136 (2010).
Barzen-Hanson, K. A. & Field, J. A. Discovery and implications of C2 and C3 perfluoroalkyl sulfonates in aqueous film-forming foams and groundwater. Environ. Sci. Technol. Lett. 2, 95–99 (2015).
Hu, X. C. et al. Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants. Environ. Sci. Technol. Lett. 3, 344–350 (2016).
Knutsen, H. K. et al. Risk to human health related to the presence of perfluorooctane sulfonic acid and perfluorooctanoic acid in food. EFSA J. 16, 5194 (2018).
Lindstrom, A. B., Strynar, M. J. & Libelo, E. L. Polyfluorinated compounds: past, present, and future. Environ. Sci. Technol. 45, 7954–7961 (2011).
Herzke, D., Olsson, E. & Posner, S. Perfluoroalkyl and polyfluoroalkyl substances (PFASs) in consumer products in Norway—a pilot study. Chemosphere 88, 980–987 (2012).
Barzen-Hanson, K. A. et al. Discovery of 40 classes of per- and polyfluoroalkyl substances in historical aqueous film-forming foams (AFFFs) and AFFF-impacted groundwater. Environ. Sci. Technol. 51, 2047–2057 (2017).
Fujii, S., Polprasert, C., Tanaka, S., Hong Lien, N. P. & Qiu, Y. New POPs in the water environment: distribution, bioaccumulation and treatment of perfluorinated compounds—a review paper. J. Water Supply Res. Technol. 56, 313–326 (2007).
Xin, Y. et al. Chlorinated polyfluoroalkylether sulfonates exhibit similar binding potency and activity to thyroid hormone transport proteins and nuclear receptors as perfluorooctanesulfonate. Environ. Sci. Technol. 52, 9412–9418 (2018).
Gomis, M. I., Vestergren, R., Borg, D. & Cousins, I. T. Comparing the toxic potency in vivo of long-chain perfluoroalkyl acids and fluorinated alternatives. Environ. Int. 113, 1–9 (2018).
Kataria, A., Trachtman, H., Malaga-Dieguez, L. & Trasande, L. Association between perfluoroalkyl acids and kidney function in a cross-sectional study of adolescents. Environ. Health 14, 89 (2015).
Gallo, V. et al. Serum perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS) concentrations and liver function biomarkers in a population with elevated PFOA exposure. Environ. Health Perspect. 120, 655–660 (2012).
Melzer, D., Rice, N., Depledge, M. H., Henley, W. E. & Galloway, T. S. Association between serum perfluorooctanoic acid (PFOA) and thyroid disease in the U.S. National Health and Nutrition Examination Survey. Environ. Health Perspect. 118, 686–692 (2010).
DeWitt, J. C., Peden-Adams, M. M., Keller, J. M. & Germolec, D. R. Immunotoxicity of perfluorinated compounds: recent developments. Toxicol. Pathol. 40, 300–311 (2012).
Kjeldsen, L. S. & Bonefeld-Jorgensen, E. C. Perfluorinated compounds affect the function of sex hormone receptors. Environ. Sci. Pollut. Res. Int. 20, 8031–8044 (2013).
Barry, V., Winquist, A. & Steenland, K. Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ. Health Perspect. 121, 1313–1318 (2013).
Castiglioni, S. et al. Sources and fate of perfluorinated compounds in the aqueous environment and in drinking water of a highly urbanized and industrialized area in Italy. J. Hazard. Mater. 282, 51–60 (2015).
Wang, T., Wang, Y., Liao, C., Cai, Y. & Jiang, G. Perspectives on the inclusion of perfluorooctane sulfonate into the Stockholm Convention on persistent organic pollutants. Environ. Sci. Technol. 43, 5171–5175 (2009).
Gao, Y. et al. Differential accumulation and elimination behavior of perfluoroalkyl acid isomers in occupational workers in a manufactory in China. Environ. Sci. Technol. 49, 6953–6962 (2015).
Zhang, X. et al. Source attribution of poly- and perfluoroalkyl substances (PFASs) in surface waters from Rhode Island and the New York Metropolitan Area. Environ. Sci. Technol. Lett. 3, 316–321 (2016).
Dufkova, V., Cabala, R., Maradova, D. & Sticha, M. A fast derivatization procedure for gas chromatographic analysis of perfluorinated organic acids. J. Chromatogr. A 1216, 8659–8664 (2009).
Liang, J., Deng, X. & Tan, K. An eosin Y-based “turn-on” fluorescent sensor for detection of perfluorooctane sulfonate. Spectrochim. Acta Part A 150, 772–777 (2015).
Niu, H. et al. Sensitive colorimetric visualization of perfluorinated compounds using poly(ethylene glycol) and perfluorinated thiols modified gold nanoparticles. Anal. Chem. 86, 4170–4177 (2014).
Cheng, Z. et al. Highly sensitive and selective detection of perfluorooctane sulfonate based on the Janus Green B resonance light scattering method. Anal. Methods 8, 8042–8048 (2016).
