
Abstract
Currently, there is considerable interest in developing advanced rechargeable batteries that boast efficient distribution of electricity and economic feasibility for use in large-scale energy storage systems. Rechargeable aqueous zinc batteries are promising alternatives to lithium-ion batteries in terms of rate performance, cost, and safety. In this investigation, we employ Cu3(HHTP)2, a two-dimensional (2D) conductive metal-organic framework (MOF) with large one-dimensional channels, as a zinc battery cathode. Owing to its unique structure, hydrated Zn2+ ions which are inserted directly into the host structure, Cu3(HHTP)2, allow high diffusion rate and low interfacial resistance which enable the Cu3(HHTP)2 cathode to follow the intercalation pseudocapacitance mechanism. Cu3(HHTP)2 exhibits a high reversible capacity of 228 mAh g−1 at 50 mA g−1. At a high current density of 4000 mA g−1 (~18 C), 75.0% of the initial capacity is maintained after 500 cycles. These results provide key insights into high-performance, 2D conductive MOF designs for battery electrodes.
Similar content being viewed by others


Introduction
Societal interest in energy storage systems (ESSs) has been increasing rapidly with the need to utilize and distribute effectively electricity generated using renewable energy sources1,2,3. Among the most suitable candidates for energy storage are lithium-ion batteries (LIBs) since they provide high performance in mobile devices, such as cellular phones and laptops. Their utilization, however, in large-scale applications, such as electric vehicles, is inhibited by high material costs and safety concerns4,5. In order to resolve the limitations of LIBs, numerous investigations4,5,6,7 have been focused on greener electrode materials and aqueous electrolytes. From these perspectives, rechargeable aqueous zinc batteries (ZBs) have recently attracted8,9,10,11,12,13 considerable attention for use in large-scale ESSs because of their high theoretical capacity (820 mAh g−1), their low toxicity, and the relatively low cost of zinc14. Furthermore, ZBs operate in aqueous electrolytes4,5, thereby gaining additional advantages related to safety, cost, and rate performance.
Despite all these advantages, rechargeable ZBs have several obstacles that need to be addressed before they can hope to replace LIBs in terms of electrochemical performance15,16. In particular, the development of a new high-performance cathode is crucial for the commercialization of ZBs. α-MnO2 with a 2 × 2 tunnel structure has been used14 as a rechargeable ZB cathode, in which the large tunnels facilitate Zn2+ ion diffusion within the host structure, providing high capacity and rate performance. These materials, however, are associated with low cyclability that can be attributed15,16 to an unstable phase transition from a tunneled to a layered structure with simultaneous Mn2+ dissolution during the discharge–charge process. Vanadium-based cathodes8,17 also provide high capacity and rate performance, although the costliness of vanadium prohibits large-scale energy storage applications. Recently, organic-based cathodes, such as quinone derivatives, have been investigated because they are low cost, ubiquitous, and lightweight compared with inorganic cathodes13,18. Dissolution issues, however, during battery cycling inhibit the use of quinone derivatives in ZBs. In an effort to improve the stability of the quinone-based materials, polymerization19, carbon composites20, and an extended analog13 have all been explored: the dissolution issues, however, of organic cathodes remain a drawback. Reflecting on all these difficulties, the development of new materials for ZB cathodes is a necessity.
Conductive metal-organic frameworks (MOFs) provide excellent platforms for resolving dissolution issues, related to organic-based cathodes. In these MOFs, the active organic species are immobilized by metal-ligand coordinate covalent bonds. In addition, their porous structures and electrical conductivities are favorable to ion and electron transport in the framework, improving high rate capability and cyclability. The potential applications of these materials in batteries has been confirmed, with high performance being achieved in electrochemical double-layer capacitors21,22 and Na+ storage23, as well as in reports of their use in various battery systems24,25,26,27.
We introduce the idea of utilizing a two-dimensional (2D) conductive MOF, Cu3(HHTP)2 (HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene)28, as the cathode material for rechargeable aqueous ZBs. Electrical conductivity (0.2 S cm−1, four-point probe, single crystal)28 and large pores (~2 nm) facilitate electron and Zn2+ ion transport to active sites. In particular, we anticipate that the redox activity of the quinoid units of HHTP28,29,30 with Zn2+ insertion will promote the performance of the cathode.
Here, on account of these properties, we have tested the electrochemical performance of the Cu3(HHTP)2 cathode. Cu3(HHTP)2 shows redox switching at 1.06 V and 0.88 V vs. Zn/Zn2+ with the highest reversible capacity of 228 mAh g−1 at 50 mA g−1 to the best of our knowledge. These reversible capacities in rechargeable aqueous ZBs are the first example in MOFs and one of the highest reported values for cathodes with open-framework structures, including Prussian Blue analogs31,32,33 that have exhibited substantially smaller values of <70 mAh g−1 at similar current densities. In addition, the high diffusion rate of Zn2+ ions and low interfacial resistance by the insertion of hydrated Zn2+ ions allows Cu3(HHTP)2 to follow the intercalation pseudocapacitance mechanism. As a consequence, Cu3(HHTP)2 achieves a high rate performance and cyclability, indicating that 75.0% of the initial capacity (124.4 mAh g−1) is maintained after 500 cycles at an extremely high current density of 4000 mA g−1 (~18 C). This work reveals the reason for the observed high rate performance and charge-storage mechanism of the Cu3(HHTP)2, which is poised to facilitate the development of 2D conductive MOFs for energy storage.
