
Abstract
Identification of genetic biomarkers associated with autism spectrum disorders (ASDs) could improve recurrence prediction for families with a child with ASD. Here, we describe clinical microarray findings for 253 longitudinally phenotyped ASD families from the Baby Siblings Research Consortium (BSRC), encompassing 288 infant siblings. By age 3, 103 siblings (35.8%) were diagnosed with ASD and 54 (18.8%) were developing atypically. Thirteen siblings have copy number variants (CNVs) involving ASD-relevant genes: 6 with ASD, 5 atypically developing, and 2 typically developing. Within these families, an ASD-related CNV in a sibling has a positive predictive value (PPV) for ASD or atypical development of 0.83; the Simons Simplex Collection of ASD families shows similar PPVs. Polygenic risk analyses suggest that common genetic variants may also contribute to ASD. CNV findings would have been pre-symptomatically predictive of ASD or atypical development in 11 (7%) of the 157 BSRC siblings who were eventually diagnosed clinically.
Similar content being viewed by others


Introduction
Behavioral assessments remain the gold standard for autism spectrum disorder (ASD) diagnosis1, and prospective analysis permits objective and longitudinal assessment for the earliest symptoms. Published estimates of sibling recurrence for ASD range from 6.9 to 19.5%2,3,4,5. Moreover, of younger siblings of autistic probands, herein referred to simply as “infant siblings”, who are not diagnosed with ASD, up to 30–40% have subclinical ASD traits and/or suboptimal developmental functioning6.
ASD and related subclinical traits show familial clustering, with a substantial portion of familial liability attributed to genetic factors7,8. Subclinical symptoms in first- and second-degree relatives support an important role for genetic factors in producing an autistic phenotype9. The genetic architecture of ASD is being resolved by studying families with different characteristics10,11,12,13,14,15,16,17,18,19,20,21, and dozens of copy number variant (CNV) loci and ASD-relevant genes and loci are known, many of which overlap those associated with other neurodevelopmental disorders14,17,18. De novo and inherited rare (<1% in a population) CNVs and other pathogenic variants are found in ~ 5–40% of individuals with ASD, depending on the cohort examined10,14,17,20. Chromosomal microarray to detect CNVs is the first-tier laboratory test for clinical genetic evaluation following an ASD diagnosis22.
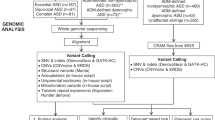
The most effective way to reduce symptoms of ASD is with early intervention, targeting behavior, and skills development23. In search of biomarkers for early indentification, we investigated whether CNVs affecting ASD-related loci correlate (pre- and post-symptomatically) with phenotypic outcomes in the Baby Siblings Research Consortium (BSRC) cohort of infant siblings whose family history is associated with a higher probability of developing ASD (Fig. 1). We analyze CNVs from 253 families registered in the BSRC24 while blinded to the infant siblings’ phenotype status. At enrollment, each family included a proband diagnosed with ASD, and at least 1 younger sibling (Supplementary Table 1). The BSRC longitudinal phenotyping design enables a predictive study of CNVs in this infant cohort (see Methods). Our analyses reveal that the detection of ASD-relevant CNVs is indeed predictive of ASD or atypical development in this sibling population and can be used to inform risk estimates for individuals and their families, with potential impact on their therapeutic trajectory.

Project flowchart. Families consisting of a proband and at least 1 infant sibling were recruited through the Baby Siblings Research Consortium. The proband was the first in the family to receive an ASD diagnosis. Psychometric data were collected for all siblings at ~36 months of age, at which point an ASD diagnosis was made if the child met clinical criteria (see Methods). All children were also assessed for ASD, cognitive and adaptive behavioral functioning at least once prior to the 36-month time point. We genotyped individuals from 253 families on the Affymetrix CytoScanTM HD Array and whole-genome sequenced 91 of these families using established pipelines12,13,14. Copy number variants determined to be ASD-relevant were confirmed with secondary methods. We scrutinized the phenotypes of high-risk infant siblings carrying these ASD-relevant CNVs to determine whether they (i) had ASD, (ii) were atypically developing, or (iii) were neurotypical/non-ASD. There were also 34 siblings who did not meet criteria for formal enrollment but for whom phenotype and microarray data were available. Following the general BSRC strategy, a separate CNV analysis was conducted on 2124 probands and 2423 non-ASD sibs from 2110 ASD-affected families part of the Simons Simplex Collection. Fourteen probands were monozygotic twins. Of the unaffected sibs, 288 were atypically developing. This analysis was performed to further assess the predictive value of chromosomal microarray. ASD autism spectrum disorder, CNV copy number variation.
Results
Clinical evaluation of infant siblings within families
The study group included the 253 probands, 288 later-born infant siblings comprising the primary diagnostic sample (Fig. 1), and 34 siblings who did not meet cohort enrollment criteria (i.e., >3 years, or status determined only through parental reports), but for whom we had phenotype designation and genotype data. Refer to “Subject recruitment and clinical evaluations” in Methods section for criteria for various atypical outcomes listed below. At age 3, 103/288 siblings (35.8%) in 94/253 families (37.2%) had a formal diagnosis of ASD; 54 were “atypically developing” (18.8%), and 131 were developing typically (45.5%) (Supplementary Table 2). Among the 54 infant siblings with atypical development (but not an ASD diagnosis) were the following phenotypic constellations: ASD symptoms (n = 5), ASD symptoms with developmental, language and/or adaptive behavior delay (n = 9); developmental delay (n = 15), language delay (n = 10), language and adaptive behavior delays (n = 5), deficits in adaptive behavior alone (n = 3), and attention deficit hyperactivity disorder (ADHD) symptoms or externalizing behaviors (n = 7). Of the 30 families with 2 or more infant siblings, 23 had 1 or more assessed as having ASD and/or atypical development and 7 had all sibs typically developing at age 3. The male:female ratio was 3.7:1 for ASD-affected infant siblings, 1.6:1 for atypically developing siblings, and 1.2:1 for non-ASD infant siblings.
