
Abstract
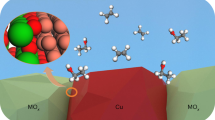
Long-chain alcohols synthesis (LAS, C5+OH) from syngas provides a promising route for the conversion of coal/biomass/natural gas into high-value chemicals. Cu-Fe binary catalysts, with the merits of cost effectiveness and high CO conversion, have attracted considerable attention. Here we report a nano-construct of a Fe5C2-Cu interfacial catalyst derived from Cu4Fe1Mg4-layered double hydroxide (Cu4Fe1Mg4-LDH) precursor, i.e., Fe5C2 clusters (~2 nm) are immobilized onto the surface of Cu nanoparticles (~25 nm). The interfacial catalyst exhibits a CO conversion of 53.2%, a selectivity of 14.8 mol% and a space time yield of 0.101 g gcat−1 h−1 for long-chain alcohols, with a surprisingly benign reaction pressure of 1 MPa. This catalytic performance, to the best of our knowledge, is comparable to the optimal level of Cu-Fe catalysts operated at much higher pressure (normally above 3 MPa).
Similar content being viewed by others

Introduction
Long-chain alcohols, containing more than five carbons, are key alternative fuels and feedstock to manufacture plasticizers, detergents, and lubricants1,2,3,4. Synthesis of long-chain alcohols (LAS) from syngas by a tandem strategy provides a facile, economical, and environment-friendly approach5,6,7. Among various binary catalyst systems (Cu–Fe1,3,8, Cu–Co9,10, Co–Mo11,12), Cu–Fe binary candidates have attracted considerable attention in the production of long-chain alcohols. Previous studies on Cu–Fe binary catalysts show that a yield of long-chain alcohol as high as 0.014–0.144 g gcat−1 h−1 could be reached, albeit a relatively harsh reaction condition (3–8 MPa) is normally required. However, there is still room for the optimization of long-chain alcohols selectivity; more importantly, it is pivotal to develop catalyst system that works under mild reaction conditions9,10,11,12,13,14,15,16,17,18,19,20,21,22. Especially, the low pressure in practical operation gives a significant reduction in pressure drop, energy, and facility costs, which takes merits of both environmental and economic benefits23,24.
To achieve this goal, a precise control over the type, dimension, and nature of Cu/Fe interface plays a key role in the production of LAS. Maintaining a high degree of Cu–Fe interface is decisive for shifting the products from hydrocarbons to long-chain alcohols. We have demonstrated previously that Fe5C2 is very active for CO dissociation and C–C bond propagation, and thus is an excellent catalyst for the FTS reaction25,26,27. Lu et al. recently synthesized a 3DOM FeCu catalyst with atomic steps on the Cu surface involving planar defects and lattice strain, which showed excellent performance toward higher alcohols synthesis3,16. If we can prepare a highly dispersed iron carbide species over Cu, where CO is activated but not dissociated, it is possible to largely enhance the density of interfacial sites. This may render an optimized rate of C–C bond propagation on iron carbide sites and CO insertion on Cu/Fe5C2 interfacial sites, and thus a high selective toward long-chain alcohols would be achieved. Layered double hydroxides (LDHs), with unique structure that metal cations are distributed in the hydroxide layers at an atomic level, have attracted extensive attention as catalyst precursors for higher alcohols synthesis10,15,18,28,29.
In this report, we used Cu–Fe–LDHs as a precursor. After a structural topological transformation followed by an activation treatment30,31,32,33,34, Fe5C2 cluster supported on Cu particle catalyst is obtained. Fe5C2 clusters (~2 nm) are highly dispersed over the surface of Cu nanoparticles (~25 nm) which creates rich interfacial sites; and the Cu4Fe1 catalyst with optimized Fe5C2–Cu interface exhibits a CO conversion of 53.2% and space time yield of 0.101 g gcat−1 h−1 for long-chain alcohols, at a low operation pressure (1 MPa). This is even comparable with the optimal space time yield level of other Cu–Fe binary catalysts at pressure of 3–8 MPa.
Results and discussion
Catalysts synthesis and characterizations
The CuxFeyMg4–LDH precursors with different Cu/Fe molar ratios (1/1, 2/1, 4/1, and 6/1, respectively) were prepared by a nucleation and aging separation method developed previously15. The XRD patterns (Fig. 1a) show characteristic diffractions corresponding to an LDH phase (JCPDS 14-0281); SEM images display a typical plate-like hexagonal morphology (Supplementary Fig. 1). Actually, Cu6Fe1Mg4–LDH could hardly give an LDH phase due to the strong Jahn–Teller effect of Cu2+. The LDHs precursors were transformed to mixed metal oxides (MMO) that inherit the plate-like morphology (Supplementary Fig. 2) after thermal treatment. A CuO phase is predominant for these three samples (Fig. 1b); while the diffractions at 35.8° and 37.1° confirm the formation of CuFe2O4 spinel in XRD patterns. From HRTEM images (Supplementary Fig. 3), some nanoparticles uniformly embedded in the platelet matrix are clearly observed. The CuFe2O4 nanoparticles are well-dispersed near CuO nanoparticles. According to the H2-TPR measurements (Fig. 1c), the main peak shifts gradually from 280 °C to 350 °C with the increase of Cu/Fe ratio, which is ascribed to the enhanced amount of CuFe2O4 spinel (a high reduction temperature) within CuO matrix (a low reduction temperature)35,36,37, as confirmed by XRD (Fig. 1b) and HRTEM (Supplementary Fig. 3). The Cu4Fe1Mg4–MMO shows the strongest interaction between CuO and CuFe2O4 species in accordance with HRTEM, possibly providing the most abundant and stable interfacial structure after activation. The precursors were reduced in syngas under optimized conditions to get the supported catalysts, which were denoted as CuxFey (x/y means the ratio between Cu and Fe). The CuxFey catalysts show similar XRD patterns (Fig. 1d) with metallic Cu. The average Cu particle sizes are 15.7, 18.4, 21.7, and 25.1 for Cu1Fe1, Cu2Fe1, Cu4Fe1, and Cu6Fe1 samples, respectively (Supplementary Table 1). Moreover, no obvious diffraction of Fe crystalline is resolved, which indicates that Fe species is highly dispersed in the catalyst.

