
Abstract
Rearrangement reactions have attracted considerable interest over the past decades due to their high bond-forming efficiency and atom economy in the construction of complex organic architectures. In contrast to the well-established [3,3]-rearrangement, [1,3] O-to-C rearrangement has been far less vigorously investigated, and stereospecific [1,3]-rearrangement is extremely rare. Here, we report a metal-free intramolecular hydroalkoxylation/[1,3]-rearrangement, leading to the practical and atom-economical assembly of various valuable medium-sized lactams with wide substrate scope and excellent diastereoselectivity. Moreover, such an asymmetric cascade cyclization has also been realized by chiral Brønsted acid-catalyzed kinetic resolution. In addition, biological tests reveal that some of these medium-sized lactams displayed their bioactivity as antitumor agents against melanoma cells, esophageal cancer cells and breast cancer cells. A mechanistic rationale for the reaction is further supported by control experiments and theoretical calculations.
Similar content being viewed by others

Introduction
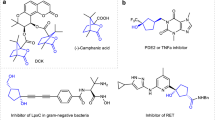
Eight-membered lactams, especially the benzo[d]azocinones, are prominent structural motifs that can be found in many natural products and bioactive molecules (Fig. 1)1,2,3,4,5. However, access to these heterocycles is challenging owing to unfavorable enthalpic and entropic barriers in transition states leading to medium-sized rings6,7,8,9,10. To date, only very limited methods have been developed, and most of them rely on noble-metal catalysis11,12,13,14,15,16,17,18,19. To this end, the development of new methods for the efficient construction of this skeleton is highly desirable, especially those with high diastereo- and enantioselectivity.

Benzo[d]azocinones in natural products and bioactive molecules. Some of representative molecules are listed
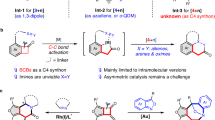
Rearrangement reactions have attracted considerable interest over the past decades due to their high bond-forming efficiency and atom economy in the construction of complex organic architectures20,21. In contrast to the well-established [3,3]-rearrangement22,23,24,25, which generally proceeds via the chair-like transition state and thus is stereospecific (Figs. 2a), [1,3] O-to-C rearrangement has been far less vigorously investigated, and stereospecific [1,3]-rearrangement is highly challenging due to the formation of presumable zwitterion pairs (Fig. 2b)26,27. Although several Lewis acid-mediated and thermal [1,3]-rearrangements that relay stereochemical information have been reported28,29, transformation in these limited cases lacks generality and significant deterioration of enantiomeric excess is observed30,31.

[3,3]-Rearrangement vs. [1,3]-rearrangement. a Typical [3,3]-rearrangement. b Typical [1,3]-rearrangement. c This work: Brønsted acid-catalyzed hydroalkoxylation/stereospecific [1,3]-rearrangement
Recently, great progress of transition metal-catalyzed intramolecular alkoxylation-initiated [1,3]-rearrangement has evoked a new round of exploration on the [1,3]-rearrangements, offering great potential to build structurally complex cyclic molecules, as elegantly established by Toste, Rhee, Hashmi, Liu, Davies, and Zhu31,32,33,34,35,36,37,38. Despite these impressive advances, these tandem reactions are limited to ether nucleophiles and rely on noble metals (Au/Pt) as the catalyst. Importantly, no direct catalytic asymmetric tandem reaction has been described to date39. Inspired by the above results and by our recent study on yttrium-catalyzed tandem intramolecular hydroalkoxylation/Claisen rearrangement40, we envisioned that the synthesis of eight-membered benzo[d]azocinones 2 might be accessed directly through catalytic intramolecular hydroalkoxylation/[1,3]-rearrangement of ynamides 141,42,43,44,45,46,47,48,49,50. Herein, we describe the realization of a metal-free tandem intramolecular hydroalkoxylation/[1,3]-rearrangement (Fig. 2c), and this method leads to the practical and atom-economical synthesis of various valuable medium-sized lactams with excellent diastereoselectivity. Moreover, such an asymmetric cascade cyclization has also been achieved via kinetic resolution by chiral spiro phosphoramide catalysis. Importantly, this [1,3]-rearrangement is highly stereospecific and proceeds with complete chirality transfer. Control experiments and density functional theory (DFT) calculations provide further evidence of the feasibility of the proposed mechanism.