Genomic findings in probands and infant siblings
From 253 probands, we identified 15 CNVs (in 13 individuals; 5.1%) that were deemed ASD-relevant (see Methods; Supplementary Tables 3 and 4). Where inheritance could be ascertained, 6 CNVs (in 5 probands) were de novo (3 deletions, 3 duplications) and 8 CNVs (5 deletions, 3 duplications) were inherited (7 maternally, 1 paternally). For proband 14-0152-001, inheritance was unknown, as parental samples were unavailable. All 8 inherited variants were shared with an infant sibling (Fig. 2), of whom 4 were diagnosed with ASD (Fig. 3a) and 3 showed atypical development without a formal ASD diagnosis (Fig. 3b).

ASD-relevant CNVs among infant siblings (n = 288). Summary of ASD-relevant genetic findings in the infant sibling cohort, stratified by family segregation and infant sibling phenotype. ASD autism spectrum disorder, CNV copy number variation.

Pedigrees demonstrating ASD-relevant CNVs in infant siblings. a Infant siblings with ASD who shared a CNV with a related index case (arrow). Targeted testing (only) for a pathogenic CNV was performed on sibling 12-8257-005. b Atypically developing infant siblings who shared a CNV with a related index case. Female sibling 4-0062-004 was positive for ASD on the ADOS (24 and 36 months) assessments, but subthreshold on the ADI-R. Clinical impression was that the child did not have ASD. She had subthreshold scores on the MSEL Early Learning Composite (ELC) (SS = 76) and VABS Adaptive Behavior Composite (ABC) (SS = 84). In family 1-0616, the second-born son (1-0616-004) displayed subthreshold ASD symptoms, vulnerabilities in language and verbal reasoning, gross and fine-motor skill delay; the third-born son (1-616-005) (not in the official cohort) experienced fine-motor delay as seen on the VABS at 24 months, and parents reported concerns regarding socialization. In family 4-0027, the female sibling displayed behavioral rigidity and transition difficulties. The male sibling in family 12-8115 was developing typically. c Infant siblings with ASD who did not share a CNV with a related index case. d Infant siblings with a CNV not shared with a related index case. Individual 4-0061-004 had language delay and cognitive regression (ELC = 56). Male sibling 12-4453-005 scored just above threshold on the ADOS (CSS = 5), but the clinical history was not consistent with a diagnosis of ASD. His ELC (SS = 88), and ABC (SS = 90) scores were within 1 standard deviation of the expected mean. The scores of male 14-0376-001 on the ADOS (CSS = 1), MSEL (ELC = 91) and the VABS (ABC = 105) reflected a typical developmental trajectory. Figure includes 5 additional non-infant siblings who were not counted as part of the formal cohort (dotted outline). CNV classification is provided in Supplementary Table 4. ASD autism spectrum disorder, CNV copy number variation, ADOS Autism Diagnostic Observation Schedule, CSS calibrated severity score, ADI-R Autism Diagnostic Interview-Revised, MSEL Mullen Scales of Early Learning, VABS Vineland Adaptive Behavior Scales, SS standard score.
Among the 288 infant siblings of the probands, 13 carried ASD-relevant CNVs (Fig. 2), of which 6 were considered pathogenic and 7 were clinically defined as variants of unknown significance overlapping genes implicated in ASD. Five infant siblings had ASD-relevant CNVs that were not shared with the related probands; 2 had ASD, 2 were atypically developing, and 1 was typically developing (Fig. 3c, d). Of the 2 typically developing children with ASD-relevant CNVs, 14-0376-004 (Fig. 3d) had a 610 kb deletion at 16p11.2 and 12-8115-004 (Fig. 3b) had a (shared) 1.7 Mb duplication at 16p13.11. Their evaluations at age 36 months did not indicate any developmental delays or ASD symptoms. Both variants are associated with recurrent and variably expressed syndromes, often involving ASD12,25,26.
Overall, among siblings of ASD probands, we found ASD-related CNVs in 6 of 103 with ASD at age 3 years, in 5 of 54 with atypical development, and in 2 of 131 with neurotypical development. Four children (Fig. 3) who did not meet study inclusion criteria had ASD-related CNVs deemed to be pathogenic. Using only CNVs considered to be pathogenic or likely pathogenic18,27, among infant siblings the positive predictive values (PPVs) of these variants were 0.50 for ASD and 0.83 for combined ASD/atypical development. Other predictive statistics are shown in Table 1.
Whole-genome sequences (WGS) were available from 91 families of our cohort. We identified no additional ASD-relevant CNVs, and of the potentially interesting sequence-level variants (Supplementary Table 5), none were found in families with previously recognized ASD-relevant CNVs (Supplementary Fig. 1).
This study adds to growing evidence that specific biomarkers might contribute to pre-symptomatic detection of infants likely to develop ASD or other developmental disorders, at least from sibships that include an ASD proband. Eleven of 157 siblings who developed ASD or were atypically developing at age 3 carried a CNV worthy of clinical follow-up (either inherited or de novo), 5 (3.2%) carried a pathogenic/likely pathogenic CNV and 6 (3.8%) carried a variant of unknown significance (VUS).