a XRD patterns of CuxFeyMg4–LDH precursors with three molar ratios of Cu/Fe (1:1, 2:1, 4:1, and 6:1, respectively). b XRD patterns of CuxFeyMg4–MMO calcined samples obtained from calcination of LDHs precursors. c H2-TPR profiles of CuxFeyMg4–MMO samples. d XRD patterns of CuxFey obtained from activation of MMO samples in syngas (25% CO+25% H2+50% CO2) under optimum conditions (300 °C (2 h)+350 °C (1 h); rate: 2 °C min−1) and passivation.
Catalytic results
The catalysts were first evaluated under a relatively high pressure (3 MPa). Figure 2a shows the alcohol distribution over catalysts with various activation treatments (Supplementary Tables 2–6), from which both the activation atmosphere (syngas containing CO2) and the two-stage activation procedure play a vital role in tuning the synergistic effect between Cu and Fe5C2. The sample activated in syngas containing CO2 (Supplementary Table 4) could slow down the rate of reduction process and facilitate carburization of Fe to produce Fe5C2 species, which would maintain the tiny size of Fe5C2 to form abundant Cu–Fe5C2 interfacial sites. The sample reduced in syngas shows diffraction peaks of both Cu and Fe5C2 phase (Supplementary Fig. 4) and rather poor catalytic behavior for the production of LA, indicating less interfacial sites merged by poor dispersion of Fe5C2 is not beneficial for the production of LA. The selectivity toward long-chain alcohols enhances gradually along with the increase of Cu/Fe ratio, reaching maximum at 54.0% for Cu4Fe1 at 3 MPa (specific data in Supplementary Table 7). Cu4Fe1 gives out the optimal space time yield (STY) (Supplementary Table 8) toward total alcohols (0.141 g gcat−1 h−1) and long-chain alcohols (0.076 g gcat−1 h−1) at 3 MPa.

a Alcohol distribution normalized data to total alcohols selectivity (a WHSV = 4800 mL gcat−1 h−1; b WHSV=1200 mL gcat−1 h−1; C5+OH: long-chain alcohols, BuOH: butanol, PrOH: propanol, EtOH: ethanol and MeOH: methanol) and b alcohols STY at different pressures over catalysts with four Cu/Fe ratios (1/1, 2/1, 4/1, and 6/1) (reaction conditions: 27% CO+55% H2+18% N2; 260 °C; WHSV of 2400 mL gcat−1 h−1). c Time-on-stream (TOS) evolution of CO conversion over the four catalysts within 100 h test at 1 MPa. d Alcohols STY of Cu4Fe1, Cu4Fe1-co, Cu4Fe1-im, and Cu10Fe1 at 1 MPa.
What attracts our attention most is the excellent long-chain alcohols production on Cu4Fe1 under 1 MPa (Fig. 2; Supplementary Fig. 5). The Cu4Fe1 has 38.6% at WHSV = 4800 mL gcat−1 h−1 with iso-conversion to other CuxFey samples, although Cu4Fe1 has higher value. The total alcohols yield (0.201 g gcat−1 h−1) of Cu4Fe1 is much higher than hydrocarbons yield (0.111 g gcat−1 h−1), demonstrating the predominant production of alcohols rather than hydrocarbons (Supplementary Table 8). This means the high LA yield is the interplay of activity and selectivity. In addition, long-chain alcohols (C5+ alcohols) give a higher yield than methanol (0.101 vs. 0.018 g gcat−1 h−1), which is among the highest level compared with previous work in Supplementary Table 9. Normally, high reaction pressure (3–8 MPa) is required for long-chain alcohols synthesis; in this work, however, a low-pressure (1 MPa) syngas conversion to long-chain alcohols is attained. Significantly, Cu4Fe1 catalyst at 1 MPa reaction condition shows a satisfactory stability: 5% decrease in activity is observed within a 100 h catalytic evaluation test (Fig. 2c) and a satisfactory reproducibility (Supplementary Table 10).