Results
Screening of reaction conditions
At the outset, ynamide 1a was used as the model substrate to demonstrate our designed cascade cyclization, as shown in Table 1 (for more details see Supplementary Table 1). To our delight, the expected benzo[d]azocinone 2a was indeed formed with exclusive cis-diastereoselectivity (diastereoselectivity (d.r.) >50:1; determined by crude proton nuclear magnetic resonance (1H NMR)), albeit in low yields, in the presence of typical gold catalysts (Table 1, entries 1 and 2). Somewhat surprisingly, further investigations revealed that the reaction also proceeded in the presence of various non-noble metals (Table 1, entries 4–7), with Zn(OTf)2 giving the best yield of the desired product 2a (Table 1, entry 7). In addition, Brønsted acids such as TsOH and MsOH could also catalyze this cascade reaction to produce 2a in 47 and 66% yields, respectively, together with significant amounts of hydration product 2a′ in both cases (Table 1, entries 8 and 9). Although the use of 10 mol% of HOTf as catalyst failed to produce the desired 2a, probably because the high acidity led to decomposition of 1a (Table 1, entry 10), the reaction efficiency was substantially improved by decreasing the loading of catalyst (Table 1, entries 11–13). With a low catalyst loading of 0.5 mol%, HOTf efficiently catalyzed the formation of 2a in 96% yield (Table 1, entry 13). These results indicate that HOTf, which was released as a hidden Brønsted acid, is presumably the true catalytic species in the above Lewis acid catalysis51,52,53.
Reaction scope study
The reaction scope was then explored under the optimized reaction conditions (Fig. 3). This metal-free tandem reaction occurred efficiently with various benzyl alcohol-tethered ynamides 1, leading to the corresponding benzo[d]azocinones 2 in good to excellent yields. Importantly, excellent diastereoselectivity (>50:1) was achieved in all cases. Ynamides with different sulfonyl-protecting groups were first investigated, and the desired products 2a–2c were formed in 73–94% yields. In addition, ynamides bearing either electron-withdrawing or electron-donating substituents, such as F, Cl, Br, Me, OMe, or even CN and CF3 on the aromatic ring (R2 = Ar), were compatible with this cyclization to produce the expected 2d–2l in generally excellent yields. This cascade cyclization was also extended to the naphthalene, thiophene, and alkenyl-substituted ynamides, delivering the desired 2m (99%), 2n (81%), and 2o (94%), respectively. Various aryl-substituted ynamides with either electron-donating or electron-withdrawing groups were then screened, and the reaction afforded the desired products 2p–2ab in 73–98% yields. Of note, in some cases better yields could be achieved by employing Zn(OTf)2 (10 mol%) as catalyst and 5 Å molecular sieve (MS) as additive (2s and 2aa). Interestingly, alkyl-substituted ynamides (R1 or R2 = alkyl) were also suitable substrates, and were converted into the desired 2ac and 2ad in good yields, and 2ae in a serviceable yield; higher temperature was needed in these cases. The molecular structures of 2a and 2ac were confirmed by X-ray diffraction (for more details see Supplementary Tables 3 and 4).

Reaction scope for the formation of 3-benzazocinones 2. Reaction conditions: 1 (0.2 mmol), HOTf (0.001 mmol), PhCl (4 mL), 80 °C, 4 h, in vials; yields are those for the isolated products. a1 mol% of HOTf was used. bUsing 10 mol% of Zn(OTf)2 as catalyst and 5 Å molecular sieve (MS) as additive. c100 °C, 60 h. d100 °C, 4 h
Notably, this cascade cyclization was also extended to the allyl alcohol-tethered ynamides, and, importantly, no competing intramolecular hydroalkoxylation/[3,3]-rearrangement was observed40. As shown in Fig. 4a, the desired benzo[d]azocinones 2af–2ah were obtained in 61–76% yields, and significantly improved yield (86%) was achieved in case of ynamide 1ah by using Zn(OTf)2 as catalyst. In addition, the reaction proceeded smoothly to produce the expected 9-membered lactam 2ai in 46% yield, and, in this case, the use of Zn(OTf)2 as catalyst also gave significantly improved yield (Fig. 4b, 2ai was confirmed by X-ray diffraction, for more details see Supplementary Table 5). Moreover, it was found that other heterocycle-linked 8-membered ring lactams 2aj–2am could also be synthesized in 41−55% yields in the presence of 20 mol% of HNTf2 as catalyst (Fig. 4c). Attempts to extend the reaction to the terminal ynamide 1an only gave a complex mixture of products, and the reaction of ynamides 1ao and 1ap also failed to produce the desired products (for more details see Supplementary Figs. 118 and 119), indicating that the formation of a stable benzylic carbocation is a key requirement for subsequent [1,3]-rearrangement (Fig. 4d).