Results
Synthesis and characterization of Cu3(HHTP)2
Cu3(HHTP)2 was synthesized according to a previously reported procedure28 and applied as the cathode material for aqueous rechargeable ZBs (Fig. 1a). PXRD analysis confirmed that the as-synthesized Cu3(HHTP)2 comprises (Fig. 1b) hexagonal 2D sheets stacked in a slipped-parallel configuration along the c axis29,34. Cu3(HHTP)2 was indexed based on a hexagonal unit cell (Fig. 2a) with the space group P6/mmm. The lattice parameters were calculated to be a = b = 21.2 Å and c = 6.6 Å with Rietveld refinement (Rp = 3.41, Rwp = 4.52, χ2 = 3.06). The morphology of Cu3(HHTP)2 was also investigated by field-emission scanning electron microscopy (FE-SEM). The shape of Cu3(HHTP)2 is similar28 (Fig. 2b and Supplementary Fig. 1a, b) to that of the uniform rods of Ni3(HHTP)2. The electrical conductivity of Cu3(HHTP)2 powder and Cu3(HHTP)2 electrode composite (60 wt% Cu3(HHTP)2, 20 wt% acetylene black, and 20 wt% PVDF) were measured on a pressed pellet using the two-point probe method. The conductivities obtained were 0.01 and 0.04 S cm−1 for Cu3(HHTP)2 powder and electrode composite, respectively. The electrical conductivity of a bulk Cu3(HHTP)2 electrode matches well the previously reported values29.

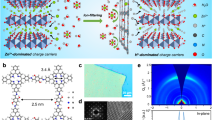
Zn-Cu3(HHTP)2 chemistry. a Schematic illustration of the rechargeable Zn-2D MOF cell. b Structure of Cu3(HHTP)2, which when viewed down the c axis, exhibits slipped-parallel stacking of 2D sheets with a honeycomb lattice. The cyan, red, and gray spheres represent Cu, O, and C atoms, respectively. The H atoms are omitted for the sake of clarity. c Expected redox process in the coordination unit of Cu3(HHTP)2

2D Chemical structure and structural analysis of Cu3(HHTP)2. a Rietveld refinement of PXRD patterns. b FE-SEM image of Cu3(HHTP)2, scale bar: 200 nm. c LD-HRTEM image of Cu3(HHTP)2 at a low resolution, scale bar: 20 nm. d LD-HRTEM image of Cu3(HHTP)2 along the [001] zone axis, indicating a hexagonal pore packing with d100 = 2.0 nm, scale bar: 2 nm. e–g LD-HRTEM images at (e) low and (g) high resolution along the [010] direction. Scale bars in (e) and (g) are 50 and 2 nm, respectively. f An FFT pattern of the yellow square in (e), scale bar: 2 nm−1
A transmission electron microscopy (TEM) image also reveals (Supplementary Fig. 2a) the one-dimensional (1D) nanorod structure of Cu3(HHTP)2. The length of the Cu3(HHTP)2 nanorods extends (Fig. 2b and Supplementary Fig. 1a, b, and 2a) a few micrometers with diameters of around 20–500 nm. In addition, a low dose—high resolution transmission electron microscopy (LD—HRTEM) image (Fig. 2d) enlarged from the selected yellow area in Fig. 2c (selected from Supplementary Fig. 2b) shows large pores with diameters of ~2.0 nm with a honeycomb arrangement viewed along the [001] direction. An enlarged LD-HRTEM image (Fig. 2g) from the selected area in Fig. 2e shows parallel Cu3(HHTP)2 nanorods along the [010] zone axis with a lattice distance of 2.0 nm for the (100) crystal plane. Fast Fourier transform (FFT) (Fig. 2f) from the selected area (Fig. 2e) indicates clearly that the Cu3(HHTP)2 nanorods have well developed (100) and (200) planes. These planes indicate28 that the as-synthesized Cu3(HHTP)2 is highly crystalline in nature with the [100] axis being the preferred orientation for the 1D nanorods. The unique structure of Cu3(HHTP)2, along with the shape of the 1D nanorods and the large pores, facilitate the diffusion of Zn2+ ions during the discharge–charge process. In addition, scanning electron microscopy-energy-dispersive X-ray spectroscopy (SEM-EDX) was used to verify (Supplementary Fig. 1c, d) the C, O, and Cu content of the Cu3(HHTP)2 particles.
Electrochemical performance of Cu3(HHTP)2
A cyclic voltammogram of Cu3(HHTP)2 thin film on SUS foil in a 3.0 M aqueous solution of Zn(CF3SO3)2 indicates (Supplementary Fig. 3) that the Zn2+ insertion and extraction reaction is reversible. The reaction of Zn2+ ions with Cu3(HHTP)2 occurs reversibly at approximately 0.65/1.10 V and 0.90/1.21 V (vs. Zn/Zn2+), respectively. Galvanostatic tests revealed that this reversibility is reflected (Fig. 3a) in the voltage profiles, with plateaus at the corresponding voltages. The first discharge plateau at ~0.90 V (vs. Zn/Zn2+) originates from the redox process between Cu2+ and Cu+. Furthermore, the second discharge plateau at 0.65 V (vs. Zn/Zn2+) may be attributed to a two-electron uptake associated with the HHTP linkers. A detailed redox reaction mechanism is discussed in the upcoming sections. The initial reversible capacity is 228 mAh g−1 at a rate of 50 mA g−1, followed by a capacity of 215 mAh g−1 in the second cycle, and the voltage profiles and capacity are retained (Fig. 3a and Supplementary Fig. 4) for 30 cycles. These reversible capacities are quite remarkable, providing some of the highest reported values for cathodes with open-framework structures, including Prussian Blue analogs31,32,33, that have been applied (Supplementary Table 1) to aqueous rechargeable ZBs.