To understand the outstanding catalytic performance of Cu4Fe1, the impact of catalyst synthesis on its catalytic performance was investigated. Control samples with the same Cu/Fe ratio of 4/1 synthesized through conventional impregnation and co-precipitation methods were prepared (Supplementary Fig. 6: termed as Cu4Fe1-im and Cu4Fe1-co, respectively), from which Cu–Fe5C2 interfaces were rarely observed over those conventional catalysts. However, both catalysts produced scarcely any long-chain alcohols (Supplementary Table 11), indicating the unique interfacial structure derived from the LDHs synthesis route definitely changes the catalytic activity. Another two control samples of pure Cu (denoted as Cu4) or pure Fe (denoted as Fe1) were prepared based on a similar LDHs approach. The Cu4 sample mainly shows methanol synthesis performance and the Fe1 gives conventional FTS performance (Supplementary Table 11), in accordance with previous work38,39,40. When separate Cu4 and Fe1 catalysts were combined with various modes, long-chain alcohols could not be synthesized. This indicates that the Cu–Fe5C2 synergistic effect is responsible for the production of long-chain alcohols. Therefore, the synergy of interfacial sites between Fe5C2 and Cu is must for the long-chain alcohols (higher alcohols) synthesis from syngas. We performed further studies on samples with higher Cu/Fe molar ratios as 10:1 (termed as Cu10Fe1, Supplementary Fig. 7). Although both Cu6Fe1 and Cu10Fe1 samples have good selectivity of long-chain alcohols (Supplementary Table 12), the STY was low (Fig. 2b, d), showing that a suitable Cu/Fe ratio is important for the best catalytic performance. This suggests that catalyst with rich Cu–Fe5C2 interfaces derived from the LDHs precursor method could result in an excellent activity and selectivity toward long-chain alcohols even at a low reaction pressure.
Fine structure characterizations
To further understand the structural property of CuxFey catalysts, the three samples (Cu1Fe1, Cu2Fe1, and Cu4Fe1) were characterized by aberration-corrected scanning transmission electron microscopy (ac-STEM). For Cu1Fe1 and Cu2Fe1 samples (Supplementary Figs. 8, 9), the lattice fringe of Cu (111) with 0.209 nm is clearly resolved; EDS mapping images of Cu, Fe, and C demonstrate the existence of Fe5C2 nanoclusters on the surface of Cu nanoparticles. In the case of Cu4Fe1 sample, Fig. 3a shows the lattice fringe of Cu (111) and Fe5C2 (11-2); corresponding EELS mapping (Fig. 3b) illustrates the modification of Cu by Fe5C2 nanoclusters within a single Cu nanoparticle, which is also confirmed in another selected region in Supplementary Fig. 10. Then, quasi-in-situ STEM measurements on the used catalyst after 1 MPa syngas conversion reaction were performed (Fig. 3). The EDX-mapping results (Fig. 3c, d) also verified the homogeneous distribution of iron species on Cu. The phase of the iron species could be identified to be Fe5C2, as the catalyst shows sextets characteristics in Mössbauer spectrum (Supplementary Fig. 11)16,41. To further confirm the existence of iron carbide species on Cu particles, the electron energy-loss spectra (EELS) of C K-edge at the surface of Cu particle (Spot A) and support (Spot B) were studied (Fig. 3e, f). For C K-edge spectra, Spot A displays a lower energy peak relative to Spot B, accompanied by a small peak at ~280 eV42, which again verifies the existence of carbide species on the surface of Cu particle.

a HRTEM image; b EELS mapping of Cu (blue), C (red), and Fe (green) of (a). quasi-in-situ STEM: c HAADF-STEM image; d EDS mapping of the selected region (white box in c) showing elemental distribution of Cu, C, and Fe; e HAADF-STEM image and f EELS curves of Fe L-edge and C K-edge selected from (e) (Spot A: red, Spot B: blue).
In addition, in situ X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) experiments for Cu4Fe1Mg4–MMO at Cu K-edge and Fe K-edge (Fig. 4a, f) were performed to reveal the structural change during the activation treatment process to obtain Cu4Fe1 catalyst (in 20 mL min−1 5% CO+5% H2+10% CO2+80% He stream). The normalized Cu K-edge XANES spectra show a gradual decrease in the energy of absorption edge relative to CuO reference, accompanied with the appearance of edge features of metallic Cu, indicating a progressive reduction of CuO phase to metallic Cu. Corresponding fitting results (Supplementary Fig. 12) show that Cu species in Cu4Fe1 is mainly metallic Cu with rather small amount of CuOx. Based on the results of previous reports43,44 and observations in this work, Cu0 is regarded as active site for CO adsorption/insertion. In the Cu K-edge EXAFS spectra, initially, the coordination environment of Cu4Fe1Mg4–MMO is consistent with CuO, in which the peaks at 1.5 Å and 2.5 Å are assigned to the first Cu–O coordination and Cu–Cu coordination, respectively45,46. During the activation process, the peak at 2.2 Å ascribed to Cu–Cu coordination shell gradually emerges and becomes predominant, further confirming the reduction to metallic Cu. In the case of Fe species, the normalized Fe K-edge XANES spectra show the reduction of CuFe2O4 spinel to iron carbide phase along with the absorption edge moving to low energy. As shown in Fe K-edge EXAFS spectra, Fe–Fe coordination shell at 2.0 Å is observed at 350 °C, confirming the formation of iron carbide3,47,48. Based on above results, it is concluded that the new interfacial structure is well established from LDHs precursor through refined activation process.

a, e In situ XANES spectra at Cu K-edge and Fe K-edge of Cu4Fe1Mg4-MMO in syngas treatment to obtain Cu4Fe1 catalyst (5% CO + 5% H2 + 10% CO2 + 80% He stream; 20 mL min−1) at different temperatures and time points, respectively. b, f Enlarged view of absorption edge selected from (a, e). c, d Fourier transform magnitude of k3-weighted in situ Cu K-edge and Fe K-edge EXAFS spectra, respectively. g CO-TPD profiles of Fe1, Cu4, and CuxFey samples. h Long-chain alcohols space time yield as a function of Fe5C2int/(Fe5C2int + Fe5C2iso). Fe5C2int: interfacial Fe5C2; Fe5C2iso: isolated Fe5C2. The error bars are between 0.002 and 0.003.