Catalytic hydroalkoxylation/[1,3]-rearrangement of other ynamides 1. a Brønsted acid-catalyzed hydroalkoxylation/[1,3]-rearrangement of allyl alcohol-tethered ynamides 1af–1ah. b Zinc-catalyzed cascade cyclization of ynamide 1ai. c HNTf2-catalyzed cascade cyclization of other heterocycle-linked ynamides 1aj–1am. d Ynamides 1an–1ap which failed to give the desired products
Screening of reaction conditions for kinetic resolution
We then considered the possibility of developing an asymmetric variant of this tandem sequence. Although no enantioselectivity was observed by the use of chiral metal catalysts, good enantioselectivity could be attained by employing chiral spiro phosphoramides as catalysts (for more details, see Supplementary Table 2 and Supplementary Fig. 120)54,55. Importantly, further studies revealed that the chiral induction was realized through kinetic resolution of racemic ynamides (for more details, see Supplementary Fig. 121). Initially, ynamide 1p, bearing an electron-donating methyl group on the aromatic ring moiety that should promote this cascade cyclization, was used as the model substrate. As shown in Table 2, the desired chiral benzo[d]azocinone 2p-ent was obtained in 42% yield with an enantiomeric ratio (e.r.) of 95:5 in the presence of chiral spiro phosphoramide Cat. 356,57,58,59, bearing two 6,6'-di(3,5-di-tert-butyl-4-methoxyphenyl) moieties (Table 2, entry 5). Interestingly, the use of the corresponding chiral binol-derived phosphoramide led to significantly decreased enantioselectivity (e.r. <60:40), indicating that the spirobiindane backbone of the phosphoramides plays a crucial role in the chiral induction step. It is notable that in this process one enantiomer ((R)-1p) favored formation of the desired chiral benzo[d]azocinone 2p-ent, while the other enantiomer ((S)-1p), which does not match with the Cat. 3, favored formation of the corresponding hydration product 2p' catalyzed by the acid (for more details, see Supplementary Figs. 122–124). Thus, it represents a rare example of parallel kinetic resolution60,61.
Scope of kinetic resolution of racemic ynamides 1
Preliminary investigations were carried out into the reaction scope by employing chiral spiro phosphoramide Cat. 3 as a catalyst (Fig. 5). Substrates with either electron-donating or electron-withdrawing groups on the aromatic ring moiety of the racemic ynamides 1 were well tolerated and resulted in 40–51% yields and good e.r. values. The absolute configuration of 2p-ent was determined by X-ray crystallographic analysis (for more details, see Supplementary Table 6). Of note, although 20 mol% catalyst loading was employed, probably because the acidity of the chiral catalyst is not high enough, the catalyst could be readily recovered and reused five times with almost unchanged enantioselectivity and reactivity (for more details, see Supplementary Fig. 125).

Reaction scope for kinetic resolution of racemic 1. Reaction conditions: 1 (0.1 mmol), Cat. 3 (0.02 mmol), Et2O (2 mL), 25 °C, 6–32 h, then Et3N (0.03 mmol), PhCl (1 mL), 60 °C, 24 h, in vials; yields are those for the isolated products; e.r.s are determined by high-performance liquid chromatography (HPLC) analysis on a chiral stationary phase
Synthetic applications and biological tests
To further demonstrate the potential utility of this reaction, we also carried out product derivatizations (Fig. 6). For example, the Ts group in benzo[d]azocinone 2a, prepared on a gram scale in 86% yield, was efficiently removed to form free amide 3a in 87% yield by the treatment with SmI2. 3a could be further methylated into the corresponding lactam 4a (89%) and oxidized into unsaturated lactam 5a (72%), respectively.