Electrochemical performance of Cu3(HHTP)2. a, b Discharge–charge voltage profiles of Cu3(HHTP)2 at a 50 mA g−1 and b various current densities. The green dots labeled with (a–e) in (a) are states where XPS analysis in Fig. 4b, c was conducted. c, d Cycling performance of Cu3(HHTP)2 at current densities of c 500 mA g−1 and d 4000 mA g−1
In order to verify the influence of the Cu3(HHTP)2 2D structure with large pores on the electrochemical performance, we conducted rate-capability tests. In these electrochemical tests, Cu3(HHTP)2 demonstrated excellent rate capability (Fig. 3b). The Cu3(HHTP)2 electrode exhibited capacities of 191.4, 189.2, 152.4, and 124.5 mAh g−1 when the current density was increased, respectively, by 2, 4, 10, and 80 times (100, 200, 500, and 4000 mA g−1) from 50 mA g−1. These results correspond to capacity retentions of 89.0%, 88.0%, 70.9%, and 57.9%, respectively, with respect to the initial capacity of 215.0 mAh g−1. Moreover, the Cu3(HHTP)2 electrodes show promising cycling stability. At a current density of 500 mA g−1 (~2 C), 75.0% of the initial capacity (152.5 mAh g−1) was maintained (Fig. 3c) after 100 cycles. In addition, by increasing the mass loading of active materials from 60 to 90%, although the initial capacity decreased slightly to 125 mAh g−1 at 500 mA g−1 (Supplementary Fig. 5a), the retention of capacity after 100 cycles was 76% of the initial capacity (Supplementary Fig. 5b). This capacity retention for a 90% active materials loading electrode is almost identical to that of a 60% active materials loading electrode. Furthermore, at an extremely high current density of 4000 mA g−1 (~18 C), 75.0% of the initial capacity (124.4 mAh g−1) was maintained (Fig. 3d) after 500 cycles. This cyclability reflects the structural stability of Cu3(HHTP)2 during repeated (de)intercalation of the Zn2+ ions.
Origin of high rate performance of Cu3(HHTP)2
In order to investigate more detailed reasons for the high rate performance of Cu3(HHTP)2, diffusion coefficient and interfacial resistance studies were carried out. The Zn2+ ion diffusion coefficient of Cu3(HHTP)2 was obtained by applying galvanostatic intermittent titration technique (GITT) measurements; See the Supplementary Note 1 for details. The overall diffusion coefficient of Zn2+ ions in Cu3(HHTP)2 over the whole potential range was 3.9 × 10−10 cm2 s−1 (Supplementary Fig. 6), which is similar to that of single crystalline Zn0.25V2O5·nH2O nanobelts8. Specifically, by excluding the loss of diffusion coefficient from the high overpotential of the copper redox region, attributed to the self-discharge, and calculating the diffusion coefficient only with the main redox region of the quinoid, the diffusion coefficient of Zn2+ ions in Cu3(HHTP)2 showed 1.2 × 10−9 cm2 s−1 (Supplementary Fig. 6), indicating fast redox reactions.
Furthermore, an interfacial resistance between the electrode and the electrolyte was studied in order to determine the rate performance of electrode materials. In order to investigate the interfacial resistance of the Cu3(HHTP)2 electrode, an electrochemical impedance spectroscopy (EIS) investigation was conducted with symmetric cells of the Cu3(HHTP)2 electrodes in aqueous or organic electrolytes. Notably, the interfacial resistance of Cu3(HHTP)2 showed 150 and 16,000 Ω cm2 (Supplementary Fig. 7), and obtained conductivities from these interfacial resistances of Zn2+ ions are 0.7 × 10−2 and 0.6 × 10−5 S cm−1 (Supplementary Fig. 7) in aqueous and organic electrolytes, respectively. In recent studies, the insertion of carrier ions with H2O molecules has been suggested as the reason for low interfacial resistance, because the H2O can decrease the desolvation energy35 and the Coulombic repulsion from the interface36,37,38. Existence of H2O in the discharged electrode was tested with thermogravimetric analysis (TGA). The TGA profile of the discharged electrode showed a 26.9% weight loss between 120 and 300 °C (Supplementary Fig. 8), indicating that the low interfacial resistance of Cu3(HHTP)2 in the aqueous electrolyte can be attributed to the insertion of H2O with Zn2+ ions during the discharge reaction. We assume the large pore size of Cu3(HHTP)2 supports the insertion of hydrated Zn2+ ions. In order to confirm the importance of H2O, the performance of Cu3(HHTP)2 was studied in an organic electrolyte. On account of the high interfacial resistance caused by the organic electrolyte, the initial discharge capacity of Cu3(HHTP)2 decreased to 144 mAh g−1 at a rate of 50 mA g−1, and the subsequent charging reaction almost did not occur because of the high overpotential (Supplementary Fig. 9a). This phenomenon which is also evident35,36,37 in the case of Mg2+ ions in organic electrolytes, is caused by the strong interaction of divalent ions with the cathode, i.e., extracting of divalent ions from the host electrode is unfavorable35,36,37. As a result of this phenomenon, the capacity retention of Cu3(HHTP)2 in the organic electrolyte was almost zero (Supplementary Fig. 9b). In total, the origin of the high rate properties of Cu3(HHTP)2 is thought to be a consequence of the high diffusion rate of Zn2+ ions in the cathode and low interfacial resistance by the hydrated Zn2+ ion insertion.