Structure-performance relationship
In addition, CO-TPD (Fig. 4g, h) was performed to quantitatively study the amount of Fe5C2 interfacial sites. As CO adsorption on Cu is rather weak and only be resolved through low-temperature TPD49,50, the observed desorption peaks are ascribed to iron species. In addition to the CO desorption from iron oxide at relatively low temperature (around 300 °C, for Cu1Fe1) and reverse Boudouard reaction (above 600 °C), two desorption peaks at 440 °C and 490 °C (for Cu1Fe1, Cu2Fe1, Cu4Fe1, and Cu6Fe1) could be attributed to CO desorption from iron carbide51,52. With Fe1 as a reference, these two peaks (440 °C and 490 °C) are attributed to interfacial Fe5C2 (Fe5C2int) and isolated Fe5C2 (Fe5C2iso) species, respectively. Clearly, Cu4Fe1 gives the largest integral peak area, indicating the most abundant total Fe5C2 sites than other samples. We fitted and deconvoluted the two peaks to roughly estimate the ratio of Fe5C2int/(Fe5C2int+Fe5C2iso) for these four CuxFey catalysts, and the results showed a volcanic correlation between the LA yield and the relative concentration of interfacial Fe5C2 (Fig. 4h). Cu4Fe1 catalyst with a moderate Fe5C2int ratio possesses the highest concentration of Fe5C2–Cu interface sites (Fe5C2int), accounting for the largest LA yield (0.101 g gcat−1 h−1). This demonstrates that the Fe5C2–Cu interfacial sites act as active center toward LA production.
It is well-known that CO does not dissociate over Cu; while Fe5C2 (or iron carbide in general) is beneficial for CO dissociation and subsequent C–C bond propagation25,26,27,47,53. The unique interfacial structure of ultrasmall Fe5C2 clusters over Cu particles has conferred the Cu4Fe1 catalyst a suitable construct for the production of long-chain alcohols production at syngas pressure as low as 1 MPa. De Jong et al. reported the size effect in supported iron carbides (2–7 nm) for Fischer–Tropsch reaction, in which a smaller particle size improved the coverage of CHx species (the monomer of carbon chain growth)54,55. In this work, long-chain alcohols synthesis at 1 MPa was achieved, which was described to the unique structure of Cu4Fe1 catalyst. The Fe5C2 nanoclusters, with an ultrasmall size (normally below 2 nm) on the surface of Cu nanoparticles, provide active sites for CO activation/dissociation and the resulting C–C bond propagation, which maintains the high activity of Cu4Fe1. As the reaction pressure decreases from 3 MPa to 1 MPa, hydrogen activation on catalyst surface is weakened (Supplementary Fig. 13), which reduces the rate of hydrogenation and hydrocarbons chain termination and thus enhances the growth of carbon chain. In contrast, the CO activation is promoted since CO molecule is prone to adsorb on a Cu-rich surface56,57. This facilitates the kinetic rate coordination between CO insertion and C–C coupling, and therefore significantly elevates the selectivity toward total alcohols and long-chain alcohols58,59,60. This is also verified by the α-ASF chain-lengthening probabilities analysis: the α-value for alcohols (0.72) at 1 MPa exceeds that for hydrocarbons (0.70), indicating the rate of CO insertion is larger than that of hydrocarbons chain termination (Supplementary Fig. 4). Therefore, a precise control over double-active-site in Cu4Fe1 catalyst accounts for the high yield toward long-chain alcohols at 1 MPa.
In summary, Cu4Fe1 catalyst was synthesized from LDH precursor which exhibits a CO conversion of 53.2% and long-chain alcohols space time yield of 0.101 g gcat−1 h−1, at a surprisingly benign pressure of 1 MPa. Combination of study verifies that highly dispersed Fe5C2 nanoclusters are confined over the surface of Cu nanoparticles, forming abundant Fe5C2–Cu interfacial sites; and the synergistic interaction between Fe5C2 clusters and Cu nanoparticle is responsible for the unique catalytic performance toward long-chain alcohol production at low pressure. This work not only discovers a catalyst with unique structure for LAS but also sheds light on the construct of binary metal catalyst.
Methods
Catalysts synthesis
The CuxFeyMg4-LDH precursors with four Cu:Fe:Mg molar ratios of 1:1:4, 2:1:4, 4:1:4, and 6:1:4 (denoted as Cu1Fe1Mg4-LDH, Cu2Fe1Mg4-LDH, Cu4Fe1Mg4-LDH, and Cu6Fe1Mg4-LDH) were prepared by using a method of nucleation and aging separation developed in our laboratory12. Solution A was a mixture of Cu(NO3)2·3H2O, Fe(NO3)3·9H2O, and Mg(NO3)2·6H2O with various Cu:Fe:Mg molar ratios dissolved in 100 mL of deionized water ([Cu2+] + [Mg2+] + [Fe3+] = 1.0 M). Solution B was obtained by dissolving NaOH and Na2CO3 in the same volume of deionized water with [NaOH] = 1.6 M and [CO32−] = 2[Fe3+]. Solution A and B were simultaneously added to a colloid mill rotating at 4000 rpm and mixed for 2 min. The resulting slurry was removed from the colloid mill and aged at 120 °C for 24 h. The final precipitate was filtered, washed thoroughly with deionized water, and dried at 60 °C for 24 h.