Gram-scale reaction and product elaboration. Gram-scale reaction of ynamide 1a and transformation of 2a into 3a, 4a, and 5a
Moreover, we tested the above-synthesized 3-benzazocinones for their bioactivity as antitumor agents. The cytotoxic effects of these compounds were evaluated against a panel of cancer cells, including melanoma cells A375, esophageal cancer cells SK-GT-4 and KYSE-450, and breast cancer cells MCF-7 and MDA-MB-231 using cell viability assay. Our preliminary studies showed that almost half of these compounds exerted significant cytotoxic effects on the A375, and a few compounds (2ac, 2ah, 2p-ent, and 3a) and compound 2am exerted cytotoxic effects on the SK-GT-4 and MCF-7 (for more details, see Supplementary Table 8), respectively, suggesting a potential application of these medium-sized lactams in medicinal chemistry.
Mechanistic investigations
We then turned our attention to mechanistic investigations (for more details, see Supplementary Figs. 126–129). First, it was found that no incorporation of 18O into the product 2a was observed when ynamide 1a was subjected to the reaction conditions with H218O, which indicates that the oxygen on the carbonyl group of 2a originates from the hydroxyl group of 1a (for more details, see Supplementary Fig. 126). In addition, hydration product 2a′ was not converted into 2a under the standard conditions, thus ruling out 2a′ as a possible intermediate (for more details, see Supplementary Fig. 127). Gratifyingly, the ketene aminal 6a (only the E isomer) could be isolated in 53% yield by quenching the reaction after 15 min (Fig. 7a). Importantly, 6a was readily converted into the desired 2a and complete chirality transfer was observed starting from chiral 6a (Fig. 7b, c). Furthermore, the acid catalyst did not work in this rearrangement process, indicating that it is an uncatalyzed thermal rearrangement (for more details, see Supplementary Fig. 128). These results strongly support that 6a is the key intermediate and stereospecific [1,3]-rearrangement is presumably involved in this tandem process.

Control experiments. a Cascade cyclization of ynamide 1a by quenching the reaction after 15 min. b Control experiments on the transformation of racemic 6a into racemic 2a. c Control experiments on the transformation of chiral 6a into chiral 2a
On the basis of the above observations, we propose a mechanism for the formation of benzo[d]azocinone 2a (Fig. 8). The reaction begins with the hydroxyl group attack of the HOTf-activated ynamide 1a to afford oxonium intermediate B presumably via a keteniminium intermediate A, thus further yielding thermodynamically stable vinyl ether intermediate 6a of E configuration and regenerating the acid catalyst. DFT calculations at B3LYP-D3/6-31G(d,p)//SMD-def2-TZVP level of theory62,63,64,65 (for more details, see Supplementary Figs. 111–113 and Supplementary Datasets 1–4) were introduced to understand the subsequent O-to-C rearrangement of (R)-6a, which is a stereospecific, rate-determining, and uncatalyzed thermal rearrangement, to produce the final cis product (R, S)-2a. Two major conformational isomers of initial compound 6a and 6a-iso can be located by calculations in terms of different Si- or Re-face at the α-carbon of amide substrate. In the following step of C-O bond cleavage, transition state TS_cis with bond breaking via Si-face side was 9.7 kcal/mol more stable than that via Re-face cleavage-formed TS_trans, suggesting the final 2a_cis from TS_cis is kinetically favorable and highly stereospecific. Furthermore, calculations indicate that the reaction mechanism is not a typical [1,3]-rearrangement (for more details, see Supplementary Figs. 114–117)66,67,68,69,70. We are able to locate the transition states of C-O bond cleavage, but failed to locate transition states of C-C bond formation, which indicate that the mechanism seems not to be stepwise. Quantitative Sensory Testing method was used and three levels of methods (B3LYP-D3, M062X, ωB97XD) were tried in our calculations. All of them give substantially similar kinetic and thermodynamic results (for more details, see Supplementary Figs. 111–117). However, more inspections for transition states imply that the developing negative charge on the α-carbon of amide substrate tends to be stabilized by aromatic rings forming formal [3,3]-rearrangement transition states. A regular spiro[5,5] product spiro-int is not accessible because of its high energy instability for 22.5 kcal/mol, compared to the energy of 2a-cis. Indeed, transition states of C-C bond formation are facile to the final product during our calculations. Based on energy profiles, the pathway to form favorable 2a-cis is kinetically irreversible and thermodynamically exothermic for 11.8 kcal/mol, which is in agreement with our experiment. When chiral Brønsted acid is employed, the resulting keteniminium intermediate A′ leads to chiral 6a via ion pairing and H-bonding interactions54,55, which undergoes stereospecific [1,3]-rearrangement to form 2a-ent with complete chirality transfer.