Of the 7 total VUSs overlapping a gene associated with a neuropsychiatric disorder, 6 were in siblings with ASD or atypical development, with none in neurotypical children, despite nearly equal numbers in each subgroup. Although not reported as pathogenic in a clinical setting, VUSs involving ASD-relevant genes may nonetheless be contributing factors for ASD or atypical development, and of interest to families14,22. Two of 131 typically developing siblings had CNVs considered as VUS or likely pathogenic. These CNVs at 16p11.2 or 16p13.11 have frequencies in the general population that range from 0.03 to 0.04% and 0.15 to 0.25%28,29,30 and have been associated with reduced cognitive and general functioning28,31 and adult-onset phenotypes32, respectively.
To further assess the predictive impact of ASD-relevant CNVs in a larger cohort, we analyzed published data from 2110 families from the Simons Simplex Collection (SSC). This cohort differs from the BRSC in being a quartet design with 4214 parents, 2124 ASD-affected probands and 2423 ASD-unaffected children, assembled to study de novo variants (single-point phenotyping; average age of diagnosis 8.9 (±3.5) years for males and 9.11 (±3.7) for females; 6.6:1 male to female ratio for probands)33. Of the SSC “unaffected” sibs, applying similar criteria as for the BSRC cohort revealed 288 (11.9%) with atypical behavioral and developmental profiles: 33 were suspected to have elevated ASD traits (Social Responsiveness Scale-Parent Report Total T-Score > 6034); 139 had emotional and behavioral problems (Child Behavioral Checklist-Parent Report Total Problems T-Score > 6035); 65 displayed mild-moderate adaptive behavior deficits (Vineland Adaptive Behavior Scales, Adaptive Behavior Composite36 < 85); 51 met criteria in more than 1 category. While blinded to proband and sibling status, we searched for ASD-relevant CNVs with the same classification criteria used for the BSRC (see Methods).
We found 118 ASD-relevant CNVs in 116 of 2124 probands (5.5%; 72.0% (85) pathogenic/likely pathogenic and 28.0% (33) VUS) and 64 CNVs in 63 of 2423 unaffected sibs (2.6%; 34.4% (22) pathogenic/likely pathogenic and 65.6% (42) VUS) (Supplementary Data File 1 and Supplementary Table 6). These results are similar to those found in probands and unaffected sibs in the BSRC cohort (5.1% and 3.8%, respectively). Seven (11.1%) of these sibs with ASD-relevant CNVs were deemed atypically developing. When considering pathogenic and likely pathogenic variants across the SSC, the PPV for ASD was 0.79, and for ASD or atypical development was 0.83 (Supplementary Table 7). From the SSC cohort, we identified 47 CNVs impacting 8 of the genes/loci harboring ASD-relevant CNVs in the siblings of the BSRC. Thirty of these CNVs were carried by probands, 2 by atypically developing sibs and 15 by unaffected sibs (Supplementary Table 6). The percentage of ASD-relevant CNVs in unaffected sibs in the SSC is similar to that observed in unaffected sibs in the BSRC. Furthermore, data analyzed from both cohorts yielded lower percentages of ASD-relevant variants in non-ASD sibs compared with probands, while noting that select variants are found in symptomatic carriers in the general population.
Polygenic transmission disequilibrium test
Recognizing emerging evidence for the role of combinations of common genetic variants in ASD susceptibility13 and their potential efficacy in predicting ASD risk, we determined the contribution of polygenic risk scores (PRS) to the phenotype in the BSRC cohort using the polygenic transmission disequilibrium test13. In families for which genotype data were available for both parents, probands and at least 1 infant sibling, we observed a statistically significant over-transmission of risk variants from parents to probands (n = 189, mean difference (cohort z-score based) = 0.13, p = 0.01). There were non-significant differences of risk transmission for unaffected siblings (n = 112, mean = −0.003, p = 0.97), atypically developing siblings (n = 44, mean = −0.004, p = 0.97) and ASD-affected siblings (n = 93, mean = 0.07, p = 0.27) (Supplementary Fig. 2). Power to reject the null hypothesis (assuming the PRS explained 2.45% of phenotypic variance37) was ~98% for the proband (n = 189) and 56% for the affected sibling (n = 93). The significant over-transmission of risk from parents to probands suggested that common genetic variants may contribute to the phenotype in the BSRC sample. This PRS analysis in the SSC revealed a similar statistical trend. Using a paired Student’s t-test, we observed a significant difference in risk of transmission for SSC probands (n = 2118, mean = 0.547, p < 1e-10) and a non-significant risk of transmission for unaffected sibs (n = 2130, mean = −0.021, p = 0.116) and atypically developing sibs (n = 286, mean = −0.012, p = 0.744).
Discussion
Early identification of the CNVs described in this study could be used to tailor recurrence risk estimates to individual families, with potential for intensified surveillance for infants at increased likelihood due to positive CNV findings. There is evidence that infants as young as 7 months with subtle features of ASD could benefit from tailored interventions23,38,39, but there are also also recent negative trial data40,41.