General activation process: the CuxFeyMg4-LDH precursors were calcined in air at 500 °C for 4 h at a heating rate of 2 °C min−1 to obtain mixed metal oxides (MMOs) (denoted as Cu1Fe1Mg4–MMO, Cu2Fe1Mg4–MMO, Cu4Fe1Mg4–MMO, and Cu6Fe1Mg4–MMO); subsequently, these MMOs materials were reduced in syngas atmosphere consisting of 25% CO+25% H2+50% CO2 with a two-step-process—300 °C for 2 h and 350 °C for another 1 h at a heating rate of 2 °C min−1 (denoted as Cu1Fe1, Cu2Fe1, Cu4Fe1, and Cu6Fe1, respectively).
In addition, based on Cu4Fe1Mg4-LDH precursor, the calcination temperature, activation procedure, and activation atmosphere were modulated to investigate their effects on catalytic performance, and the results were shown in Supplementary Tables 2–4. Cu4, Fe1, and Cu10Fe1 were prepared following above synthesis and activation process, which were derived from Cu4Al1Mg4–LDH, Fe1Mg8–LDH, and Cu10Fe1Mg4–LDH changing the ratio to Cu:Al:Mg = 4:1:4, Fe:Mg = 1:8, and Cu:Fe:Mg = 10:1:4, respectively (calcined samples: Cu4Al1Mg4–MMO, Fe1Mg8–MMO, and Cu10Fe1Mg4–MMO).
Cu4Fe1-co as a reference sample was prepared by co-precipitated method10. A mixture of copper and iron nitrate (Cu/Fe molar ratio of 4/1) was precipitated with an aqueous solution of Na2CO3. After aging for 4 h, the precipitate was washed thoroughly with distilled water until pH = 7–8, and was dried at 60 °C for 12 h. Then, the sample was calcined and reduced via the same procedure described above. Cu4Fe1-im as a reference sample was prepared by incipient-wetness impregnation method9. In all, 8.0 g of CuO powdered sample was added into an aqueous solution containing 0.025 M iron nitrate. After impregnation for 4 h, the catalyst precursor was dried in air at 60 °C for 12 h followed by calcination and reduction in general activation process.
Catalytic evaluation
The catalytic evaluation was carried out in a 10 -mm fixed-bed stainless-steel reactor. In total, 1.0 g of MMO precursor was loaded on the catalyst bed, and the remaining volume of the reactor tube was filled with quartz beads of 20–40 mesh. Before reaction, the catalyst was activated in situ as mentioned above with a flow rate of 40 mL min−1. After the reactor was cooled to room temperature, syngas with a flow rate of 40 mL min−1 (27% CO+55% H2+18% N2) was introduced to purge the reaction line and reach the required pressure with nitrogen as an internal standard gas. The reaction was conducted at 260 °C. The outlet gas components (CO, H2, CH4, CO2, and N2) after passing through a hot trap (180 °C) and a cold trap (5 °C) were determined using an online GC-2014C Shimadzu gas chromatograph with TCD detector (TDX-1 column) and FID detector (Al2O3 packed column) using He as carrier gas. The liquid products were collected from the hot trap and cold trap, followed by determination offline with FID detector (RTX-5). 1,4-dioxane was used as internal standard for the aqueous products; and ethyl cyclohexane was used as an internal standard after extraction by cyclohexane.
Where F is the moles of CO and product Ci (CO2, hydrocarbon, or alcohols) containing i carbon atoms. The mass balance and carbon balance have been calculated at each product and kept between 85 and 90%.
The ASF chain growth probability α is calculated according to the equation: ln (Wn/n) = nlnα + ln(1 − α)2/α, where n is the number of carbon atoms in products; Wn is the weight fraction of products containing n carbon atoms; and 1 − α is the probability of chain termination.
