
Abstract
More than 7 million individuals have been conceived by Assisted Reproductive Technologies (ART) and there is clear evidence that ART is associated with a range of adverse early life outcomes, including rare imprinting disorders. The periconception period and early embryogenesis are associated with widespread epigenetic remodeling, which can be influenced by ART, with effects on the developmental trajectory in utero, and potentially on health throughout life. Here we profile genome-wide DNA methylation in blood collected in the newborn period and in adulthood (age 22–35 years) from a unique longitudinal cohort of ART-conceived individuals, previously shown to have no differences in health outcomes in early adulthood compared with non-ART-conceived individuals. We show evidence for specific ART-associated variation in methylation around birth, most of which occurred independently of embryo culturing. Importantly, ART-associated epigenetic variation at birth largely resolves by adulthood with no direct evidence that it impacts on development and health.
Similar content being viewed by others


Introduction
Assisted Reproductive Technologies (ART) have resulted in more than 7 million births since 19781. Today, ART procedures are diverse, spanning the relatively less invasive intervention of gamete intra-fallopian transfer (GIFT) and intra-uterine insemination (IUI)2, through fertilization of gametes in vitro with culturing (in vitro fertilization, IVF), to the more recent direct injection of a sperm into an ovum (intracytoplasmic sperm injection, ICSI), followed by culturing, with or without subsequent embryo freeze/thawing3,4.
Mounting evidence suggests that early periconceptional exposures (such as ART) may influence developmental trajectories in offspring5,6. ART conception is associated with an approximately two-fold increased risk of preterm birth, low birth weight, being small for gestational age or perinatal mortality7,8,9. However, despite the continuing expansion of ART worldwide10,11,12, few studies have investigated the potential long-term health outcomes associated with assisted conception, or the potential underlying molecular and cellular variations. Some but not all studies of children and adolescents born following ART report possible increased cardiovascular13,14 and metabolic risks13,15, raised plasma lipids, and higher rates of obesity15. Large epidemiological studies also suggest an increased risk of rare imprinting disorders in association with epigenetic disruption early in development16. Notwithstanding, meta-analyses and systematic reviews suggests a dearth of compelling data supporting any consistent ART-associated adverse outcomes in either children or adults17,18,19,20,21,22,23,24,25.
The periconceptional period is associated with widespread epigenetic remodeling in gametes and the early embryo26. It is therefore plausible that the early epigenetic profile is influenced by ART processes, with potential to alter the developmental trajectory in utero and throughout life27,28. For example, the hormonal milieu created by ovarian stimulation and the in vitro culturing of the embryo have both been suggested as processes that can alter epigenetic profile in ART progeny29,30, however published data are circumstantial, limited, and at times contradictory31. A recent review summarizes the potential adverse effects on long-term health associated with ART, some of which may be attributable to epigenetic variation induced in the periconceptional period6. Further evidence suggests that variation in the developing epigenetic profile may occur at repetitive elements that make up a large proportion of the human genome32.
Given the rising rates of ART pregnancies internationally4, limited evidence of potential adverse short to medium term health outcomes, and the relatively limited number of studies of epigenetic variation in association with ART, it is imperative that any underlying epigenetic variation induced by ART is fully explored in humans, particularly as this population ages. This is especially important given emerging links between epigenetic variation and a range of adverse adult-onset cardiometabolic, neurodevelopmental, and respiratory conditions33,34.
We previously established a cohort of singleton ART-conceived young adults (aged 18–28 years) and a matched non-ART group from the same source population, and using a telephone interview found an increased rate of maternally reported hospital admissions, atopic respiratory conditions, and metabolic/endocrine/nutritional disease (ICD-10 coding category) in the ART-conceived group35. More recently, we assessed vascular, cardiometabolic, anthropometric, and respiratory health clinically in a subset of the original cohort, now aged 22–35 years, and found no evidence of adverse health outcomes associated with ART conception36. In the current study, we perform a longitudinal Epigenome-wide Association Study (EWAS) of these ART and non-ART-conceived individuals from the neonatal period through to adulthood, spanning up to 35 years since birth.
Results
ART-associated differential methylation at birth is largely attenuated in adulthood
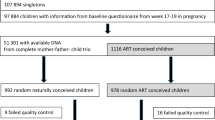
To investigate whether DNA methylation levels in blood differ between ART-conceived individuals relative to non-ART conceived individuals, we analyzed epigenome-wide methylation data in neonatal (Guthrie spot) and adult peripheral whole blood using the EPIC array. DNA methylation status was generated for 149 neonatal (84♀ 65♂) and 158 adult (87♀ 71♂) ART-conceived individuals and for 58 neonatal (37♀, 21♂) and 75 adult (51♀, 24♂) non-ART conceived individuals (Fig. 1a).

Study design and identification of ART-associated differentially methylated probes in neonatal and adult blood. a Summary of the longitudinal EWAS. b Number of DMPs that pass an adjusted p-value cut-off of <0.05 and a Δβ cut-off of ≥0.05. c Correlation plot of mean Δβ between ART and non-ART groups in neonatal (x axis) and adult (y axis) blood at neonatal DMPs. Δβ is always calculated as mean ART DNA methylation minus mean non-ART DNA methylation. Red dots represent probes that show a Δβ ≥ 5% between ART and controls in both neonatal and adult blood (red dotted line and blue dotted line designate the 0.05 mark in all directions), black dots represent probes that only fulfill DMP criteria at birth. d. Bar plot of neonatal (red) and adult (blue) blood Δβ values for top ranked probes based on methylation change in neonatal blood, with accompanying probe ID, name of nearest gene and distance to gene TSS in brackets. While most probes no longer show differences in adult blood (at adjusted p-value <0.05 (Bayesian levene’s test)), there are several probes that show a persistent change in methylation. n = 207 biologically independent birth samples, n = 233 biologically independent adult samples
In neonatal blood, we identified 2340 (out of total 724,897 probes) differentially methylated probes (DMPs) between ART and non-ART groups following FDR correction for multiple testing, but none in the adult samples (out of total 766,247 probes) (Fig. 1b). The mean (SD) methylation difference (Δβ) of the 2340 DMPs between the groups was 0.026 ± 0.013 (largest effect of 0.129 (i.e., 12.9%)). The majority of DMPs (79.1%), showed a higher DNA methylation level among ART offspring in neonatal blood compared with non-ART offspring. Restricting DMPs to those that showed greater than 5% difference between groups (−0.05 ≥ Δβ ≥ 0.05) revealed 116 DMPs (85%) with higher, and 20 (15%) with lower methylation in the ART group relative to the non-ART group in neonatal blood (Fig. 1b, Supplementary Data 1). Despite not reaching significance following FDR correction in adulthood, six of these 136 DMPs were also differentially methylated by ≥5% in adulthood (Fig. 1c), albeit attenuated in magnitude (Fig. 1d, Supplementary Data 2). We did not observe any anti-correlating probes (e.g., hypomethylated in ART neonates but hypermethylated in ART adults). Of the 136 DMPs showing greater than 5% difference between groups, all but one were within 1 Mb of a gene transcription start site, with 4 genes having 2 DMPs in their vicinity (Supplementary Data 1).