This study had possible limitations. First, given the 37% recurrence of ASD in our BSRC families, compared with 6.9–19.5% found previously2,3,4,5, this cohort had selective over-recruitment of infant siblings with ASD. A similar over-recruitment was noted for atypically developing sibs, which partially accounts for the difference in rate between cohorts (29.2% in BSRC compared to 11.9% in SSC). This oversampling, not uncommon in genetic studies, may bias estimates of positive and negative predictive value of biomarkers. As well, PPV estimates related to CNV detection must be considered relative to overall ASD rates among participants. Further impacting the predictive value statistics, we only considered diagnoses made by 3 years of age. Despite the stability of an ASD diagnosis at 36 months, clinical phenotypes are known to be fluid throughout development and apparent non-ASD siblings might later demonstrate ASD or another disorder as they age42,43. Likewise, longitudinal studies following high-probability siblings from 3 years of age to middle childhood observed that an ASD diagnosis was revoked in 5.6–20% of children44,45. Second, considering the massive effort in phenotyping, the BSRC sample would be considered comparably large, but participants with ASD-relevant CNVs were still few, limiting power of the analysis. The primary BSRC study involved a cohort with increased probability of ASD, and while the findings are generally consistent with those from the SSC, other family structures may yield different findings.
The recurrent CNVs described here often display variable expressivity and reduced penetrance for ASD, particularly when sex is considered46. For these reasons, we explored inclusion of siblings with atypical forms of ASD when determining PPV. We attribute the low sensitivity (0.03 in BSRC) of these markers to the vast heterogeneity in ASD etiology, but this parameter may improve with clearer genotype-phenotype correlations17,47. We can also consider coupling these already useful CNV indicators of ASD with metrics captured by other genetic approaches such as WGS and PRS discussed here, as well as other techniques, such as brain imaging48 and other early phenotypic assessment49.
Families must receive adequate counseling when such variants are found, at any stage of life, including the prenatal setting, so that they fully understand the potential ramifications of these findings. This sibling cohort also provided evidence for certain CNVs as potential early biological predictors of ASD or other developmental difficulties, whether or not they shared these with their respective probands (Fig. 2), which will also influence interpretation.
Methods
Subject recruitment and clinical evaluations
From 9 research sites registered with the BSRC (The Center for Applied Genomics, Genetics, and Genome Biology, The Hospital for Sick Children; Autism Research Center, Bloorview Research Institute; Autism Research Center, IWK Health Center and Dalhousie University; Department of Psychology, UC San Diego; Center for Autism and Related Disorders, Kennedy Krieger Institute; Department of Psychology, University of Miami; MIND Institute, Department of Psychiatry, UC Davis; Department of Psychology, University of Washington; Vanderbilt Kennedy Center Treatment and Research Institute for Autism Spectrum Disorders, Vanderbilt Kennedy Center; Autism Research Center, University of Alberta), participants from 253 families enrolled (134 from USA sites and 119 from Canada). Recruitment was through organizations serving individuals with ASD and their families, referrals from medical professionals, web-based media, or word-of-mouth, as previously described2. This study was approved by the Research Ethics Board (REB) at The Hospital for Sick Children (REB # 0019980189) and informed consent was obtained from participants or their legal guardians, when appropriate. A total of 253 probands, 322 siblings (including 288 infant siblings), and 447 parents (242 mothers; 205 fathers) constituted the final cohort and were analyzed.
All families had at least 1 child (i.e., the proband) diagnosed with ASD according to the Diagnostic and Statistical Manual for Mental Disorders, 4th Edition (DSM-IV). We excluded probands with a genetic syndrome that could account for their ASD (e.g., Fragile-X, Rett Syndrome)50. Younger siblings were recruited at a mean age of 10.2 ± 8.4 months; we followed them, and determined their clinical outcomes with respect to ASD at a mean age of 37.4 ± 2.3 months. An expert clinician determined diagnostic status by clinical best-estimate, informed by the Autism Diagnostic Observation Schedule (ADOS) (calibrated severity score (CSS) ≥ 4 as threshold)51, DSM-IV criteria, psychometric assessment of language and cognitive development (Mullen Scales of Early Learning (MSEL)52), adaptive functioning (Vineland Adaptive Behavioral Scales (VABS)36), and overall clinical impression.
Adapting criteria from published reports of this cohort6, from among infant siblings not diagnosed with ASD, we defined another subgroup as “atypically developing”. This classification was based on the presence of elevated ASD symptoms (CSS ≥ 3, requisite scores on the Autism Diagnostic Index-Revised and clinical impression), developmental delay (MSEL composite score > 1 SD below the mean), language delay (MSEL expressive and/or receptive language subscale score > 1 SD below the mean), deficits in adaptive behavior (VABS composite score or subscale score > 1 SD below the mean) or other atypical behavior patterns (e.g., externalizing behaviors) or disorders (e.g., ADHD), as indicated by the clinician best-estimate diagnoses and/or scores above cutoff.
From 253 families, 144 mothers and 127 fathers self-reported the Broader Autism Phenotype Questionnaire (BAPQ) (Supplementary Table 8). The BAPQ includes items mapping onto 3 broader scales (Aloof, Rigid, and Pragmatic Language), which comprise ASD-related subclinical traits previously reported in a subset of parents. A cutoff of 3.15 was shown to optimize agreement with expert clinical assessment of the Broader Autism Phenotype (BAP)53.