Electronic states analysis during discharge–charge
With a view to investigating changes in the electronic states of Cu3(HHTP)2 during discharge-charge, X-ray photoelectron spectroscopy (XPS) was conducted on the Zn, O, and Cu elements. After inserting Zn2+ ions into the Cu3(HHTP)2, the Zn 2p peaks appear and disappear (Fig. 4a and Supplementary Fig. 10) at the discharged and charged states, respectively; this behavior is a consequence of the reversible insertion/extraction of Zn2+ into/from the Cu3(HHTP)2 cathodes. The quinoid peak at 532 eV shifts (Fig. 4b) to a benzoid peak at 533 eV in the O 1s spectrum, while discharging from 0.8 V (point b in Fig. 3a) to a fully discharged state (point c in Fig. 3a). The peaks which had shifted returned to their original positions, while charging from the fully discharged state (point c in Fig. 3a) to 1.15 V (point d in Fig. 3a). This shift reveals that the second plateau (Fig. 3a), which exists during the discharge process, originates from the quinoid structure acting as a redox center. Based on these XPS results, we infer that the quinoid structure is involved in the redox reaction; a similar redox mechanism was reported26 for Cu(2,7-AQDC) MOF (2,7-H2AQDC = 2,7-anthraquinonedicarboxylic acid), where oxygen and copper are the redox centers for LIBs. Similarly, the presence of transition metals involved in the redox reaction in our system causes the peaks of Cu2+ satellites in the pristine state to disappear (Fig. 4c). The Cu 2p peaks then separate into lower binding-energy peaks between the pristine state (point a in Fig. 3a) and 0.8 V (point b in Fig. 3a) in the Cu 2p spectrum (Fig. 4c). There is then no further shift in the Cu 2p peaks that lie between 0.8 V (point b in Fig. 3a) and 1.15 V (point d in Fig. 3a). As expected, the initial Cu 2p spectrum was reinstated, including its original profiles, between 1.15 V (point d in Fig. 3a) and the fully charged state (point e in Fig. 3a). From these changes in the Cu 2p peaks, the first plateau (Fig. 3a) that appears during the discharge process can be attributed to a partial redox reaction from Cu2+ to Cu+. Consequently, these XPS analyses suggest that both the quinoid component and the copper in Cu3(HHTP)2 participate as redox centers during the discharge–charge process. The theoretical capacity of Cu3(HHTP)2 should be 197 mAh g−1, when using the quinoid structure as the redox center and inserting Zn2+ ions with two electrons. The initial capacity determined (Fig. 3a), however, for Cu3(HHTP)2 is 228 mAh g−1, revealing that these Cu3(HHTP)2 cathodes can obtain 2.3 electrons (Fig. 1c). In light of these XPS results, the additional discharge capacity of Cu3(HHTP)2, equivalent to 0.3 electrons, can be derived from the redox events of Cu2+. In order to identify the redox center of Cu3(HHTP)2, density functional theory (DFT) calculations were performed. When supplying 6.9 extra electrons to Cu3(HHTP)2, Cu atom, as well as to the linker, takes of the additional electron (Fig. 4d), indicating that Cu atoms participate in the reduction reaction; See the Supplementary Note 2 for details. In the density of states (DOS) analysis (Supplementary Fig. 11), the electronic states just above the Fermi level consist of O, C, and Cu. This result supports the observed redox events occurring at these atoms. Furthermore, both peaks of O 1s and Cu 2p of the charged electrode after 500 cycles at a rate of 4000 mA g−1 (Fig. 4b, c) are more or less similar to those of the pristine electrode, indicating that the redox reaction of Cu3(HHTP)2 is highly reversible.

Electronic states analysis during discharge–charge. a–c Ex situ XPS spectra of a Zn 2p, b O 1 s, and c Cu 2p. d Changes of electron density upon the reduction of Cu3(HHTP)2
Structure analysis during discharge–charge
The PXRD patterns of Cu3(HHTP)2 in the discharged (inserting Zn2+ ions into Cu3(HHTP)2) electrode demonstrate that the (100) peak has a slight right-side shift from 4.70° to 4.85°, revealing (Fig. 5a) that the pore size in Cu3(HHTP)2 decreases from 19.3 to 18.7 Å. This change indicates that inserting Zn2+ ions into Cu3(HHTP)2 decreases the pore size of Cu3(HHTP)2 as a result of the electrostatic interaction between divalent Zn2+ cations and the oxygen anion of the host structure. With the exception of peak shifts following Zn2+ insertion, no changes (appearance or disappearance of peaks) are observed, indicating that the discharge process does not include H+ insertion accompanied by the formation of the Zn(OH)2 analog39. After the charge process (extracting Zn2+ ions from Cu3(HHTP)2), the PXRD peaks in the charged electrode return fully (Fig. 5a) to the position of the original pristine state. In addition, after 500 cycles at a rate of 4000 mA g−1, the PXRD patterns of Cu3(HHTP)2 are identical (Fig. 5a) to those of the pristine state. This observation implies that the inserted Zn2+ ions only affect the pore size of the host structure and that the structure of Cu3(HHTP)2 is maintained robustly when Zn2+ ions are inserted/extracted into/from Cu3(HHTP)2. Similarly, the morphology of the Cu3(HHTP)2, after Zn2+ ion insertion (Supplementary Fig. 12b, d), is almost the same (Supplementary Fig. 12a, c) as that of Cu3(HHTP)2 in a pristine state. In addition, ion-exchange from Cu2+ to Zn2+ ions is endothermic by 0.8 eV per ion, according to DFT calculations (Supplementary Fig. 13), indicating the high stability of Cu3(HHTP)2 against Zn2+ substitution. Consequently, the PXRD results lead us to infer that the Zn2+ ions are accommodated in the large pores of Cu3(HHTP)2, thus enabling high long-term stability while cycling at a high rate.