Next we examined differentially methylated regions (DMRs), which contain multiple DMPs that show correlative methylation. DMRs, defined as a region containing ≥3 DMPs, at least one of which have a Δβ ≥ 5%, were identified using DMRcate (Fig. 2a). In total 18 DMRs (comprising 106 total probes) were identified in neonatal blood (Fig. 2b) and 4 DMRs (comprising 27 probes) were identified in adulthood (Fig. 2c). Three DMRs common to both time-points were found near the genes CHRNE (7 probes), PRSS16 (3 probes), and TMEM18 (9 probes), with the same direction and similar level of DNA methylation change (Fig. 2d, e). The full list of significant DMPs in neonatal blood, the highest ranked in adult blood, and the DMRs at both time-points, are listed in Supplementary Data 1–4.

Identification of ART-associated differentially methylated regions in neonatal and adult blood. a. Overview of the strategy used to identify DMRs. For each probe with an adjusted p-value <0.05 (Bayesian levene’s test) identified during linear regression analysis, DMRcate was used to scan the surrounding region for probes that show the same general DNA methylation change. Once regions were identified by DMRcate, the following cut-offs were used: at least 3 probes in the region, at least 1 of which has a Δβ ≥ 5%. b. Scatterplot of DMR size and number of probes within a DMR in neonatal blood. A total of 106 probes within 18 DMRs were identified, with the size ranging from 100 to 1700 bp and number of probes per DMR ranging from 3 to 13. c Scatterplot of DMR size and number of probes within a DMR in adult blood. A total of 27 probes within 4 DMRs were identified, with the size ranging from 300 to 1900 bp and number of probes per DMR ranging from 3 to 9. d Venn diagram showing that three DMRs which overlap between neonatal and adult blood. The name of nearest gene, location of DMR, and distance to gene TSS is shown for the common DMRs. e Column graph showing Δβ between mean ART and mean control for individual probes within the three common DMRs for neonatal (red) and adult (blue) blood. Probe ID and gene name are shown on the x axis
CHRNE exhibits both age-specific and ART-specific differential methylation
In order to further explore the specificity of the observed ART-associated differential methylation, we examined methylation profiles more broadly around the identified DMRs of interest, at both time-points. Probes around all three DMRs identified at birth showed a complex pattern of ART-associated and age-associated differential methylation, with little difference between the ART and non-ART groups at probes outside the identified DMRs (Fig. 3, Supplementary Figs. 2 and 3).

Detailed DNA methylation map of the CHRNE gene. a (i) Boxplot and dot-plot of DNA methylation for individual neonatal control and ART samples at the two probes within the CHRNE gene that showed a significant difference between groups (adjusted p-value <0.05 (Bayesian levene’s test)). n = 207 biologically independent birth samples, n = 233 biologically independent adult samples. (ii) Boxplot and dot-plot of the same two probes in individual adult control and ART samples. The change is no longer significant in adult samples after correction for multiple testing, but the direction of methylation change persists. Boxplot elements are: center line-median; box limits-upper (Q3) and lower (Q1) quartiles; whiskers–smallest and largest non-outlier; points-outliers. b. Map of the CHRNE gene in hg19, showing EPIC probe locations. c Mean DNA methylation level at CHRNE for neonatal and adult non-ART and ART groups. Error bars are 95% confidence intervals. DMR1 is split into two: DMR1a that shows both ART and age specific DNA methylation differences and DMR1b that only shows ART-specific DNA methylation change. In addition, an age-specific DMR (DMR2) and a DMP are highlighted
Initial identification of DMPs associated with ART revealed two probes in close proximity to each other within the CHRNE gene, displaying a loss of methylation at genome-wide significance at birth (cg10553748, Δβ –0.063; cg24768135, Δβ –0.11), (Fig. 3a(i)). The same DMPs showed a slightly reduced methylation difference in adulthood (Δβ –0.053 and −0.062, respectively) and did not reach FDR significance (Fig. 3a(ii)). DMR analysis revealed a regional loss of methylation in association with ART, encompassing 7 probes in total (DMR1) and spanning approximately 1.89 kb. Average loss of methylation in the ART group at probes within this DMR was Δβ −0.053 and −0.056 in neonatal and adult blood, respectively.
Evidence of differential methylation in association with increasing age was also apparent within CHRNE, both at the DMP and DMR level (Fig. 3b, c). A sub-region of the ART-associated DMR1 (DMR1a), showed evidence of higher methylation in adulthood relative to infancy (Δβ = 0.108, p = 3.4 × 10−24 (Bayesian levene’s test) for non-ART neonatal vs. non-ART adult), with a reduction of methylation in ART relative to non-ART at both ages (p = 0.003 and p = 2.47 × 10−10 (Bayesian levene’s test)), whereas a second region of the same DMR (DMR1b) was differentially methylated specifically in association with ART. In addition, within CHRNE, an age-specific (aDMR, DMR2) encompassing 5 probes, and a single aDMP (cg20814095), showed an-age specific methylation difference not sensitive to ART at either age (Fig. 3c).