Baby Siblings Research Consortium (BSRC) sample collection
For microarray analysis, biological samples were obtained from 251 probands, 321 siblings and 444 parents (241 mothers; 203 fathers) at their respective recruiting sites (total samples = 1016). Blood samples from U.S. sites (n = 517) were submitted to the DNA and Cell Repository at Rutgers University, and extracted DNA was subsequently sent to The Center for Applied Genomics (TCAG). Genomic DNA used for genotyping on microarray was extracted from whole blood (86.0%; 874/1016 samples), saliva (0.4%; 4/1016), lymphoblastoid cell lines (10.4%;106/1016), or source undocumented (3.1%; 32/1016).
Microarray genotyping quality control metrics
Genomic DNA samples were processed on the high-density Affymetrix CytoScanTM HD microarray platform at TCAG (2.67 million copy number markers) following protocols in our other published studies10,17,18,54. Quality control thresholds were imposed, as specified by the manufacturer: Waviness standard deviation (Waviness SD) ≤ 0.12; median absolute pairwise difference (MAPD) ≤ 0.25; and single-nucleotide polymorphism (SNP) quality control (SNP QC) ≥ 15.0. One father’s sample did not meet these criteria, but the sample was retained in the analysis to help identify CNV segregation. We used PLINK55 software to identify loss of heterozygosity (LOH) and Mendelian inconsistencies using the 750,000 informative SNPs available on the array. We observed Mendelian inconsistencies in 3 families, and eliminated them from the study.
Variant detection and characterization
We detected CNVs using 4 algorithms: Chromosome Analysis Suite (ChAS) (Affymetrix Inc., USA), iPattern56, Nexus57, and Partek58. We sequentially applied a series of data constraints to ascertain high-confidence, rare CNVs27. We considered only stringent CNVs, called by ≥2 algorithms, at least 1 of which was ChAS or iPattern, for downstream analyses. CNVs on the X chromosome were called uniquely by ChAS and iPattern. We eliminated all calls on the Y chromosome. We retained CNVs ≥ 15 kb in length and overlapping ≥10 consecutive probes to reduce the detection of false-positive calls. They were then restricted to those in which ≤70% spanned a segmental duplication and ≥75% of the variant was present in a copy number stable region as previously defined59. We used a platform-matched control dataset consisting of 873 individuals with no reported psychiatric history, from the Ontario Populations Genomics Platform (OPGP)60, to classify CNVs as rare. We considered a CNV as rare if it did not exceed 50% reciprocal overlap with a CNV found in <0.1% of the control dataset. To corroborate a CNV’s status as rare, we compared to an additional 4 unrelated control populations totaling 9978 individuals: the Collaborative Genetic Study of Nicotine Dependence (COGEND)61 and KORA62, genotyped on the Illumina Omni 2.5 M; the SAGE consortium controls63, Ontario Colorectal Cancer case-control study cohort64,65 and the Health, Aging, and Body Composition (Health ABC) Study66, genotyped on the Illumina 1 M; the Ottawa Heart Institute controls67, and POPGEN68, both genotyped on the Affymetrix 6.0 microarray. We sequenced whole genomes of 84 probands, 118 infant siblings and 158 parents (86 mothers; 72 fathers), from 91 families, using DNA from whole blood17.
We used criteria adapted from the American College of Medical Genetics classification69 and an established annotation strategy14,18,70 to classify CNVs as ASD-relevant and pathogenic, likely pathogenic or variant of unknown significance (VUS). A CNV was considered pathogenic or likely pathogenic if (a) it was associated with an established genomic disorder of which ASD is a characteristic (e.g., 16p11.2 microdeletion), or (b) it overlapped a coding exon of a high-confidence ASD-susceptibility gene (e.g., SHANK3; Supplementary Data File 3). We considered whether the CNV overlapping the gene was de novo (pathogenic) or inherited (likely pathogenic). Variants overlapping exons of long noncoding RNA PTCHD1-AS71, and specific noncoding exons of the MBD5 gene72, which constitute the critical region of 2q23.1 microdeletion syndrome, were retained in the analysis. We further defined a class of VUS as ASD-relevant if they overlapped exons of candidate ASD-susceptibility genes or related neuropsychiatric disorder genes (Supplementary Data File 3) and had a frequency ~0.1% in the Database of Genomic Variants. We assessed the efficacy of the variants so identified, as markers for ASD or atypical phenotype status, using the epiR package in R73 to calculate positive predictive value, negative predictive value, sensitivity and specificity. All CNVs in infant siblings were classified while blinded to the phenotype status of the individual.
Whole-genome sequence data were processed as previously described17. We defined rare loss of function and de novo damaging missense variants as in Yuen et al.10. We prioritized single-nucleotide variants and indels that overlapped genes associated with ASD and other related neurodevelopment disorders, and considered whether variants with similar transcriptional consequences were found in other ASD cases.
Molecular validation and characterization of CNVs
The presence of de novo and ASD-relevant CNVs was confirmed via real-time quantitative PCR (qPCR) using the TaqMan© Copy Number Assay and SYBR® Green methods; all experiments were conducted in triplicate. For SYBR® Green assays, an amplicon 90–140 base pairs in length was amplified using 2 sets of primers positioned ≥ 500 bp from both reported breakpoints. A similar amplicon designed within the FOXP2 locus served as a 2-copy control74. All TaqMan© Copy Number Assays involved predesigned probes located in the gene of interest and RNaseP, which served as an endogenous control. All experiments included both male and female control samples (HapMap samples: NA10851 (male) and NA15510 (female)).