Structure analysis during discharge–charge. a PXRD patterns of the Cu3(HHTP)2 electrode in the pristine, first fully discharged/charged states at a rate of 50 mA g−1, and 500th fully charged states at a rate of 4000 mA g−1. b Scanning transmission electron microscopy (STEM) image of the fully discharged Cu3(HHTP)2 alongside its EDX elemental mapping with respect to C, Cu, O, and Zn, suggesting uniform Zn insertion over the electrode, scale bar: 100 nm. c An LD-HRTEM image of discharged Cu3(HHTP)2 viewed down the [010] zone axis. An inset in (c) shows a magnified area depicting the (100) plane, scale bar: 20 nm. d Measurements of the (100) interplanar distances from the white boxed area in (c) indicate the average d100 = 1.87 nm. e, f SAD patterns from Cu3(HHTP)2 at (e) pristine and (f) discharged states used to confirm the interplanar distances of (100). The arrows and scale bar indicate the [100] direction and 2 nm−1, respectively
Confirmation of inserting Zn2+ ions into the pore structure of Cu3(HHTP)2
The uniform presence of Zn2+ ions in Cu3(HHTP)2 nanorods was confirmed (Fig. 5b) by EDX chemical mapping which shows uniform distribution of Zn ions over the entire electrode area at the fully discharged state. In order to elucidate the consequences of the insertion of Zn2+ ions into the pores of Cu3(HHTP)2, the lattice parameter changes were analyzed (Fig. 5c) with LD-HRTEM in the discharged state. Significantly, after inserting Zn2+ ions into Cu3(HHTP)2 nanorods, the lattice distance of the (100) plane (inset of Fig. 5c, d) decreases slightly to 1.87 nm, demonstrating the same tendency observed (Fig. 5a) in the PXRD patterns. In addition, selected-area diffraction patterns from pristine and discharged samples (Fig. 5e, f) demonstrate that the (100) lattice distance decreases from 2.01(±0.01) nm to 1.90(±0.01) nm, in the consequent interaction of divalent cations inserted into the pores of the framework. This result verifies the fact that Zn2+ ions are inserted into the pores in MOFs in a battery system.
Charge-storage mechanism of Cu3(HHTP)2
In order to understand the charge-storage mechanism of Cu3(HHTP)2, CV measurements were carried out using various scan rates (Fig. 6a). Currents depending on the scan rates study enables determining b-values from the equation of a power law40,41,42,43,44,45: i = aνb where i is the current (A), v is the potential scan rate (V s−1), a and b are arbitrary coefficients. Generally, battery electrode materials are characterized by b = 0.5, indicating a semi-infinite diffusion process40,41,42,43,44,45, whereas the closer the b-values are to 1, the closer to the capacitive contribution. The b-values are the slope obtained by plotting the peak currents (i) and scan rates (ν) in a log plot (Supplementary Fig. 14a) with an assumption that the current obeys the power-law relationship. The b-values of Cu3(HHTP)2 are above 0.85 within all operating voltage ranges (Fig. 6b), indicating the operating mechanism is not dominated by diffusion.

Charge-storage mechanism of Cu3(HHTP)2. a Cyclic voltammograms of Cu3(HHTP)2 recorded at different scan rates. b b-values for the Cu3(HHTP)2 electrodes plotted as a function of the potential for cathodic scans. c Capacitive and diffusion currents contributed to the charge-storage of Cu3(HHTP)2 at the rate of 0.5 mV s−1. d A self-discharge profile of Cu3(HHTP)2. The inset shows voltage profiles for the self-discharge test before and after storage
Furthermore, for quantitative analysis of capacitance, the scan rate dependence of the current was plotted (Supplementary Fig. 14b). The capacitive effect (k1ν) and diffusion-controlled insertion (k2ν1/2) could be calculated with the plot, see the Supplementary Note 3 for details. The capacitive contribution was 83% (Fig. 6c) out of the total current, at a scan rate of 0.5 mV s−1, indicating the total energy storage in Cu3(HHTP)2 arises from a capacitive process rather than the solid-state diffusion of Zn2+ in Cu3(HHTP)2. Unlike a non-Faradaic surface adsorption present in the typical responses of a capacitor, reversible redox peaks on CV profiles (Fig. 6a, c) and the reversible shifts of (100) peaks in PXRD (Fig. 5a) during the discharge–charge process were observed, indicating that the Cu3(HHTP)2 follows an intercalation pseudocapacitance charge-storage mechanism41,42,43,44,45. In this mechanism, charge-storage occurs by intercalation/de-intercalation of cations in the bulk active materials, and its kinetics are not limited by the diffusion of the cations. As a consequence, the advantage of batteries (high capacitance) and supercapacitors (high rate) are integrated into one system. In addition, a self-discharge test46 was carried out to confirm the ability of charge-storage, and Cu3(HHTP)2 showed (Fig. 6d) remarkably low self-discharge rate of 0.003 V h−1. The loss of the capacity during self-discharge mainly occurs near the Cu redox region above 0.9 V, agreeing well with the GITT study (Supplementary Fig. 6). Furthermore, after 5 days of storage, 83% of the initial capacity was still maintained (inset of Fig. 6d) and therefore proving its outstanding stability in the fully charged state.
Discussion
In summary, we have demonstrated a Cu3(HHTP)2 2D conductive MOF that may be utilized as a ZB cathode. The solid-state structure of Cu3(HHTP)2, with a high diffusion rate of Zn2+ ions, and low interfacial resistance caused by the insertion of hydrated Zn2+ ions, as a result of the large open channel structures, provides an increased rate performance and cyclability compared with those of conventional organic-based materials. In addition, the kinetic analyses of the electrochemical behavior of Cu3(HHTP)2 obtained by CV suggest that the charge-storage mechanism of Cu3(HTTP)2 is intercalation pseudocapacitance, indicating that the mechanism is not determined by diffusion. Furthermore, XPS measurements and DFT calculations suggest that Cu3(HHTP)2 utilizes both copper and the quinoid structure as redox-active sites, increasing the specific capacity of the material. In addition, the PXRD and LD-HRTEM data indicate that inserted Zn2+ ions are stored in the Cu3(HHTP)2 pores. These findings point to the potential of these cathodes for use in large-scale applications. This investigation paves the way for the further exploration of 2D conductive MOFs with other transition metals that could increase their redox potential, thus improving the performance of 2D conductive MOF-based ZB cathodes.