Independent replication of ART-associated differential methylation in infancy
In order to test our ART-associated specific differential methylation in an unrelated cohort, we analyzed a dataset previously published by Estill et al. that was acquired using the Illumina Infinium HumanMethylation450K array (from now on referred to as the ‘450K dataset’) (GSE79257) generated from 94 ART and 43 non-ART neonatal blood spots37. The disadvantage of using the 450K array for validation is that it does not include all EPIC probes and therefore generates a lower resolution picture of DNA methylation profile. Despite this, there is a strong correlation between methylation values generated by the 450K and EPIC arrays, which allows us to confirm a subset of our findings38. Of the 136 ART-associated DMPs we identified in neonatal blood on the EPIC array, data for 50 probes were also present in the 450K dataset, of which 14 (28%) showed evidence of differential methylation in association with ART (6 of 50 probes at p-value < 0.05 and a further 8 at p-value < 0.10 (Bayesian levene’s test); Supplementary Data 5). Further examination of methylation of probes from the 3 strongest DMRs in our dataset (Fig. 4a), also covered by probes in the 450K dataset, revealed a replication of the CHRNE and PRSS16 ART-associated DMRs, both in terms of direction of effect and magnitude of difference, with little supporting data obtained for TMEM18 (Fig. 4b, Supplementary Data 6).

Validation of ART-associated differentially methylated regions in an unrelated cohort. a. Summary of three ART-DMRs that are detected in both neonatal and adult blood. b Scatterplot of individual probes within the three DMRs at CHRNE, PRSS16 and TMEM18 genes, with mean Δβ for neonatal blood in our cohort (CHART) shown on y axis and mean Δβ for neonatal blood in the 450K cohort shown on x axis. DMRs at CHRNE and PRSS16 correlate well between the two studies. c Map of the CHRNE gene in hg19, showing EPIC probe locations and mean DNA methylation level at the ART-DMR in the 450K study. d Map of the PRSS16 gene in hg19, showing EPIC probe locations and mean DNA methylation level at the ART-DMR in the 4 study. Error bars are 95% CI. n = 133 biologically independent birth samples
The samples used in the 450K dataset were separated into three groups: IVF with fresh embryo, IVF with thawed frozen embryo, and intra uterine insemination (IUI) (i.e., no IVF or embryo culturing)37. A change in methylation at the CHRNE and PRSS16 DMRs was observed in all ART sub-types (Fig. 4c, d), suggesting that ART-associated effects are not associated with embryo culture specifically, but with other steps in the ART process, or potentially infertility in general. Conversely, an exploration of the top differentially methylated region in the 450K dataset, (SPATC1L) in our dataset revealed some supporting evidence of an ART-associated DMR. Previous analysis identified two DMRs in SPATC1L, one at the promoter (7 probes with mean Δβ 0.088) and one in the gene body (6 probes with mean Δβ −0.14) (Supplementary Fig. 4A). We only observed a difference between the non-ART and ART groups in our data at the promoter DMR, with a mean change in DNA methylation of −0.03 Δβ (Supplementary Fig. 4B), which is the same direction, but significantly smaller, compared with the change previously reported.
Subtypes of ART associate with specific differential methylation in neonates
The identification of differential methylation in CHRNE in association with IUI, without embryo culture, in the 450K dataset prompted us to explore the potential for different stages of ART processes to induce differential methylation in our CHART cohort. We categorized ART into three groups (i) ovarian stimulation only (gamete intrafallopian transfer, GIFT, n = 35), (ii) IVF with fresh embryos (n = 75) and (iii) IVF with thawed frozen embryos (n = 30) (Supplementary Fig. 5A). Plotting of differential methylation between groups confirmed a loss of methylation in CHRNE in all ART groups, including GIFT (no embryo culturing) (Supplementary Fig. 5B). Overall, there was no evidence that a subgroup was associated with a larger change in DNA methylation, though frozen IVF had a slightly lower median change in methylation relative to non-ART individuals (Supplementary Fig. 5C). Finally, we were interested to specifically compare culture (all IVF n = 105) and no-culture conditions (GIFT), but found no significant differences between these two subgroups (Supplementary Fig. 5D).
Imprinted regions show limited evidence of differential methylation in association with ART
Previous studies have demonstrated a relationship between ART conception and rare imprinted disorders39, associated with aberrant DNA methylation (summarized in a systemic review and meta-analysis)16. In addition, several studies have reported locus specific variation in imprinting associated regions in various tissues in association with ART conception, including in placenta40,41 and cord blood40. We carried out a focussed analysis of 706 EPIC array probes that are located within 50 DMRs previously identified as being associated with imprinting42 in our longitudinal dataset (Fig. 5a; Supplementary Data 7). In our original EWAS, only 2 imprinting associated probes showed evidence of ART-associated differential methylation (at birth), with a Δβ of 0.025 (cg12054318; adjusted p-value = 0.037) and Δβ of 0.027 (cg26104781; adjusted p-value = 0.0483). Nevertheless, we examined whether there was any evidence for enrichment of imprinted regions within the larger set of probes showing unadjusted p-value <0.05 (Bayesian levene’s test), relative to non-imprinted regions. Whereas approximately 9% of all 722,000 probes showed some evidence of differential methylation in neonatal blood using this relaxed threshold, only 4% of imprinting-associated probes fell into this category (Fig. 5b). There was similarly no evidence of enrichment for imprinted regions in the ART-associated differential methylation results in adult blood (Fig. 5b). Where differential methylation at imprinting-associated probes was observed between ART and non-ART groups, the magnitude of difference was very modest (Δβ under 5% in all instances; Fig. 5c). Nevertheless, several imprinted regions showed weak evidence of coordinated gain or loss of methylation in DMRs at birth in association with ART, though in most instances this was not apparent in adulthood (Fig. 5d). For example, 4 DMPs associated with KCNQ1QT1 imprinting showed higher methylation in ART vs. non-ART blood, specifically in the neonatal period (Fig. 5d). These probes are adjacent and are the only ones in the broader KCNQ1QT1 locus to show ART-associated differential methylation (Supplementary Fig. 6). In contrast, a single DMP in the IGF1R and two in the NAP1L5 locus showed slightly greater methylation differences in adult blood between ART and non-ART groups relative to neonatal blood (Fig. 5d).