Simons Simplex Collection (SSC) to assess CNV false discovery
In order to assess the possible false discovery for CNVs associated with ASD, we performed a separate CNV analysis on 2110 families from the SSC33. This included 2107 mothers, 2107 fathers, 2124 ASD probands, and 2,425 siblings. Families were recruited as previously described (Fischbach and Lord, 2010). Of the 2425 sibs, 2093 are designated unaffected. Of the remaining 332 siblings, 2 have ASD and have thus been excluded from the analysis, leaving 2423 sibs unaffected by ASD available for CNV analysis.
As we did for unaffected sibs in the BSRC cohort of infant siblings, we looked for atypical developmental outcomes in these non-ASD sibs using the psychometric tools available. We considered the potential presence of ASD traits using the Social Responsiveness Scale (SRS)-Parent Report (n = 2298)34; mild-moderate developmental delay by identifying adaptive behavior deficits using the VABS (n = 2368) and other emotional and behavioral concerns as identified through the Child Behavioral Checklist (CBCL)-Parent Report35 (n = 448 for ages 1.5–5 test; n = 1833 for ages 6–18 test). To define atypical development, we applied a cutoff of >1 SD below the mean for the total scores for the SRS and the CBCL, expressed as T-scores (i.e., T-scores > 60), and the Adaptive Behavior Composite from the VABS, expressed as a standard score (i.e., scores < 85). Sibs had to meet this cutoff on at least 1 of the 3 tests to qualify as atypically developing.
We received microarray intensity data in the form of .IDAT files for 8761 individuals genotyped on 3 different microarray platforms: 1246 on the Illumina Human1Mv1; 3826 on the Illumina Human1M-Duov3; and 3689 on the Illumina HumanOmni2.5–4v1. We called CNVs using the same pipeline as for the infant sibling cohort, with the exception that the following 3 algorithms were employed, as previously described:18 PennCNV75, QuantiSNP76, and iPattern. Stringent CNVs were called by a minimum of 2 of these algorithms, with at least one being iPattern. We applied criteria for filtering and prioritizing variants as described above. CNVs were not molecularly characterized, as no DNA for this cohort was available.
Polygenic transmission disequilibrium test
We analyzed the contribution of common genetic variants to ASD risk for families from the infant sibling cohort for which microarray data were available for the proband (n = 189), at least 1 infant sib (n = 193) and both parents. We generated PRS using PRSice77 (clump-kb 250, clump-p 1.000000, clump-r2 0.100000, info-base 0.9), using a p-value threshold of 0.1 as suggested by Grove et al.78 in Supplemental Fig. 4.4.1. In total, 246,607 SNPs were included in both the SNP genotypes from the Affymetrix CytoScanTM HD array and the iPSYCH-PGC_ASD_Nov2017 GWAS summary statistic file, with 9,112,386 variants, of which 17,362 were removed due to having an INFO score less than 0.90. For the study genotypes, we began with 749,157 SNPs from the Affymetrix CytoScanTM HD array, removing 83,849 SNPs with a call rate >90% and 97,770 SNPs with minor allele frequency < 5%. Using PRSice, 87,215 genotype array variants were removed (e.g., A- > T), with 228,151 intersecting the ASD GWAS variants. A total of 52,923 SNPs remained after linkage disequlibrium-based clumping, of which 11,401 met the specified p-value threshold, and were thus used for PRS computation.
We performed the same analysis for 2104 families from the Simons Simplex Collection. All families consisted of 2x parents (n = 2104 mothers; n = 2104 fathers), a proband (n = 2118) and at least 1 unaffected sibling (n = 2416). Using identical criteria for inclusion of SNPs, 53,803 SNPs remained after linkage disequilibrium-based clumping, and 11,161 SNPs were enriched for in cases versus controls as per a p-value of 0.1, and therefore used for the generation of PRS.
Previous estimates for narrow-sense heritability (h2g) for ASD are varied, depending on phenotype definition, study design, genotyping method, and other factors. Estimates include 12%37, 52%79, and 83%80; we used h2g = 60% in the power calculation. Power was calculated under the liability threshold model assuming prevalence of ASD of 1.5%81.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All raw microarray data for 1016 individuals that were genotyped on the Affymetrix CytoScanTM HD Array were submitted to dbGaP and can be accessed via a data access committee using dbGaP accession number phs001876.v1.p1. Whole-genome sequence data are accessible through the Autism Speaks MSSNG database (https://research.mss.ng).
References
Falkmer, T., Anderson, K., Falkmer, M. & Horlin, C. Diagnostic procedures in autism spectrum disorders: a systematic literature review. Eur. Child Adolesc. Psychiatry 22, 329–340 (2013).
Ozonoff, S. et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128, e488–e495 (2011).
Grønborg, T. K., Schendel, D. E. & Parner, E. T. Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: a population-based cohort study. JAMA Pediatr. 167, 947–953 (2013).
Risch, N. et al. Familial recurrence of autism spectrum disorder: evaluating genetic and environmental contributions. Am. J. Psychiatry 171, 1206–1213 (2014).
Messinger, D. S. et al. Early sex differences are not autism-specific: A Baby Siblings Research Consortium (BSRC) study. Mol. Autism 6, 32 (2015).
Charman, T. et al. Non-ASD outcomes at 36 months in siblings at familial risk for autism spectrum disorder (ASD): a baby siblings research consortium (BSRC) study. Autism Res. J. Int. Soc. Autism Res. 10, 169–178 (2017).
Colvert, E. et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72, 415–423 (2015).
Bai, D. et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry (2019). https://doi.org/10.1001/jamapsychiatry.2019.1411.
Sandin, S. et al. The familial risk of autism. JAMA 311, 1770–1777 (2014).