Mechanistic hypothesis. a Plausible catalytic cycle. b Density functional theory (DFT) calculations on the O-to-C rearrangement of (R)-6a
Discussion
In summary, we have achieved a metal-free intramolecular hydroalkoxylation/[1,3]-rearrangement, and significantly, this [1,3]-rearrangement is highly stereospecific, and a mechanistic rationale for this stereospecificity is also strongly supported by DFT calculation. This method leads to the practical and atom-economical synthesis of a diverse array of valuable medium-sized lactams from readily available ynamides in high yields with broad substrate scope and excellent diastereoselectivity. Furthermore, this asymmetric cascade cyclization has also been realized via kinetic resolution by chiral spiro phosphoramide catalysis, thus constituting a rare example of chiral Brønsted acid-catalyzed kinetic resolution. In addition, biological tests reveal that some of these medium-sized lactams displayed their bioactivity as antitumor agents against melanoma cells, esophageal cancer cells, and breast cancer cells. We anticipate that the mechanistic insights of this chemistry may provoke new developments in related stereospecific [1,3]-rearrangement and chiral Brønsted acid-catalyzed kinetic resolution, and the present protocol will find broad applications in synthetic and medicinal chemistry.
Methods
Materials
Unless otherwise noted, materials were obtained commercially and used without further purification. All the solvents were treated according to general methods. Flash column chromatography was performed over silica gel (300–400 mesh). See Supplementary Methods for experimental details.
General methods
1H NMR spectra and carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker AV-400 spectrometer and a Bruker AV-500 spectrometer in chloroform-d3. For 1H NMR spectra, chemical shifts are reported in p.p.m. with the internal tetramethylsilane signal at 0.0 p.p.m. as a standard. For 13C NMR spectra, chemical shifts are reported in p.p.m. with the internal chloroform signal at 77.0 p.p.m. as a standard. Infrared spectra were recorded on a Nicolet AVATER FTIR330 spectrometer as thin film and are reported in reciprocal centimeter (cm−1). Mass spectra were recorded with Micromass QTOF2 Quadrupole/Time-of-Flight Tandem mass spectrometer using electron spray ionization. 1H NMR, 13C NMR, and HPLC spectra (for chiral compounds) are supplied for all compounds: see Supplementary Figs. 1–110. See Supplementary Methods for the characterization data of compounds not listed in this part.
General procedure for the synthesis of 3-benzazocinones 2
To a mixture of the ynamide 1 (0.20 mmol) in PhCl (3.75 mL) at room temperature, HOTf (0.001 mmol/0.25 mL) in 0.25 mL PhCl was added. Then, the reaction mixture was stirred at 80 °C and the progress of the reaction was monitored by thin layer chromatography (TLC). The reaction typically took 4 h. Upon completion, the mixture was concentrated and the residue was purified by chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired 3-benzazocinone 2.
General procedure for the synthesis of chiral 2-ent
To a mixture of the ynamide 1 (0.1 mmol) and 5 Å MS (60 mg) in Et2O (2 mL) at room temperature, Cat. 3 (0.02 mmol, 17.6 mg) was added during stiring. Then, the reaction mixture was stirred at 25 °C and the progress of the reaction was monitored by TLC. After the corresponding reaction time (6–32 h), Et3N (0.03 mmol, 4.2 μL) and PhCl (1 mL) was added to the reaction mixure to quench the Cat. 3. The resulting reaction solution was stirred at 60 °C for another 24 h. The mixture was concentrated and the residue was purified by chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired chiral 3-benzazocinone 2-ent.
Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Center (CCDC) under the deposition numbers CCDC 1880379 (2a), 1880411 (2ac), 1880414 (2ai), and 1887308 (2p-ent). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files, or from the corresponding authors on request.
References
Kang, G., Yamagami, M., Vellalath, S. & Romo, D. Enantioselective synthesis of medium-sized lactams via chiral α,β-unsaturated acylammonium salts. Angew. Chem. Int. Ed. 57, 6527–6531 (2018).
Li, J. et al. Asymmetric synthesis and biological activities of natural product (+)-balasubramide and its derivatives. Nat. Prod. Res. 30, 800–805 (2016).