No evidence for ART-associated DNA methylation change at imprinted genes. a Strategy for analysis of DNA methylation at imprinted gene regions. A total of 706 probes mapped within 50 imprinted regions. b Column plot showing the percentage of all probes and probes at imprinted regions showing a difference between ART and non-ART groups at an unadjusted p-value <0.05 (Bayesian levene’s test). This analysis shows that imprinted regions are less likely to have an ART-associated DNA methylation change than the average gene region. c Correlation plot of mean DNA methylation change between ART and non-ART in neonatal (x-axis) and adult (y-axis) blood at all imprinted probes. This shows that no probes show a Δβ ≥ 4% in adults, while only two probes meets this criteria in neonatal blood. d Bar plot of neonatal (red) and adult (blue) blood Δβ values for top ranked imprinted probes based on methylation change in neonatal or adult blood, with name of nearest gene
No evidence for repeat-based ‘global’ methylation change in association with ART
One of the most common proxy approaches to assess global DNA methylation is to focus on highly repetitive Alu and LINE1 elements that comprise 11% and 17% of the human genome, respectively (Fig. 6a)43. Taking a mean value of methylation of probes in these elements provides a summary measure of ‘global’ methylation. The EPIC array platform contains >23,000 probes with homology to Alu and >29,000 to LINE143. When the combined mean of these probes was compared across ART and non-ART groups, there was clear evidence of a gain in methylation with age (Fig. 6b, d), consistent with previous findings in buccal cells from birth to 7 years of age44 but no evidence of a significant effect of ART at either age, despite a slightly higher median methylation level in both elements in neonatal blood (Fig. 6b, d). To further explore the potential for large scale altered genomic methylation of small magnitude, we compared the genome-wide average methylation level (GWAM45) of all 722,000 probes between ART and non-ART offspring at both ages (Fig. 6c). Unlike for Alu and LINE1, there was weak evidence for an effect of ART conception affecting this measure in adults (Δβ = 0.003, p = 0.002 (Student’s t-test)) (Fig. 6c, d).

No evidence for ART-associated DNA methylation change at repetitive elements. a Global DNA methylation was assessed by grouping EPIC probes into Alu or LINE1 repetitive element regions using the REMP tool. b Boxplot of mean DNA methylation level of all probes at Alu and LINE1 in neonatal and adult blood. An age-effect is observed, but no significant differences between control and ART groups are detected. Boxplot elements are: center line-median; box limits-upper (Q3) and lower (Q1) quartiles; whiskers–smallest and largest non-outlier; points-outliers. c. Mean DNA methylation across all 722,301 probes in neonatal and adult blood. d P-values based on mean Alu, LINE1 and all probe DNA methylation for each group, using a Student’s t-test
Discussion
We performed a longitudinal analysis of DNA methylation profile in whole blood from early infancy to adulthood in a cohort of individuals conceived by ART and compared findings with non-ART conceived individuals. We found compelling evidence for specific ART-associated methylation variation around birth, some of which replicated in an independent cohort, with less evidence for persistence of differential methylation into adulthood. We found no evidence for an association between ART conception and DNA methylation at imprinting-associated regions, nor measures of global methylation relative to non-ART conception at either birth or adulthood. These findings demonstrate consistent ART-associated epigenetic variation by genome-wide analysis across two independent cohorts.
Given that the reported increase in rare imprinting disorders following ART conception is associated with variation in DNA methylation16, it is logical that other genomic regions may also be sensitive to epigenetic disruption following ART. Several studies have directly tested this hypothesis using a combination of locus-fspecific, global and genome-wide approaches, with some finding no evidence of ART-associated epigenetic variation40,46, while others report evidence of associations across different tissues and time-points (discussed below). Studies to date have generally been heterogeneous in design, have focussed on different tissues, ages, or genomic regions, used a variety of measurement approaches and have small sample sizes—all factors that likely contribute to the lack of replication of findings.
The effects of ART on DNA methylation have been directly tested in cultured human embryos with ART-associated aberrant methylation found at imprinting regions, including H19/IGF247. No ART-associated DNA methylation change at imprinted regions was detected in neonatal blood spots in our analysis, suggesting that either the original studies reporting imprinting changes were underpowered, or that this DNA methylation signature is lost by birth. Other studies have similarly focused on imprinting regions, as well as specific genes, or genome-wide analyses in samples collected at birth or in childhood, revealing differential methylation in human and mouse placenta48 and neonatal blood37,49,50,51. Such studies suggest that ART-induced epigenetic variation may be stable throughout pregnancy52 and potentially even childhood41. It is important to note that, although we found no evidence for a specific effect of ART conception on imprinting associated DNA methylation, or on a commonly used proxy measure of global DNA methylation, our analysis cannot conclusively exclude that such effects may be revealed using alternative approaches.
The potential for ART to affect ‘global’ methylation has also been assessed. This is generally tested using proxy measures such as repeat-based methylation at LINE1 and/or Alu elements, or average methylation across thousands of individual probes within these elements. Using the latter approach, we found no evidence of a difference in ‘global’ DNA methylation in blood of ART and non-ART conception groups at birth or adulthood, despite clear evidence of an age effect (increasing with age). Our findings are in keeping with a previous analysis of first trimester chorionic villous tissue, where no evidence of an effect of ART on global methylation was found53. However, others have reported variation in LINE1 and/or Alu elements in blood54 and/or placenta of ART offspring32,55, with ART generally associated with lower methylation at these regions. Other comparable studies found no differences in LINE1 methylation in either tissue56.