Catalyst characterization
Powder XRD measurements were performed on a Rigaku XRD-6000 diffractometer, using Cu Kα radiation (λ = 0.15418 nm) at 40 kv and 30 mA, with a scanning rate of 5° min−1 and a 2θ angle ranging from 3° to 90°. The phases of components were identified based on JCPDS standard cards. Scanning electron microscope (SEM; Zeiss SUPRA 55) with an accelerating voltage of 20 kV was performed. Aberration-corrected scanning transmission electron microscopy (ac-STEM), electron energy-loss spectroscopy (EELS), and element energy-dispersive spectroscopy (EDS) mapping measurements were carried out on a JEOL JEM-ARM200F instrument. The quasi-in-situ scanning transmission electron microscopy (STEM) and EDX-mapping measurements were performed on a FEI Tecnai G2 F20 microscope with an accelerating voltage of 120 kV. The sample was hold in glovebox in Ar atmosphere and transferred by a vacuum transfer TEM holder. The specific surface area determination and pore volume analysis were performed by Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halenda (BJH) methods using a Quantachrome Autosorb-1C-VP Analyzer. Elemental analysis for Cu and Fe was performed using a Shimadzu ICPS-75000 inductively coupled plasma atomic emission spectrometer (ICP-AES). Hydrogen temperature-programmed reduction (H2-TPR) was measured on a Micromeritics ChemiSorb 2070 with a thermal conductivity detector (TCD). In a typical process, 100 mg of sample was sealed in a quartz tube reactor and pretreated in a Ar atmosphere at 150 °C for 2 h, followed by reduction in a stream of H2/Ar (1/9, v/v; a total flow rate of 40 mL min−1) at a heating rate of 10 °C min−1 up to 800 °C. Mössbauer spectrum experiments of the as-prepared catalysts were carried out at −268.8 °C. The spectrometer was calibrated using a standard α-Fe foil and was fitted with five sextets, which reflect Fe5C2 and Fe3O4 with different hyperfine parameters. The spectra components were identified according to their isomer shift, quadruple splitting, and magnetic hyperfine field. Magnetic hyperfine fields were calibrated with the 330 kOe field of α-Fe. In situ X-ray absorption fine structure spectroscopy (XAFS) at the Fe and Cu K-edge were acquired at 1W2 beamline of at Beijing Synchrotron Radiation Facility (BSRF) under transmission mode. The typical energy of the storage ring was 2.5 GeV with a maximum current of 250 mA. The Si (111) double-crystal monochromator was used. The powdered sample was first pressed into sheet and loaded into a reactor cell equipped with polyimide windows. The sample sheet was reduced in 5% CO+5% H2+10% CO2 + 80% He stream with 20 mL min−1, and underwent a heat treatment at 300 °C for 120 min then at 350 °C for 180 min (rate: 2 °C min−1). The XAFS spectra were collected 60 times h−1 in the whole process. All the collected spectra were processed and analyzed using Athena code within Ifeffit package. CO- or H2-temperature-programmed desorption (CO-TPD or H2-TPD) experiments were carried out in a fixed-bed reactor and detected by MS. 100 mg catalyst after reaction was exposed to CO or H2 for 120 min in room temperature. The catalyst was switched to He exposure until the baseline of the CO or H2 signal leveled off. Finally, the temperature was increased to 800 °C at 10 °C min−1. The mass signal of 28 or 2 was monitored by quadruple mass spectrometer. The apparent kinetic order of H2 was measured as follows: the catalytic test was performed at 260 °C with Cu4Fe1 catalyst (0.1 g) in a gas flow rate of 40 mL min−1 in order to keep the H2 conversion under 10%. For determining the order of H2, the partial pressure of H2 was controlled from 22.5 to 45%.
References
Luk, H. T., Mondelli, C., Ferre, D. C., Stewart, J. A. & Pérez-Ramírez, J. Status and prospects in higher alcohols synthesis from syngas. Chem. Soc. Rev. 46, 1358–1426 (2017).
Xiang, Y. Z. & Kruse, N. Tuning the catalytic CO hydrogenation to straight- and long-chain aldehydes/alcohols and olefins/paraffins. Nat. Commun. 7, 13058–13064 (2016).
Lu, Y. W. et al. Elucidating the Copper–Hägg iron carbide synergistic interactions for selective CO hydrogenation to higher alcohols. ACS Catal. 7, 5500–5512 (2017).
Prieto, G. et al. Design and synthesis of copper–cobalt catalysts for the selective conversion of synthesis gas to ethanol and higher alcohols. Angew. Chem. Int. Ed. 53, 6397–6401 (2014).
Sun, J. et al. Promotional effects of cesium promoter on higher alcohol synthesis from syngas over cesium-promoted Cu/ZnO/Al2O3 catalysts. ACS Catal. 6, 5771–5785 (2016).
Li, L. & Sholl, D. S. Computational identification of descriptors for selectivity in syngas reactions on a Mo2C catalyst. ACS Catal. 5, 5174–5185 (2015).
Pei, Y. P. et al. High alcohols synthesis via Fischer-Tropsch reaction at cobalt metal/carbide interface. ACS Catal. 5, 3620–3624 (2015).
Zhao, Y. H., Li, S. G. & Sun, Y. H. CO dissociation mechanism on Cu-doped Fe(100) surfaces. J. Phys. Chem. C. 117, 24920–24931 (2013).
Guo, H. J. et al. Effects of Cu/Fe ratio on structure and performance of attapulgite supported CuFeCo-based catalyst for mixed alcohols synthesis from syngas. Appl. Catal. A 503, 51–61 (2015).
Gao, W. et al. Core–shell Cu@(CuCo-alloy)/Al2O3 catalysts for the synthesis of higher alcohols from syngas. Green. Chem. 17, 1525–1534 (2015).
Wu, X. M. et al. Co-decorated carbon nanotubes as a promoter of Co–Mo–K oxide catalyst for synthesis of higher alcohols from syngas. Appl. Catal., A 340, 87–97 (2008).
Toyoda, T., Minami, T. & Qian, E. W. Mixed alcohol synthesis over sulfided molybdenum-based catalysts. Energy Fuels 27, 3769–3777 (2013).
Lin, M. G., Fang, K. G., Li, D. B. & Sun, Y. H. CO hydrogenation to mixed alcohols over co-precipitated Cu–Fe catalysts. Catal. Commun. 9, 1869–1873 (2008).
Wang, N. et al. Synthesis of higher alcohols from syngas over Fe/K/β-Mo2C catalyst. Catal. Lett. 136, 9–13 (2010).
Gao, W. et al. Catalytic conversion of syngas to mixed alcohols over CuFe-based catalysts derived from layered double hydroxides. Catal. Sci. Technol. 3, 1324–1332 (2013).