Methods
Materials
All commercially available reagents and solvents were purchased from Sigma-Aldrich and used as received without further purification. Zn and SUS films were purchased from Goodfellow. All the parts for making coin cells were obtained from Pred Materials International. Cu3(HHTP)2 was prepared according to a previously reported procedure28, washed with deionized H2O and Me2CO, respectively, and dried in air.
Characterization
The morphology of powder and elementary analysis was obtained by field-emission scanning electron microscopy (FE-SEM, Hitachi S-4800) with implemented energy-dispersive X-ray spectroscopy (EDX, Oxford Aztec X-max 80 SDD EDX detector). Images were acquired at a working distance of 7 mm with an electron beam energy of 20 kV and emission current of 20 µA. In order to investigate the H2O content after the discharge process, thermogravimetric analysis (TGA, Netzsch Jupiter) was performed by raising the temperature from room temperature to 300 °C at a ramp rate of 5 °C min−1 under an Ar flow. Powder X-ray diffraction (PXRD, STOE STADI-P) with Cu-Kα1 radiation was measured through transmission geometry for crystal structure analysis by scanning in the 2θ range of 2°–90° with scan steps of 0.015° with accelerating voltage and current of 40 kV and 40 mA. For the characterization of Cu3(HHTP)2 at different charge and discharge states, the cells were opened and rinsed with deionized H2O inside a glove-box. The oxidation states of electrodes were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo scientific ESCALAB 250Xi). Each sample was dried under vacuum for 1 h prior to XPS measurements. For the ex situ XPS characterization of Cu3(HHTP)2 at different charge and discharge states, the cells were opened and rinsed with deionized H2O inside a glove-box. The electrical conductivity of Cu3(HHTP)2 was measured by the two-point probe method at 25 °C. A pellet was placed on a home-built in situ pellet press47 and connected to an electrometer (Keithley 4200-SCS). The current-voltage (I–V) measurements were performed at 25 °C by sweeping the voltage.
Transmission electron microscopy
Pristine and discharged Cu3(HHTP)2 MOF samples were dispersed in EtOH and drop-cast on lacey carbon Mo-based TEM grids. LD-HRTEM was performed using a JEOL Grand ARM instrument operated at 300 kV. Data were collected using a Gatan K3-IS direct electron detector. In order to avoid MOF structure degradation under electron beams, images were collected at dose rates below 20 e−/pixel/s and the cumulative dose in the range of 15–20 e−/A[2 48. For selected-area diffraction (SAD), the electron beam was spread out and with data acquired at low magnification to avoid sample damage. SAD Patterns were collected using a Gatan OneView camera. EDX data were collected using an SDD EDX detector.
Electrochemical tests
In order to investigate the electrochemical performance of Cu3(HHTP)2 as a cathode in zinc batteries, coin cells with a two-electrode configuration—which comprise a Cu3(HHTP)2 cathode and a Zn-film anode (100 μm in thickness)—were assembled. The Cu3(HHTP)2 electrode was first of all prepared by making a slurry containing Cu3(HHTP)2:acetylene black:poly(vinylidene difluoride) (PVDF) in the ratio of 60:20:20 or 90:5:5 in 1-methyl-2-pyrrolidinone (NMP), respectively. The slurry was then cast onto stainless steel (SUS 304) foil, followed by drying at 70 °C in a vacuum oven. The mass loading of the active material in each electrode was 2 mg cm−2. The electrolyte solution was 3 M and 0.25 M zinc trifluoromethanesulfonate (Zn(CF3SO3)2) in deionized H2O and acetonitrile (MeCN), respectively. All cells were aged for 1 h prior to initiating electrochemical processes to ensure good soaking of the electrolyte solution into the electrodes. The cells were cycled in the voltage range of 0.5–1.3 V (vs. Zn/Zn2+). All measurements were made at 25 °C using a battery tester (BST8-300-CST, MTI, USA). All galvanostatic measurements were recorded in the constant current mode (no constant voltage steps). CV was carried out using coin cells with a two-electrode configuration, which comprise the Cu3(HHTP)2 cathode and the Zn-film anode (Reference 600 potentiostat, Gamry Instruments, USA). EIS measurements were performed on symmetric cells over the frequency range of 0.01 Hz–1 MHz with an input voltage amplitude of 10 mV (Reference 600 potentiostat, Gamry Instruments, USA).
DFT calculations
These calculations were performed using the Perdew–Burke–Ernzhof (PBE) exchange-correlation functional49 and the projector-augmented wave (PAW) method50 as implemented in the VASP51. An energy cutoff of 520 eV was used and the gamma centered single k-point was sampled for integration because of the large cell size. A Grimme’s dispersion correction (D3) with a zero damping was also applied52. The convergence criteria were 10−6 eV and 0.02 eV Å−1 for the electronic and ionic cycles, respectively. The monolayer of Cu3(HHTP)2 was assumed because the long-range order of Cu3(HHTP)2 has not yet been identified. In order to avoid a fictitious interaction between layers, the vacuum layer along the z-direction was set to be ~20 Å so that the lattice size was 21 × 21 × 20 Å. In order to represent the reduction of Cu3(HHTP)2, we supplied extra electrons to the pristine state and the charge-density difference between the reduced and pristine states was illustrated using the VESTA software53. In order to estimate the ion-substitution energy, we employed the hydrated Zn2+ and Cu2+ states as the reference. To this end, an implicit solvent model54 was applied and a higher energy cutoff (650 eV) was used.
Data availability
The authors declare that all the relevant data are available within the paper and its Supplementary Information file or from the corresponding author upon reasonable request.
References
Dunn, B., Kamath, H. & Tarascon, J.-M. Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011).