Few studies to date have implicated different aspects of ART procedures, rather than underlying infertility, in inducing epigenetic variation in the embryo, including ovarian stimulation57 and embryo culturing58. For example, the specific use of ICSI has been linked to a higher SNRPN gene methylation relative to spontaneous or IVF conception44. Specific components within culture media may contribute to altered epigenetic status59. A recent study of genome wide methylation of placentas from pregnancies conceived with IVF/ICSI showed distinct epigenetic profiles relative to those conceived with less invasive procedures (ovulation induction, intrauterine insemination)60.
The specific DMR within CHRNE that appears sensitive to ART-procedures has recently been demonstrated to show parent of origin allele-specific differential methylation. Specifically, analysis of 250 adult blood methylomes and more than 1100 transcriptomes identified significantly higher methylation on the maternally inherited allele with evidence for inter-individual variation associated with a specific genetic variant (methylation quantitative trait loci)61. Furthermore, the nearby age-associated DMR2 we identified is consistent with previous data in children (aged zero to 5 years) that showed a loss of methylation at this region in infancy62. In combination, these data suggest a complex interplay of age, genetic, sex specific and environment in regulating CHRNE gene methylation, the functional consequences of which will require further investigation.
An interesting observation is that the CHRNE DMR at birth in both cohorts, was present in both IVF (with embryo culturing) and those who underwent IUI and GIFT procedures in the absence of culturing. This implies an effect of the ovarian stimulation or subfertility itself, rather than any of the additional embryo culturing processes associated with IVF. A similar direct effects of ovarian stimulation on offspring epigenetic profile have been reported for maternally imprinted regions63,64. and for LINE1 methylation, which was decreased in association with high-dose hormone treatment65.
Mounting evidence links epigenetic variation, primarily differential DNA methylation, to a range of human phenotypes and health conditions66. Despite this, the relevance of the ART-associated methylation variation at birth described here remains unclear, particularly as we recently reported no evidence of adverse health outcomes in the same population of ART conceived individuals following extensive phenotypic examination in adulthood36.
There are several unique strengths of the current study. These include (i) the longitudinal analysis of blood collected soon after birth and in adulthood, (ii) the relatively large sample size compared with previous similar studies, (iii) the availability of information about the type of ART procedure employed, that allowed the effects of ovarian stimulation and culturing to be assessed and, (iv) the independent replication of ART-associated epigenetic variation in a previously published cohort. Limitations include (i) the lack of any functional assessment of the impact of the small ART-associated DNA methylation variation (5–12.9% difference) on gene expression, (ii) an inability to directly assess the effects of ovarian stimulation as a contributor to the identified epigenetic variants, (iii) limited data on other pregnancy and postnatal exposures that may affect DNA methylation of ART (e.g., medications, smoking and alcohol consumption), (iv) the use of de-convoluted whole blood for analysis does not allow us to make any comment about changes in specific blood cell types, (v) the possibility that adult participants (ART and non-ART) were self-selected as a comparatively healthy cohort and therefore less resolution of DNA methylation may be more evident in a less healthy cohort, and (vi) the number of samples, while being the largest of its type, is small relative to contemporary EWAS studies in other fields, which limits our power to detect associations between DNA methylation and specific ART procedures.
In summary, ART conception is associated with limited epigenetic variation at birth that largely attenuates by adulthood. The epigenetic variation may be associated in part with ovarian stimulation, or infertility per se. Additional studies of larger sample size in both animal models and humans are required in order to replicate our findings. Even if the transient epigenetic changes associated with ART are replicated, the potential health implications should not be over-interpreted given the absence of any direct evidence for downstream functional consequences of the observed epigenetic change, and the lack of compelling evidence for altered health outcomes in adulthood.
Methods
Participants
The protocol and details of measurements in the ‘Clinical review of the Health of adults conceived following Assisted Reproductive Technologies’ (CHART) study have been published previously67. A total of 193 ART-conceived and 86 non-ART-conceived adults gave informed consent to a have a detailed phenotypic analysis, including providing a venous blood sample and researcher access to previously collected neonatal blood spots for DNA isolation and epigenetic analysis35,68. Amongst the ART participants there were 147 IVF, 43 GIFT, and 3 with an unknown type of ART36. Matched data for both time points were available for 131 ART and 55 non-ART individuals. The study was approved by The Royal Children’s Hospital Human Research Ethics Committee (RCH HREC Project 33163).
Blood collection
Up to 9 mL of peripheral whole blood was collected from ART and non-ART adults in Sarstedt EDTA tubes. Blood tubes were spun at 500 × g for 10 min at 20 °C with no brake and full acceleration, and six 0.5 mL plasma aliquots were taken for storage. The remaining buffy coat layer was collected and mixed with Fetal Bovine Serum (FBS), 10% Dimethyl Sulfoxide (DMSO) and EDTA. The samples were aliquoted in a volume of 500 µL into barcoded cryotubes. Tubes were frozen at a controlled rate (decrease of 1 °C/min) and stored in the vapor phase of a liquid nitrogen tank until thawed for DNA extraction.
Guthrie spot retrieval
Neonatal blood spots (Guthrie spots) were prepared between 48 to 72 h post birth with parental informed consent and stored at room temperature. HREC approval (RCH HREC Project 33163) was obtained and informed consent from participants was sought to retrieve the Guthrie spots from New Born Screening (NBS) at Victorian Clinical Genetics Services (VCGS) of Murdoch Children’s Research Institute (MCRI). Approximately nine 3 mm punches were obtained from each card.
DNA isolation and quality control
Whole 3 mm diameter Guthrie spots were lysed with proteinase K (Bioline Cat. No. BIO-37037) overnight then macerated with beads using the Qiagen TissueLyser II at frequency 30 for 40 s to separate blood from filter paper69. DNA was extracted using the Zymo Research ZR DNA-Card Extraction Kit (Cat. No. D6040), according to the manufacturer’s protocol. Adult buffy coats were lysed with proteinase K for 2 h and the DNA was extracted using the Qiagen QIAamp® DNA Mini spin kit (Ref 56304). DNA was quantified using Nanodrop and quality was checked using gel electrophoresis69.