Yuen, R. K. C. et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191 (2015).
Ye, K. et al. Measuring shared variants in cohorts of discordant siblings with applications to autism. Proc. Natl Acad. Sci. U.S.A. 114, 7073–7076 (2017).
Weiss, L. A. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675 (2008).
Weiner, D. J. et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017).
Tammimies, K. et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903 (2015).
Sebat, J. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007).
Sanders, S. J. et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011).
C Yuen, R. K. et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611 (2017).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694 (2014).
Marshall, C. R. et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488 (2008).
Levy, D. et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897 (2011).
Leppa, V. M. et al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet. 99, 540–554 (2016).
Miller, D. T. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86, 749–764 (2010).
Zwaigenbaum, L. et al. Early intervention for children with autism spectrum disorder under 3 years of age: recommendations for practice and research. Pediatrics 136(Suppl 1), S60–S81 (2015).
Messinger, D. et al. Beyond autism: a baby siblings research consortium study of high-risk children at three years of age. J. Am. Acad. Child Adolesc. Psychiatry 52, 300–308.e1 (2013).
Ramalingam, A. et al. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J. Hum. Genet. 56, 541–544 (2011).
Hanson, E. et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry 77, 785–793 (2015).
Marshall, C. R. & Scherer, S. W. Detection and characterization of copy number variation in autism spectrum disorder. Methods Mol. Biol. Clifton NJ 838, 115–135 (2012).
Stefansson, H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014).
Männik, K. et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA 313, 2044–2054 (2015).
Kendall, K. M. et al. Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol. Psychiatry 82, 103–110 (2017).
Kendall, K. M. et al. Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br. J. Psychiatry J. Ment. Sci. 214, 297–304 (2019).
Ingason, A. et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol. Psychiatry 16, 17–25 (2011).
Fischbach, G. D. & Lord, C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195 (2010).
Constantino, J. N. & Gruber, C. P. Social Responsiveness Scale: Manual. Los Angeles, CA: Western Psychological Services (2005).
Achenback, T. M. in The Use of Psychological Testing for Treatment Planning and Outcomes Assessment. (ed. Maruish, M. E.) 429–466 (Lawrence Erlbaum Associates Publishers, 1999).
Sparrow, S. S. et al. Vineland adaptive behavior scales. Circle Pines, MN: American Guidance Service Inc (1984).
Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Dawson, G. et al. Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125, e17–e23 (2010).
Rogers, S. J. et al. Autism treatment in the first year of life: a pilot study of infant start, a parent-implemented intervention for symptomatic infants. J. Autism Dev. Disord. 44, 2981–2995 (2014).
Kasari, C. Time to rethink pre-emptive interventions for infants with early signs of autism spectrum disorder. Lancet Child Adolesc. Health (2019). https://doi.org/10.1016/S2352-4642(19)30234-2.
Whitehouse, A. J. O. et al. Pre-emptive intervention versus treatment as usual for infants showing early behavioural risk signs of autism spectrum disorder: a single-blind, randomised controlled trial. Lancet Child Adolesc. Health (2019). https://doi.org/10.1016/S2352-4642(19)30184-1.
Ozonoff, S. et al. Diagnosis of autism spectrum disorder after age 5 in children evaluated longitudinally since infancy. J. Am. Acad. Child Adolesc. Psychiatry 57, 849–857.e2 (2018).
Miller, M. et al. School-age outcomes of infants at risk for autism spectrum disorder. Autism Res. J. Int. Soc. Autism Res. 9, 632–642 (2016).
Brian, J. et al. Stability and change in autism spectrum disorder diagnosis from age 3 to middle childhood in a high-risk sibling cohort. Autism Int. J. Res. Pract. 20, 888–892 (2016).
Shephard, E. et al. Mid-childhood outcomes of infant siblings at familial high-risk of autism spectrum disorder. Autism Res. J. Int. Soc. Autism Res. 10, 546–557 (2017).
Kirov, G. et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 75, 378–385 (2014).
Kosmicki, J. A. et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017).
Hazlett, H. C. et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 (2017).
Zwaigenbaum, L. & Penner, M. Autism spectrum disorder: advances in diagnosis and evaluation. BMJ 361, k1674 (2018).
Autism Genome Project Consortium. et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328 (2007).
Gotham, K., Pickles, A. & Lord, C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord. 39, 693–705 (2009).
Mullen, E. M. Mullen scales of early learning. Circle Pines, MN: American Guidance Service Inc (1995). (AGS ed.).
Hurley, R. S. E., Losh, M., Parlier, M., Reznick, J. S. & Piven, J. The broad autism phenotype questionnaire. J. Autism Dev. Disord. 37, 1679–1690 (2007).
Zarrei, M. et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet. Med. J. Am. Coll. Med. Genet. 20, 172–180 (2018).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Pinto, D. et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 29, 512–520 (2011).
Darvishi, K. Application of Nexus copy number software for CNV detection and analysis. Curr. Protoc. Hum. Genet. Chapter 4, Unit 4.14.1–28 (2010).
Downey, T. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 411, 256–270 (2006).
Zarrei, M., MacDonald, J. R., Merico, D. & Scherer, S. W. A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183 (2015).
Uddin, M. et al. A high-resolution copy-number variation resource for clinical and population genetics. Genet. Med. J. Am. Coll. Med. Genet. 17, 747–752 (2015).
Bierut, L. J. et al. Variants in nicotinic receptors and risk for nicotine dependence. Am. J. Psychiatry 165, 1163–1171 (2008).