Lu, Y. W. et al. High selectivity higher alcohols synthesis from syngas over three-dimensionally ordered macroporous Cu-Fe catalysts. ChemCatChem 6, 473–478 (2014).
Ding, M. Y. et al. Enhancement of conversion frombio-syngas to higher alcohols fuels over K-promoted Cu-Fe bimodal pore catalysts. Fuel Process. Technol. 159, 436–441 (2017).
Han, X., Fang, K., Zhou, J., Zhao, L. & Sun, Y. Synthesis of higher alcohols over highly dispersed Cu–Fe based catalysts derived from layered double hydroxides. J. Colloid Interface Sci. 470, 162–171 (2016).
Xu, R. et al. Fe-modified CuMnZrO2 catalysts for higher alcohols synthesis from syngas. J. Mol. Catal. A: Chem. 221, 51–58 (2004).
Su, Y. L., Wang, Y. L. & Liu, Z. M. Preparation and characterization of ultrafine Fe-Cu-based catalysts for CO hydrogenation. J. Nat. Gas. Chem. 17, 327–331 (2008).
Luk, H. T. et al. Carbon nanofibres-supported KCoMo catalysts for syngas conversion into higher alcohols. Catal. Sci. Technol. 8, 187–200 (2018).
Luk, H. T. et al. Role of carbonaceous supports and potassium promoter on higher alcohols synthesis over copper-iron catalysts. ACS Catal. 8, 9604–9618 (2018).
Dasari, M. A. et al. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal., A 281, 225–231 (2005).
Paulik, F. E. & Roth, J. F. Novel catalysts for the low-pressure carbonylation of methanol to acetic acid. Chem. Commun. 24, 1578 (1968).
Zhai, P. et al. Highly tunable selectivity for syngas-derived alkenes over zinc and sodium-modulated Fe5C2 catalyst. Angew. Chem. Int. Ed. 55, 9902–9907 (2016).
Gao, W. et al. Photo-driven syngas conversion to lower olefins over oxygen-decorated Fe5C2 catalyst. Chem 4, 2917–2928 (2018).
Yang, C. et al. Construction of synergistic Fe5C2/Co heterostructured nanoparticles as an enhanced low temperature Fischer–Tropsch synthesis catalyst. ACS Catal. 7, 5661–5667 (2017).
Ning, X., An, Z. & He, J. Remarkably efficient CoGa catalyst with uniformly dispersed and trapped structure for ethanol and higher alcohol synthesis from syngas. J. Catal. 340, 236–247 (2016).
An, Z., Ning, X. & He, J. Ga-promoted CO insertion and C–C coupling on Co catalysts for the synthesis of ethanol and higher alcohols from syngas. J. Catal. 356, 157–164 (2017).
Zhang, S. T. et al. DFT-based simulation and experimental validation of the topotactic transformation of MgAl layered double hydroxides. ChemPhysChem 17, 2754–2766 (2016).
Chen, H. et al. Promoted synergic catalysis between metal Ni and acid–base sites toward oxidant-free dehydrogenation of alcohols. ACS Catal. 7, 2735–2743 (2017).
Chen, H. et al. Ru-cluster-modified Ni surface defects toward selective bond breaking between C–O and C–C. Chem. Mater. 28, 4751–4761 (2016).
Zhao, Y. F. et al. Oxide-modified nickel photocatalysts for the production of hydrocarbons in visible light. Angew. Chem. Int. Ed. 55, 4215–4219 (2016).
Xu, M. et al. TiO2–x-modified Ni nanocatalyst with tunable metal–support interaction for water–gas shift reaction. ACS Catal. 7, 7600–7609 (2017).
Kameoka, S., Tanabe, T. & Tsai, A. P. Spinel CuFe2O4: a precursor for copper catalyst with high thermal stability and activity. Catal. Lett. 100, 89–93 (2005).
Faungnawakij, K. et al. Hydrogen production from dimethyl ether steam reforming over composite catalysts of copper ferrite spinel and alumina. Appl. Catal. B 74, 144–151 (2007).
Hirunsit, P. & Faungnawakij, K. Cu–Cr, Cu–Mn, and Cu–Fe spinel-oxide-type catalysts for reforming of oxygenated hydrocarbons. J. Phys. Chem. C. 117, 23757–23765 (2013).
Behrens, M. et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 336, 893–897 (2012).
Kuld, S. et al. Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis. Science 352, 969–974 (2016).
Xu, K. et al. ε-Iron carbide as a low-temperature Fischer–Tropsch synthesis catalyst. Nat. Commun. 5, 5783–5790 (2014).
Chun, D. H. et al. Highly selective iron-based Fischer–Tropsch catalysts activated by CO2-containing syngas. J. Catal. 317, 135–143 (2014).
Liu, X. et al. Environmental TEM studies of single iron nanoparticle carburization in synthesis gas. ACS Catal. 7, 4867–4875 (2017).
Goulas, K. A. et al. Synergistic effects in bimetallic palladium–copper catalysts improve selectivity in oxygenate coupling reactions. J. Am. Chem. Soc. 138, 6805–6812 (2016).
Su, J. et al. Higher alcohols synthesis from syngas over CoCu/SiO2 catalysts: dynamic structure and the role of Cu. J. Catal. 336, 94–106 (2016).