DNA methylation profiling
Genomic DNA (200 to 500 ng) from Guthrie spots and whole adult blood were randomized into 96-well plates and sent to GenomeScan (Netherlands) for sodium bisulfite treatment and genome-wide methylation analysis using Illumina InfiniumMethylationEPIC BeadChips (referred to from now as ‘EPIC array’)70. The EPIC array measures DNA methylation level at more than 850,000 CpG sites (referred to as ‘EPIC probes’), and covers all gene promoters, gene bodies and ENCODE-assigned distal regulatory elements71. Raw IDAT files were received on a hard-disk from Service XS and used for data analysis.
Data cleaning, normalization, and statistical analysis
Raw IDAT files were processed and analyzed using the MissMethyl and minfi packages for R72,73, both available from Bioconductor74. Samples were checked for quality and those with a mean detection p-value of >0.01 were removed (5 neonatal and 4 adult samples), leaving 207 neonatal blood (n = 149 ART, n = 58 non-ART) and 233 whole adult blood (n = 158 ART, n = 75 non-ART) samples for analysis. Data were normalized for both within and between array technical variation using SWAN (Subset-quantile Within Array Normalization)75. Probes with poor average quality scores (detection p-value > 0.01), those associated with SNPs (MAF > 0%) and cross-reactive probes71 were removed from further analysis. This left a total of 724,897 probes for neonatal blood spot analysis and 766,247 probes for adult blood analysis, of which 722,301 probes were common to both datasets. Cell composition was determined using the estimateCellCounts tool, with the ‘CordBlood’ reference data used for neonatal blood spot analysis76 and the ‘Blood’ reference data used for adult peripheral blood analysis77. Differential methylation analysis by linear regression modeling was performed using limma78. Confounders and covariates were identified using principal component analysis (shown in Supplementary Fig. 1) and were incorporated in the analysis models as required. The final model incorporated the following covariates: Sex, EPIC array position (plate well and chip position) and cell composition proportions (CD8 T cells, CD4 T cells, B cells, Monocytes, Eosonophils, Neutrophils). Differentially methylated probes (DMPs) were those that showed an adjusted p-value of <0.05 (Benjamini Hochberg) and a change in methylation (delta beta or Δβ) of ≥5%. Differentially methylated regions (DMRs) were identified using the DMRcate tool79 and Bedtools were used to intersect DMRs with individual probes80. DMPs were assigned to the nearest gene within 1 megabase (1 Mb) using the GREAT tool81. DNA methylation at repetitive elements was calculated using the REMP (Repetitive Element Methylation Prediction) package43.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data sets generated and analyzed for the current study are deposited in the Gene Expression Omnibus repository with the accession number GSE131433.
References
Adamson, G. D. et al. International committee for monitoring assisted reproductive technology: world report on assisted reproductive technology, 2011. Fertil. Steril. 110, 1067–1080 (2018).
Asch, R. H., Ellsworth, L. R., Balmaceda, J. P. & Wong, P. C. Pregnancy after translaparoscopic gamete intrafallopian transfer. Lancet 2, 1034–1035 (1984).
Farquhar, C. & Marjoribanks, J. Assisted reproductive technology: an overview of cochrane reviews. Cochrane Database Syst. Rev. 8, CD010537 (2018).
Kushnir, V. A., Barad, D. H., Albertini, D. F., Darmon, S. K. & Gleicher, N. Systematic review of worldwide trends in assisted reproductive technology 2004–2013. Reprod. Biol. Endocrinol. 15, 6 (2017).
Roseboom, T. J. Developmental plasticity and its relevance to assisted human reproduction. Hum. Reprod. 33, 546–552 (2018).
Fleming, T. P. et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet 391, 1842–1852 (2018).
Helmerhorst, F. M., Perquin, D. A., Donker, D. & Keirse, M. J. Perinatal outcome of singletons and twins after assisted conception: a systematic review of controlled studies. BMJ 328, 261 (2004).
Kalra, S. K. & Barnhart, K. T. In vitro fertilization and adverse childhood outcomes: what we know, where we are going, and how we will get there. A glimpse into what lies behind and beckons ahead. Fertil. Steril. 95, 1887–1889 (2011).
Pandey, S., Shetty, A., Hamilton, M., Bhattacharya, S. & Maheshwari, A. Obstetric and perinatal outcomes in singleton pregnancies resulting from IVF/ICSI: a systematic review and meta-analysis. Hum. Reprod. Update 18, 485–503 (2012).
Levine, A. D., Boulet, S. L. & Kissin, D. M. Contribution of assisted reproductive technology to overall births by maternal age in the United States, 2012-2014. JAMA 317, 1272–1273 (2017).
European, I. V. F. m. C. et al. Assisted reproductive technology in Europe, 2013: results generated from European registers by ESHRE. Hum. Reprod. 32, 1957–1973 (2017).
ASRM. Latest Data from SART Show Increasing Use of Cryopreservation for Fertility Preservation. ASRM Press Release Bull. (2019).
Ceelen, M., van Weissenbruch, M. M., Vermeiden, J. P., van Leeuwen, F. E. & Delemarre-van de Waal, H. A. Cardiometabolic differences in children born after in vitro fertilization: follow-up study. J. Clin. Endocrinol. Metab. 93, 1682–1688 (2008).
Valenzuela-Alcaraz, B. et al. Assisted reproductive technologies are associated with cardiovascular remodeling in utero that persists postnatally. Circulation 128, 1442–1450 (2013).
Gkourogianni, A. et al. Plasma metabolomic profiling suggests early indications for predisposition to latent insulin resistance in children conceived by ICSI. PloS ONE 9, e94001 (2014).
Lazaraviciute, G., Kauser, M., Bhattacharya, S., Haggarty, P. & Bhattacharya, S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum. Reprod. Update 20, 840–852 (2014).
Carson, C. et al. Asthma in children born after infertility treatment: findings from the UK Millennium Cohort Study. Hum. Reprod. 28, 471–479 (2013).