Takayose, M., Akamatsu, K., Nawafune, H., Murashima, T. & Matsui, J. Colorimetric detection of perfluorooctanoic acid (PFOA) utilizing polystyrene-modified gold nanoparticles. Anal. Lett. 45, 2856–2864 (2012).
Lin, H., Niu, J., Ding, S. & Zhang, L. Electrochemical degradation of perfluorooctanoic acid (PFOA) by Ti/SnO2–Sb, Ti/SnO2–Sb/PbO2 and Ti/SnO2–Sb/MnO2 anodes. Water Res. 46, 2281–2289 (2012).
Xiao, L. et al. beta-Cyclodextrin polymer network sequesters perfluorooctanoic acid at environmentally relevant concentrations. J. Am. Chem. Soc. 139, 7689–7692 (2017).
Yu, Q., Zhang, R., Deng, S., Huang, J. & Yu, G. Sorption of perfluorooctane sulfonate and perfluorooctanoate on activated carbons and resin: kinetic and isotherm study. Water Res. 43, 1150–1158 (2009).
Li, X., Chen, S., Quan, X. & Zhang, Y. Enhanced adsorption of PFOA and PFOS on multiwalled carbon nanotubes under electrochemical assistance. Environ. Sci. Technol. 45, 8498–8505 (2011).
Koda, Y., Terashima, T. & Sawamoto, M. Fluorous microgel star polymers: selective recognition and separation of polyfluorinated surfactants and compounds in water. J. Am. Chem. Soc. 136, 15742–15748 (2014).
Karoyo, A. H. & Wilson, L. D. Nano-sized cyclodextrin-based molecularly imprinted polymer adsorbents for perfluorinated compounds-a mini-review. Nanomaterials 5, 981–1003 (2015).
Zhang, W., Zhang, D. & Liang, Y. Nanotechnology in remediation of water contaminated by poly- and perfluoroalkyl substances: a review. Environ. Pollut. 247, 266–276 (2019).
Webber, M. J., Appel, E. A., Meijer, E. W. & Langer, R. Supramolecular biomaterials. Nat. Mater. 15, 13–26 (2015).
Xu, Z. et al. Heteromultivalent peptide recognition by co-assembly of cyclodextrin and calixarene amphiphiles enables inhibition of amyloid fibrillation. Nat. Chem. 11, 86–93 (2019).
Liu, Z., Nalluri, S. K. M. & Stoddart, J. F. Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes. Chem. Soc. Rev. 46, 2459–2478 (2017).
Zhang, J. & Ma, P. X. Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv. Drug Deliv. Rev. 65, 1215–1233 (2013).
Guo, D.-S. & Liu, Y. Supramolecular chemistry of p-sulfonatocalix[n]arenes and its biological applications. Acc. Chem. Res. 47, 1925–1934 (2014).
Beatty, M. A., Borges-González, J., Sinclair, N. J., Pye, A. T. & Hof, F. Analyte-driven disassembly and turn-on fluorescent sensing in competitive biological media. J. Am. Chem. Soc. 140, 3500–3504 (2018).
Geng, W.-C. et al. A noncovalent fluorescence turn-on strategy for hypoxia imaging. Angew. Chem. Int. Ed. 58, 2377–2381 (2019).
Omorodion, H. et al. A rationally designed perfluorinated host for the extraction of PFOA from water utilising non-covalent interactions. N. J. Chem. 42, 7956–7968 (2018).
Jie, K., Zhou, Y., Li, E. & Huang, F. Nonporous adaptive crystals of pillararenes. Acc. Chem. Res. 51, 2064–2072 (2018).
Li, B. et al. A pH responsive complexation-based drug delivery system for oxaliplatin. Chem. Sci. 8, 4458–4464 (2017).
Lazar, A. I. et al. Nanomolar binding of steroids to cucurbit[n]urils: selectivity and applications. J. Am. Chem. Soc. 138, 13022–13029 (2016).
Assaf, K. I. & Nau, W. M. Cucurbiturils as fluorophilic receptors. Supramol. Chem. 26, 657–669 (2014).
Li, B. et al. Terphen[n]arenes and quaterphen[n]arenes (n=3–6): one-pot synthesis, self-assembly into supramolecular gels, and iodine capture. Angew. Chem., Int. Ed. 58, 3885–3889 (2019).
Gavette, J. V., McGrath, J. M., Spuches, A. M., Sargent, A. L. & Allen, W. E. Fluorous effects in amide-based receptors for anions. J. Org. Chem. 74, 3706–3710 (2009).
Serio, N., Moyano, D. F., Rotello, V. M. & Levine, M. Array-based detection of persistent organic pollutants via cyclodextrin promoted energy transfer. Chem. Commun. 51, 11615–11618 (2015).
Alsbaiee, A. et al. Rapid removal of organic micropollutants from water by a porous beta-cyclodextrin polymer. Nature 529, 190–194 (2016).