Wang, X. Q., Hanson, J. C., Frenkel, A. I., Kim, J. Y. & Rodriguez, J. A. Time-resolved studies for the mechanism of reduction of copper oxides with carbon monoxide: complex behavior of lattice oxygen and the formation of suboxides. J. Phys. Chem. B 108, 13667–13673 (2004).
Frenkel, A. I. et al. Combining X-ray absorption and X-ray diffraction techniques for in situ studies of chemical transformations in heterogeneous catalysis: advantages and limitations. J. Phys. Chem. C 115, 17884–17890 (2011).
Yang, C., Zhao, H. B., Hou, Y. L. & Ma, D. Fe5C2 nanoparticles: a facile bromide-induced synthesis and as an active phase for Fischer-Tropsch synthesis. J. Am. Chem. Soc. 134, 15814–15821 (2012).
de Smit, E. et al. Stability and reactivity of ε-χ-θ iron carbide catalyst phases in Fischer-Tropsch synthesis: controlling μ C. J. Am. Chem. Soc. 132, 14928–14941 (2010).
Saw, E. et al. Bimetallic Ni–Cu catalyst supported on CeO2 for high-temperature water–gas shift reaction: methane suppression via enhanced CO adsorption. J. Catal. 314, 32–46 (2014).
Nie, C. et al. Effects of Ce addition on Fe–Cu catalyst for Fischer–Tropsch synthesis. Catal. Lett. 149, 1375–1382 (2019).
Ding, M. Y. et al. Study on reduction and carburization behaviors of iron phases for iron-based Fischer–Tropsch synthesis catalyst. Appl. Energy 160, 982–989 (2015).
Qin, Z. H., Lewandowski, M., Sun, Y. N., Shaikhutdinov, S. & Freund, H. Encapsulation of Pt nanoparticles as a result of strong metal-support interaction with Fe3O4 (111). J. Phys. Chem. C 112, 10209–10213 (2008).
Wang, P. et al. Synthesis of stable and low-CO2 selective ε-iron carbide Fischer-Tropsch catalysts. Sci. Adv. 4, https://doi.org/10.1126/sciadv.aau2947 (2018).
Xie, J. et al. Size and promoter effects in supported iron Fischer–Tropsch catalysts: insights from experiment and theory. ACS Catal. 6, 3147–3157 (2016).
Xie, J. et al. Size and promoter effects on stability of carbon-nanofiber-supported iron-based fischer–tropsch catalysts. ACS Catal. 6, 4017–4024 (2016).
van Dijk, H. A. J., Hoebink, J. & Schouten, J. C. A mechanistic study of the Fischer–Tropsch synthesis using transient isotopic tracing. Part 2: model quantification. Top. Catal. 26, 163–171 (2003).
Johnson, G. R., Werner, S. & Bell, A. T. An investigation into the effects of Mn promotion on the activity and selectivity of Co/SiO2 for Fischer–Tropsch synthesis: evidence for enhanced CO adsorption and dissociation. ACS Catal. 5, 100–114 (2015).
Xiang, Y. Z., Barbosa, R. & Kruse, N. Higher alcohols through CO hydrogenation over CoCu catalysts: influence of precursor activation. ACS Catal. 4, 2792–2800 (2014).
Xiang, Y. Z., Barbosa, R., Li, X. & Kruse, N. Ternary cobalt–copper–niobium catalysts for the selective CO hydrogenation to higher alcohols. ACS Catal. 5, 2929–2934 (2015).
Xiang, Y. Z. et al. Long-chain terminal alcohols through catalytic CO hydrogenation. J. Am. Chem. Soc. 135, 7114–7117 (2013).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC: 21871021, 21521005, 21725301, 21932002, 21821004, and 91645115), the National Key Research and Development Program (2017YFA0206804 and 2017YFB0602200), and the Fundamental Research Funds for the Central Universities (XK1802-6 and buctylkxj01). We acknowledge the Beijing Synchrotron Radiation Facility (BSRF) for providing the experimental resources and materials of the XAS experiments.
Author information
Authors and Affiliations
Contributions
Y.W.L., M.W., and D.M. designed the experiments. Y.W.L. conducted material synthesis and carried out the catalytic performance test. Y.W.L. and M.P. conducted the X-ray absorption fine structure spectroscopy measurements and analyzed the data. X.L. and B.S.Z. contributed to the scanning transmission electron microscopy. W.G., J.B.Z., J.L.S., and Y.X. performed some of the experiments. S.H. contributed to the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy. X.W.L. conducted the Mössbauer spectrum measurements. This paper was primarily written by Y.W.L. All authors contributed to discussions. M.W., B.S.Z., and D.M. conceived the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Gao, W., Peng, M. et al. Interfacial Fe5C2-Cu catalysts toward low-pressure syngas conversion to long-chain alcohols. Nat Commun 11, 61 (2020). https://doi.org/10.1038/s41467-019-13691-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13691-4
This article is cited by
-
Stabilized ε-Fe2C catalyst with Mn tuning to suppress C1 byproduct selectivity for high-temperature olefin synthesis
Nature Communications (2024)
-
Upgrading CO2 to sustainable aromatics via perovskite-mediated tandem catalysis
Nature Communications (2024)
-
Active learning streamlines development of high performance catalysts for higher alcohol synthesis
Nature Communications (2024)
-
Ruthenium-cobalt single atom alloy for CO photo-hydrogenation to liquid fuels at ambient pressures
Nature Communications (2023)
-
Enabling direct-growth route for highly efficient ethanol upgrading to long-chain alcohols in aqueous phase
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