Cetinkaya, F., Gelen, S. A., Kervancioglu, E. & Oral, E. Prevalence of asthma and other allergic diseases in children born after in vitro fertilisation. Allergol. Immunopathol. 37, 11–13 (2009).
Ericson, A., Nygren, K. G., Olausson, P. O. & Kallen, B. Hospital care utilization of infants born after IVF. Hum. Reprod. 17, 929–932 (2002).
Guo, X. Y. et al. Cardiovascular and metabolic profiles of offspring conceived by assisted reproductive technologies: a systematic review and meta-analysis. Fertil. Steril. 107, 622–631 e625 (2017).
Hart, R. & Norman, R. J. The longer-term health outcomes for children born as a result of IVF treatment: Part I–General health outcomes. Hum. Reprod. Update 19, 232–243 (2013).
Kallen, B., Finnstrom, O., Nygren, K. G. & Otterblad Olausson, P. Asthma in Swedish children conceived by in vitro fertilisation. Arch. Dis. Child 98, 92–96 (2013).
Kuiper, D. B. et al. Asthma and asthma medication use among 4-year-old offspring of subfertile couples–association with IVF? Reprod. Biomed. Online 31, 711–714 (2015).
Sicignano, N., Beydoun, H. A., Russell, H., Jones, H. Jr. & Oehninger, S. A descriptive study of asthma in young adults conceived by IVF. Reprod. Biomed. Online 21, 812–818 (2010).
Vrooman, L. A. & Bartolomei, M. S. Can assisted reproductive technologies cause adult-onset disease? Evidence from human and mouse. Reprod. Toxicol. 68, 72–84 (2017).
Cantone, I. & Fisher, A. G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 20, 282–289 (2013).
van Montfoort, A. P. et al. Assisted reproduction treatment and epigenetic inheritance. Hum. Reprod. Update 18, 171–197 (2012).
La Bastide-Van Gemert, S. et al. Is ovarian hyperstimulation associated with higher blood pressure in 4-year-old IVF offspring? Part II: an explorative causal inference approach. Hum. Reprod. 29, 510–517 (2014).
Maheshwari, A., Pandey, S., Shetty, A., Hamilton, M. & Bhattacharya, S. Obstetric and perinatal outcomes in singleton pregnancies resulting from the transfer of frozen thawed versus fresh embryos generated through in vitro fertilization treatment: a systematic review and meta-analysis. Fertil. Steril. 98, 368–377, e361-369 (2012).
Healy, D. L. et al. Prevalence and risk factors for obstetric haemorrhage in 6730 singleton births after assisted reproductive technology in Victoria Australia. Hum. Reprod. 25, 265–274 (2010).
Camprubi, C. et al. Stability of genomic imprinting and gestational-age dynamic methylation in complicated pregnancies conceived following assisted reproductive technologies. Biol. Reprod. 89, 50 (2013).
Choux, C. et al. The epigenetic control of transposable elements and imprinted genes in newborns is affected by the mode of conception: ART versus spontaneous conception without underlying infertility. Hum. Reprod. 33, 331–340 (2018).
Tobi, E. W. et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 5, 5592 (2014).
Kelstrup, L. et al. Gene expression and DNA methylation of PPARGC1A in muscle and adipose tissue from adult offspring of women with diabetes in pregnancy. Diabetes 65, 2900–2910 (2016).
Halliday, J. et al. Comparing indicators of health and development of singleton young adults conceived with and without assisted reproductive technology. Fertil. Steril. 101, 1055–1063 (2014).
Halliday, J. et al. Health of 22–35 year olds conceived by Assisted Reproductive Technology. Fertil. Steril. 112, 130–139, https://doi.org/10.1016/j.fertnstert.2019.03.001 (2019).
Estill, M. S. et al. Assisted reproductive technology alters deoxyribonucleic acid methylation profiles in bloodspots of newborn infants. Fertil. Steril. 106, 629–639 e610 (2016).
Solomon, O. et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics 13, 655–664 (2018).
Halliday, J., Oke, K., Breheny, S., Algar, E. & D, J. A. Beckwith-Wiedemann syndrome and IVF: a case-control study. Am. J. Hum. Genet 75, 526–528 (2004).
Rancourt, R. C., Harris, H. R. & Michels, K. B. Methylation levels at imprinting control regions are not altered with ovulation induction or in vitro fertilization in a birth cohort. Hum. Reprod. 27, 2208–2216 (2012).
Turan, N. et al. Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet. 6, e1001033 (2010).
Hernandez Mora, J. R. et al. Characterization of parent-of-origin methylation using the Illumina Infinium MethylationEPIC array platform. Epigenomics 10, 941–954 (2018).
Zheng, Y. et al. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. 45, 8697–8711 (2017).
Whitelaw, N. et al. Epigenetic status in the offspring of spontaneous and assisted conception. Hum. Reprod. 29, 1452–1458 (2014).
Li, S. et al. Genome-wide average DNA methylation is determined in utero. Int. J. Epidemiol. https://doi.org/10.1093/ije/dyy028 (2018).
Puumala, S. E. et al. Similar DNA methylation levels in specific imprinting control regions in children conceived with and without assisted reproductive technology: a cross-sectional study. BMC Pediat. 12, 33 (2012).
Chen, S. L., Shi, X. Y., Zheng, H. Y., Wu, F. R. & Luo, C. Aberrant DNA methylation of imprinted H19 gene in human preimplantation embryos. Fertil. Steril. 94, 2356–2358, 2358 e2351 (2010).
de Waal, E. et al. In vitro culture increases the frequency of stochastic epigenetic errors at imprinted genes in placental tissues from mouse concepti produced through assisted reproductive technologies. Biol. Reprod. 90, 22 (2014).
Katari, S. et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum. Mol. Genet. 18, 3769–3778 (2009).
Melamed, N., Choufani, S., Wilkins-Haug, L. E., Koren, G. & Weksberg, R. Comparison of genome-wide and gene-specific DNA methylation between ART and naturally conceived pregnancies. Epigenetics 10, 474–483 (2015).