Verhoeven, V. J. M. et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 45, 314–318 (2013).
Bierut, L. J. et al. A genome-wide association study of alcohol dependence. Proc. Natl. Acad. Sci. U.S.A. 107, 5082–5087 (2010).
Figueiredo, J. C. et al. Genotype-environment interactions in microsatellite stable/microsatellite instability-low colorectal cancer: results from a genome-wide association study. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 20, 758–766 (2011).
Newcomb, P. A. et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 16, 2331–2343 (2007).
Goodpaster, B. H. et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J. Gerontol. A. Biol. Sci. Med. Sci. 61, 1059–1064 (2006).
Stewart, A. F. R. et al. Kinesin family member 6 variant Trp719Arg does not associate with angiographically defined coronary artery disease in the Ottawa Heart Genomics Study. J. Am. Coll. Cardiol. 53, 1471–1472 (2009).
Krawczak, M. et al. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 9, 55–61 (2006).
Kearney, H. M. et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. J. Am. Coll. Med. Genet. 13, 680–685 (2011).
Yuen, R. K. C. et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 1, 160271–1602710 (2016).
Ross, P. J. et al. Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol. Psychiatry S0006322319315471 (2019). https://doi.org/10.1016/j.biopsych.2019.07.014.
Talkowski, M. E. et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 89, 551–563 (2011).
Team, R. C. R: A language and environment for statistical computing. (Team, R. C, 2013).
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951 (2004).
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674 (2007).
Colella, S. et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucl. Acids Res. 35, 2013–2025 (2007).
Euesden, J., Lewis, C. M. & O’Reilly, P. F. PRSice: polygenic risk score software. Bioinforma. Oxf. Engl. 31, 1466–1468 (2015).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014).
Sandin, S. et al. The heritability of autism spectrum disorder. JAMA 318, 1182–1184 (2017).
Christensen, D. L. et al. Prevalence and characteristics of autism spectrum disorder among 4-year-old children in the autism and developmental disabilities monitoring network. J. Dev. Behav. Pediatr. JDBP 37, 1–8 (2016).
Acknowledgements
We would like to thank the families who took part in this study, and the affiliated clinicians and research staff who assisted in recruitment and clinical evaluations of the participants. The Center for Applied Genomics provided technical and conceptual support in the form of genotyping, sequencing, CNV calling and data processing. This study was funded jointly by Autism Speaks, Autism Speaks Canada, the Simons Foundation Autism Research Initiative, Canadian Institutes of Health Research (CIHR), Canada Foundation for Innovation (CFI), The Hospital for Sick Children Foundation, Genome Canada/Ontario Genomics, Kids Brain Health Network, Canadian Institutes for Advanced Research (CIFAR), Ontario Brain Institute, Women and Children’s Health Research Institute at the University of Alberta, the Government of Ontario, and the University of Toronto McLaughlin Center at the University of Toronto. The authors wish to acknowledge the resources of Autism Speaks, MSSNG (www.mss.ng), as well as the generosity of the donors who supported this program. We also thank the participating families for their time and contributions to this database. Data collection was supported in part in the USA by National Institutes of Health (NIH) R01 HD047417 (D.M.), NIH R01 HD057284 (W.L.S., D.M.) and NIMH R01 MH059630 (R.L.). L.D.A. is funded by the Research Training Competition Doctoral Scholarships awarded by the Hospital for Sick Children. R.K.C.Y. is funded by the CIHR Postdoctoral Fellowship, NARSAD Young Investigator Award and Thrasher Early Career Award. L.Z holds the Stollery Children’s Foundation Chair in Autism. S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children.
Author information
Authors and Affiliations
Contributions
L.D.A., L.Z. and S.W.S. conceived the project design and methodology. L.D.A. analyzed the data with S.W., R.K.C.Y., K.T., J.W., B.T. and J.H. from The Center for Applied Genomics. R.D. performed the polygenic transmission disequilibrium test. J.B., S.B., K.D., R.L., J.L., D.M., S.O., I.M.S., W.L.S., Z.E.W. and L.Z. recruited families from their respective clinical sites and shared all phenotype information on study subjects. DNA samples were processed by J.H. G.Y. compiled the phenotype information and created the comprehensive BSRC clinical database. The manuscript was written by L.D.A, J.A.B. and S.W.S. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.W.S. is on Scientific Advisory Committees for Population Bio and Deep Genomics, and his institution, The Hospital for Sick Children, has licensed to Lineagen software code he co-developed: “Method of determining disease causality of genome mutations”.
Additional information
Peer review information Nature Communications thanks David Dimmock and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Abate, L., Walker, S., Yuen, R.K.C. et al. Predictive impact of rare genomic copy number variations in siblings of individuals with autism spectrum disorders. Nat Commun 10, 5519 (2019). https://doi.org/10.1038/s41467-019-13380-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13380-2
This article is cited by
-
Disruption of DDX53 coding sequence has limited impact on iPSC-derived human NGN2 neurons
BMC Medical Genomics (2023)
-
Oscillatory biomarkers of autism: evidence from the innate visual fear evoking paradigm
Cognitive Neurodynamics (2023)
-
Towards accurate and reliable resolution of structural variants for clinical diagnosis
Genome Biology (2022)
-
Novel clinical, molecular and bioinformatics insights into the genetic background of autism
Human Genomics (2022)
-
Mutations in trpγ, the homologue of TRPC6 autism candidate gene, causes autism-like behavioral deficits in Drosophila
Molecular Psychiatry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