Yang, Z. et al. Electrochemical energy storage for green grid. Chem. Rev. 111, 3577–3613 (2011).
Larcher, D. & Tarascon, J. M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 7, 19–29 (2015).
Li, W., Dahn, J. R. & Wainwright, D. S. Rechargeable lithium batteries with aqueous electrolytes. Science 264, 1115–1118 (1994).
Luo, J.-Y., Cui, W.-J., He, P. & Xia, Y.-Y. Raising the cycling stability of aqueous lithium-ion batteries by eliminating oxygen in the electrolyte. Nat. Chem. 2, 760–765 (2010).
Zheng, M. et al. Tungsten-based materials for lithium-ion batteries. Adv. Func. Mater. 28, 1707500 (2018).
Zhou, H., Li, X., Li, Y., Zheng, M. & Pang, H. Applications of MxSey (M = Fe, Co, Ni) and their composites in electrochemical energy storage and conversion. Nano-Micro Lett. 11, 40 (2019).
Kundu, D., Adams, B. D., Duffort, V., Vajargah, S. H. & Nazar, L. F. A high-capacity and long-life aqueous rechargeable zinc battery using a metal oxide intercalation cathode. Nat. Energy 1, 16119 (2016).
Pan, H. et al. Reversible aqueous zinc/manganese oxide energy storage from conversion reactions. Nat. Energy 1, 16039 (2016).
Zhang, N. et al. Rechargeable aqueous zinc-manganese dioxide batteries with high energy and power densities. Nat. Commun. 8, 405 (2017).
Hu, E. & Yang, X.-Q. Rejuvenating zinc batteries. Nat. Mater. 17, 480–481 (2018).
Wang, F. et al. Highly reversible zinc metal anode for aqueous batteries. Nat. Mater. 17, 543–549 (2018).
Zhao, Q. et al. High-capacity aqueous zinc batteries using sustainable quinone electrodes. Sci. Adv. 4, eaao1761 (2018).
Xu, C., Li, B., Du, H. & Kang, F. Energetic zinc ion chemistry: the rechargeable zinc ion battery. Angew. Chem. Int. Ed. 51, 933–935 (2012).
Lee, B. et al. Electrochemically-induced reversible transition from the tunneled to layered polymorphs of manganese dioxide. Sci. Rep. 4, 6066 (2014).
Lee, B. et al. Elucidating the intercalation mechanism of zinc ions into α-MnO2 for rechargeable zinc batteries. Chem. Commun. 51, 9265–9268 (2015).
Xia, C., Guo, J., Li, P., Zhang, X. & Alshareef, H. N. Highly stable aqueous zinc-ion storage using a layered calcium vanadium oxide bronze cathode. Angew. Chem. Int. Ed. 57, 3943–3948 (2018).
Liang, Y. et al. Universal quinone electrodes for long cycle life aqueous rechargeable batteries. Nat. Mater. 16, 841–848 (2017).
Dawut, G., Lu, Y., Miao, L. & Chen, J. High-performance rechargeable aqueous Zn-ion batteries with a poly(benzoquinonyl sulfide) cathode. Inorg. Chem. Front. 5, 1391–1396 (2018).
Kundu, D. et al. Organic cathode for aqueous Zn-ion batteries: taming a unique phase evolution toward stable electrochemical cycling. Chem. Mater. 30, 3874–3881 (2018).
Sheberla, D. et al. Conductive MOF electrodes for stable supercapacitors with high areal capacitance. Nat. Mater. 16, 220–224 (2016).
Feng, D. et al. Robust and conductive two-dimensional metal-organic frameworks with exceptionally high volumetric and areal capacitance. Nat. Energy 3, 30–36 (2018).
Park, J. et al. Stabilization of hexaaminobenzene in a 2D conductive metal-organic framework for high power sodium storage. J. Am. Chem. Soc. 140, 10315–10323 (2018).
Férey, G. et al. Mixed-valence Li/Fe-based metal-organic frameworks with both reversible redox and sorption properties. Angew. Chem. Int. Ed. 46, 3259–3263 (2007).
Aubrey, M. L. & Long, J. R. A dual-ion battery cathode via oxidative insertion of anions in a metal-organic framework. J. Am. Chem. Soc. 137, 13594–13602 (2015).
Zhang, Z., Yoshikawa, H. & Awaga, K. Monitoring the solid-state electrochemistry of Cu(2,7-AQDC) (AQDC = anthraquinone dicarboxylate) in a lithium battery: coexistence of metal and ligand redox activities in a metal-organic framework. J. Am. Chem. Soc. 136, 16112–16115 (2014).
Wada, K., Sakaushi, K., Sasaki, S. & Nishihara, H. Multielectron-transfer-based rechargeable energy storage of two-dimensional coordination frameworks with non-innocent ligands. Angew. Chem. Int. Ed. 57, 8886–8890 (2018).
Hmadeh, M. et al. New porous crystals of extended metal-catecholates. Chem. Mater. 24, 3511–3513 (2012).
Campbell, M. G., Liu, S. F., Swager, T. M. & Dincă, M. Chemiresistive sensor arrays from conductive 2D metal-organic frameworks. J. Am. Chem. Soc. 137, 13780–13783 (2015).
Zhu, R. et al. π-Conjugated molecule boosts metal-organic frameworks as efficient oxygen evolution reaction catalysts. Small 14, 1803576 (2018).
Zhang, L., Chen, L., Zhou, X. & Liu, Z. Morphology-dependent electrochemical performance of zinc hexacyanoferrate cathode for zinc-ion battery. Sci. Rep. 5, 18263 (2015).
Zhang, L., Chen, L., Zhou, X. & Liu, Z. Towards high-voltage aqueous metal-ion batteries beyond 1.5 V: The zinc/zinc hexacyanoferrate system. Adv. Energy Mater. 5, 1400930 (2015).