Castillo-Fernandez, J. E. et al. DNA methylation changes at infertility genes in newborn twins conceived by in vitro fertilisation. Genome Med. 9, 28 (2017).
Lou, H. et al. Assisted reproductive technologies impair the expression and methylation of insulin-induced gene 1 and sterol regulatory element-binding factor 1 in the fetus and placenta. Fertil. Steril. 101, 974–980 e972 (2014).
Xu, N. et al. Comparison of genome-wide and gene-specific DNA methylation profiling in first-trimester chorionic villi from pregnancies conceived with infertility treatments. Reprod. Sci. 24, 996–1004 (2017).
Loke, Y. J., Galati, J. C., Saffery, R. & Craig, J. M. Association of in vitro fertilization with global and IGF2/H19 methylation variation in newborn twins. J. Dev. Orig. Health Dis. 6, 115–124 (2015).
Ghosh, J., Coutifaris, C., Sapienza, C. & Mainigi, M. Global DNA methylation levels are altered by modifiable clinical manipulations in assisted reproductive technologies. Clin. Epigenetics 9, 14 (2017).
Vincent, R. N., Gooding, L. D., Louie, K., Chan Wong, E. & Ma, S. Altered DNA methylation and expression of PLAGL1 in cord blood from assisted reproductive technology pregnancies compared with natural conceptions. Fertil. Steril. 106, 739–748 e733 (2016).
Song, S. et al. DNA methylation differences between in vitro- and in vivo-conceived children are associated with ART procedures rather than infertility. Clin. Epigenet. 7, 41 (2015).
Rivera, R. M. et al. Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 9.5 of development. Hum. Mol. Genet. 17, 1–14 (2008).
Khosla, S., Dean, W., Brown, D., Reik, W. & Feil, R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol. Reprod. 64, 918–926 (2001).
Choufani, S. et al. Impact of assisted reproduction, infertility, sex and paternal factors on the placental DNA methylome. Hum. Mol. Genet. 28, 372–385 (2019).
Zink, F. et al. Insights into imprinting from parent-of-origin phased methylomes and transcriptomes. Nat. Genet. 50, 1542–1552 (2018).
Martino, D. J. et al. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics 6, 1085–1094 (2011).
Sato, A., Otsu, E., Negishi, H., Utsunomiya, T. & Arima, T. Aberrant DNA methylation of imprinted loci in superovulated oocytes. Hum. Reprod. 22, 26–35 (2007).
Velker, B. A. M., Denomme, M. M., Krafty, R. T. & Mann, M. R. W. Maintenance of Mest imprinted methylation in blastocyst-stage mouse embryos is less stable than other imprinted loci following superovulation or embryo culture. Environ. Epigenet. 3, dvx015 (2017).
Liang, X. W. et al. Superovulation induces defective methylation in line-1 retrotransposon elements in blastocyst. Reprod. Biol. Endocrinol. 11, 69 (2013).
Lappalainen, T. & Greally, J. M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451 (2017).
Lewis, S. et al. Clinical review of 24-35 year olds conceived with and without in vitro fertilization: study protocol. Reprod. Health 14, 117 (2017).
Wilson, C. et al. Health and development of ART conceived young adults: a study protocol for the follow-up of a cohort. Reprod. Health 10, 15 (2013).
Joo, J. E. et al. The use of DNA from archival dried blood spots with the Infinium HumanMethylation450 array. BMC Biotechnol. 13, 23 (2013).
Martino, D. et al. Epigenetic dysregulation of naive CD4+ T-cell activation genes in childhood food allergy. Nat. Commun. 9, 3308 (2018).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208 (2016).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Aryee, M. J. et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Bakulski, K. M. et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 11, 354–362 (2016).
Houseman, E. A., Molitor, J. & Marsit, C. J. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 30, 1431–1439 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 8, 6 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Acknowledgements
The authors acknowledge the participants who generously gave their time to the study. This work was made possible through the Victorian State Government Operational Infrastructure Support and the Australian Government NHMRC IRIISS. This study was funded by a National Health and Medical Research Council Project Grant (APP1099641; 2016–2017), The Royal Children’s Hospital Research Foundation, Monash IVF Research and Education Foundation, and Reproductive Biology Unit Sperm Fund, Melbourne IVF. B.N. is supported by an NHMRC (Australia) CJ Martin Fellowship (1072966). DPB is supported by an NHMRC Senior Research Fellowship (1064629).
Author information
Authors and Affiliations
Contributions
R.S. and J.H. conceived and designed this study. B.N. performed differential DNA methylation analysis. B.N. and A.S. performed quality control of DNA methylation data. S.L. and J.K. collected and stored biological samples from the CHART cohort. A.C. and B.K. performed molecular biology sample processing. J.H., D.P.B., M.J., K.H., D.J.A., L.W.D., S.R., L.W., M.C., J.M. and R.M. were involved in subject recruitment, data collection, and clinical phenotyping of the CHART cohort. R.S. supervised the study. B.N. and R.S. drafted the paper, which was reviewed by all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks David Albertini, Richard David Leslie and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Novakovic, B., Lewis, S., Halliday, J. et al. Assisted reproductive technologies are associated with limited epigenetic variation at birth that largely resolves by adulthood. Nat Commun 10, 3922 (2019). https://doi.org/10.1038/s41467-019-11929-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11929-9
This article is cited by
-
Frequency, morbidity and equity — the case for increased research on male fertility
Nature Reviews Urology (2024)
-
Association of chorioamnionitis with infertility treatment and subsequent neonatal outcomes in the US: a population-based cohort study
BMC Pregnancy and Childbirth (2023)
-
Genome-wide assessment of DNA methylation alterations induced by superovulation, sexual immaturity and in vitro follicle growth in mouse blastocysts
Clinical Epigenetics (2023)
-
The X-factor in ART: does the use of assisted reproductive technologies influence DNA methylation on the X chromosome?
Human Genomics (2023)
-
Cryopreservation effect on DNA methylation profile in rainbow trout spermatozoa
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