Dai, D. et al. Supramolecular assembly-induced emission enhancement for efficient mercury(II) detection and removal. J. Am. Chem. Soc. 141, 4756–4763 (2019).
Palepu, R. & Reinsborough, V. C. Solution inclusion complexes of cyclodextrins with sodium perfluorooctanoate. Can. J. Chem. 67, 1550–1553 (1989).
Karoyo, A. H., Sidhu, P., Wilson, L. D. & Hazendonk, P. Characterization and dynamic properties for the solid inclusion complexes of beta-cyclodextrin and perfluorooctanoic acid. J. Phys. Chem. B 117, 8269–8282 (2013).
Klemes, M. J. et al. Reduction of a tetrafluoroterephthalonitrile-beta-cyclodextrin polymer to remove anionic micropollutants and perfluorinated alkyl substances from water. Angew. Chem. Int. Ed. 58, 12049–12053 (2019).
Zheng, Z. et al. Ultrasensitive and specific fluorescence detection of a cancer biomarker via nanomolar binding to a guanidinium-modified calixarene. Chem. Sci. 9, 2087–2091 (2018).
Hehre, W. J., Ditchfield, R. & Pople, J. A. Self—consistent molecular orbital methods. XII. further extensions of gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261 (1972).
Hariharan, P. C. & Pople, J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222 (1973).
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Murray, J. S. & Politzer, P. The electrostatic potential: an overview. WIREs Comput. Mol. Sci. 1, 153–163 (2011).
Schneider, H.-J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 3, 1381–1394 (2012).
You, L., Zha, D. & Anslyn, E. V. Recent advances in supramolecular analytical chemistry using optical sensing. Chem. Rev. 115, 7840–7892 (2015).
MacDougall, D. & Crummett, W. B. Guidelines for data acquisition and data quality evaluation in environmental chemistry. Anal. Chem. 52, 2242–2249 (2002).
Pobozy, E., Krol, E., Wojcik, L., Wachowicz, M. & Trojanowicz, M. HPLC determination of perfluorinated carboxylic acids with fluorescence detection. Microchim. Acta 172, 409–417 (2011).
Hoffman, K. et al. Private drinking water wells as a source of exposure to perfluorooctanoic acid (PFOA) in communities surrounding a fluoropolymer production facility. Environ. Health Perspect. 119, 92–97 (2011).
Wang, Y. et al. Distribution of perfluorooctane sulfonate and other perfluorochemicals in the ambient environment around a manufacturing facility in China. Environ. Sci. Technol. 44, 8062–8067 (2010).
Gao, J. et al. Biomarker displacement activation: a general host-guest strategy for targeted phototheranostics in vivo. J. Am. Chem. Soc. 140, 4945–4953 (2018).
Geng, W.-C. et al. A self-assembled white-light-emitting system in aqueous medium based on a macrocyclic amphiphile. Chem. Commun. 53, 392–395 (2017).
Zhang, Z. et al. Expanded porphyrin-anion supramolecular assemblies: environmentally responsive sensors for organic solvents and anions. J. Am. Chem. Soc. 137, 7769–7774 (2015).
Acknowledgements
This work was supported by NSFC (21672112 and 51873090) and the Fundamental Research Funds for the Central Universities, which are gratefully acknowledged. The simulation studies were performed at the LvLiang Cloud Computing Center of China, and the calculations were performed on TianHe-2, which are gratefully acknowledged. The authors also thank Tielong Li at Nankai University for providing the waste water samples and Xianrui Wang at Tianjin University of Traditional Chinese Medicine for the UPLC–ESI–MS/MS experiments.
Author information
Authors and Affiliations
Contributions
Z.Z. and D.S.G. conceived the experiments. W.C.G. synthesized the GC5A-6C and GC5A-12C. W.C.G. and Z.L. performed the DFT calculations. Z.Z., H.Y., X.Y.H. and Y.Y.W. performed all the other experiments. Z.Z., X.Y.H., Y.W. and D.S.G. contributed to writing of this paper and all authors commented on it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Robert Baker and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, Z., Yu, H., Geng, WC. et al. Guanidinocalix[5]arene for sensitive fluorescence detection and magnetic removal of perfluorinated pollutants. Nat Commun 10, 5762 (2019). https://doi.org/10.1038/s41467-019-13775-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13775-1
This article is cited by
-
Elimination of chloramphenicol through electro-fenton-like reaction: Reaction mechanism and electron transfer pathway
npj Clean Water (2023)
-
Repurposing host-guest chemistry to sequester virulence and eradicate biofilms in multidrug resistant Pseudomonas aeruginosa and Acinetobacter baumannii
Nature Communications (2023)
-
Sol-gel synthesis of metal ions doped TiO2 catalyst with efficient photodegradation of dye pollutant
Journal of Central South University (2023)
-
Nucleotide Recognition by a Guanidinocalixarene Receptor in Aqueous Solution
Chemical Research in Chinese Universities (2023)
-
Installation of synergistic binding sites onto porous organic polymers for efficient removal of perfluorooctanoic acid
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