Trócoli, R. & La Mantia, F. An aqueous zinc-ion battery based on copper hexacyanoferrate. ChemSusChem 8, 481–485 (2015).
Miner, E. M., Wang, L. & Dincă, M. Modular O2 electroreduction activity in triphenylene-based metal-organic frameworks. Chem. Sci. 9, 6286–6291 (2018).
Okoshi, M., Yamada, Y., Yamada, A. & Nakai, H. Theoretical analysis on de-solvation of lithium, sodium, and magnesium cations to organic electrolyte solvents. J. Electrochem. Soc. 160, A2160–A2165 (2013).
Mizuno, Y. et al. Suppressed activation energy for interfacial charge transfer of a prussian blue analog thin film electrode with hydrated ions (Li+, Na+, and Mg2+). J. Phys. Chem. C. 117, 10877–10882 (2013).
Nam, K. W. et al. The high performance of crystal water containing manganese birnessite cathodes for magnesium batteries. Nano. Lett. 15, 4071–4079 (2015).
Nam, K. W., Kim, H., Choi, J. H. & Choi, J. W. Crystal water for high performance layered manganese oxide cathodes in aqueous rechargeable zinc batteries. Energy Environ. Sci. 12, 1999–2009 (2019).
Huang, J. et al. Polyaniline-intercalated manganese dioxide nanolayers as a high-performance cathode material for an aqueous zinc-ion battery. Nat. Commun. 9, 2906 (2018).
Wang, J., Polleux, J., Lim, J. & Dunn, B. Pseudocapacitive contributions to electrochemical energy storage in TiO2 (Anatase) nanoparticles. J. Phys. Chem. C. 111, 14925–14931 (2007).
Augustyn, V. et al. High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance. Nat. Mater. 12, 518–522 (2013).
Brezesinski, K. et al. Pseudocapacitive contributions to charge storage in highly ordered mesoporous group V transition metal oxides with iso-oriented layered nanocrystalline domains. J. Am. Chem. Soc. 132, 6982–6990 (2010).
Brezesinski, T., Wang, J., Tolbert, S. H. & Dunn, B. Ordered mesoporous α-MoO3 with iso-oriented nanocrystalline walls for thin-film pseudocapacitors. Nat. Mater. 9, 146–151 (2010).
Zukalová, M., Kalbáč, M., Kavan, L., Exnar, I. & Graetzel, M. Pseudocapacitive lithium storage in TiO2(B). Chem. Mater. 17, 1248–1255 (2005).
Come, J. et al. Electrochemical kinetics of nanostructured Nb2O5 electrodes. J. Electrochem. Soc. 161, A718–A725 (2014).
Wang, Z. et al. A MOF-based single-ion Zn2+ solid electrolyte leading to dendrite-free rechargeable Zn batteries. Nano Energy 56, 92–99 (2019).
Sun, L., Park, S. S., Sheberla, D. & Dincă, M. Measuring and reporting electrical conductivity in metal–organic frameworks: Cd2(TTFTB) as a case study. J. Am. Chem. Soc. 138, 14772–14782 (2016).
Zhang, D. et al. Atomic-resolution transmission electron microscopy of electron beam–sensitive crystalline materials. Science 359, 675–679 (2018).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 44, 1272–1276 (2011).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Aras, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Acknowledgements
This research was conducted as part of the Joint Center of Excellence in Integrated Nanosystems at King Abdulaziz City for Science and Technology (KACST) and Northwestern University. The authors thank both KACST and NU for their financial support of this research. The Integrated Molecular Structure Education and Research Center (IMSERC) at NU is recognized for the use of its instrumentation. This work made use of the EPIC facility in Northwestern University’s NUANCE Center, which receives support from (1) the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), (2) the MRSEC program (NSF DMR-1720139) at the Materials Research Center, (3) the International Institute for Nanotechnology (IIN), and (4) the Keck Foundation; and the State of Illinois, through the IIN. C.A.M. and S.S.P. acknowledge support by the Air Force Office of Scientific Research under Award FA9550-17-1-0348 and the National Science Foundation under Grant CHE-1709888. We thank Dr. L. Sun and Dr. M.E. Schott for helpful discussions. The structural study by TEM is partially based on research sponsored by the Air Force Research laboratory under agreement number is FA8650-15-2-5518. The U.S. Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of Air Force Research Laboratory or the U.S. Government.
Author information
Authors and Affiliations
Contributions
K.W.N., S.S.P., C.A.M., and J.F.S. designed the research. S.S.P. carried out the synthesis of the active materials. K.W.N. worked on the electrochemical measurements and analysis. R.dR. performed the TEM measurements. H.K. conducted DFT calculations. K.W.N., S.S.P., R.dR., V.P.D., H.K., C.A.M., and J.F.S. co-wrote the paper. J.F.S. supervised the research. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nam, K.W., Park, S.S., dos Reis, R. et al. Conductive 2D metal-organic framework for high-performance cathodes in aqueous rechargeable zinc batteries. Nat Commun 10, 4948 (2019). https://doi.org/10.1038/s41467-019-12857-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-12857-4
This article is cited by
-
A solubility limited pyrene-4,5,9,10-tetraone-based covalent organic framework for high-performance aqueous zinc-organic batteries
Nano Research (2024)
-
Unveiling Organic Electrode Materials in Aqueous Zinc-Ion Batteries: From Structural Design to Electrochemical Performance
Nano-Micro Letters (2024)
-
A mixed-valence polyoxometalate-based 3D inorganic framework cathode material for high-efficiency rechargeable AZIBs
Rare Metals (2024)
-
Design strategies for rechargeable aqueous metal-ion batteries
Science China Chemistry (2024)
-
Metal/covalent organic frameworks for aqueous rechargeable zinc-ion batteries
Science China Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