Boonya-udtayan, S., Eno, M., Ruchirawat, S., Mahidol, C. & Thasana, N. Palladium-catalyzed intramolecular C–H amidation: synthesis and biological activities of indolobenzazocin-8-ones. Tetrahedron 68, 10293–10301 (2012).
Yang, L. et al. Highly efficient and stereoselective N-vinylation of oxiranecarboxamides and unprecedented 8-endo-epoxy-arene cyclization: expedient and biomimetic synthesis of some Clausena alkaloids. Org. Lett. 9, 1387–1390 (2007).
Johansen, M. B., Leduc, A. B. & Kerr, M. A. Concise biomimetic total syntheses of both antipodes of balasubramide. Synlett 16, 2593–2595 (2007).
Yet, L. Metal-mediated synthesis of medium-sized rings. Chem. Rev. 100, 2963–3008 (2000).
Molander, G. A. Diverse methods for medium ring synthesis. Acc. Chem. Res. 31, 603–609 (1998).
Iwai, T., Okochi, H., Ito, H. & Sawamura, M. Construction of eight-membered carbocycles through gold catalysis with acetylene-tethered silyl enol ethers. Angew. Chem. Int. Ed. 52, 4239–4242 (2013).
Zhao, W., Li, Z. & Sun, J. A new strategy for efficient synthesis of medium and large ring lactones without high dilution or slow addition. J. Am. Chem. Soc. 135, 4680–4683 (2013).
Zhao, W., Wang, Z. & Sun, J. Synthesis of eight-membered lactones: intermolecular [6+2] cyclization of amphoteric molecules with siloxy alkynes. Angew. Chem. Int. Ed. 51, 6209–6213 (2012).
Wang, N. et al. Direct photocatalytic synthesis of medium-sized lactams by C−C bond cleavage. Angew. Chem. Int. Ed. 57, 14225–14229 (2018).
Mancuso, R. et al. A palladium-catalyzed carbonylation approach to eight-membered lactam derivatives with antitumor activity. Chem. Eur. J. 22, 3053–3064 (2016).
Shaw, M. H., Croft, R. A., Whittingham, W. G. & Bower, J. F. Modular access to substituted azocanes via a rhodium-catalyzed cycloaddition–fragmentation strategy. J. Am. Chem. Soc. 137, 8054–8057 (2015).
Wu, S. et al. Rhodium-catalyzed C–H functionalization-based approach to eight-membered lactams. Chem. Sci. 6, 2275–2285 (2015).
Scully, S. S., Zheng, S.-L., Wagner, B. K. & Schreiber, S. L. Synthesis of oxazocenones via gold(I)-catalyzed 8-endo-dig hydroalkoxylation of alkynamides. Org. Lett. 17, 418–421 (2015).
Yu, R. T., Friedman, R. K. & Rovis, T. Enantioselective rhodium-catalyzed [4+2+2] cycloaddition of dienyl isocyanates for the synthesis of bicyclic azocine rings. J. Am. Chem. Soc. 131, 13250–13251 (2009).
Lu, S.-M. & Alper, H. Sequence of intramolecular carbonylation and asymmetric hydrogenation reactions: highly regio- and enantioselective synthesis of medium ring tricyclic lactams. J. Am. Chem. Soc. 130, 6451–6455 (2008).
Zhou, Y., Wei, Y.-L., Rodriguez, J. & Coquerel, Y. Enantioselective organocatalytic four-atom ring expansion of cyclobutanones: synthesis of benzazocinones. Angew. Chem. Int. Ed. 58, 456–460 (2019).
Zhao, W., Qian, H., Li, Z. & Sun, J. Catalytic ring expansion of cyclic hemiaminals for the synthesis of medium-ring lactams. Angew. Chem. Int. Ed. 54, 10005–10008 (2015).
Rojas, C. M. Molecular Rearrangements in Organic Synthesis (Wiley, New York, 2015).
Jones, A. C., May, J. A., Sarpong, R. & Stoltz, B. M. Toward a symphony of reactivity: cascades involving catalysis and sigmatropic rearrangements. Angew. Chem. Int. Ed. 53, 2556–2591 (2014).
Evano, G., Lecomte, M., Thilmany, P. & Theunissen, C. Keteniminium ions: unique and versatile reactive intermediates for chemical synthesis. Synthesis 49, 3183–3214 (2017).
Adcock, H. V. & Davies, P. W. π-Acid mediated insertion of alkynes into carbon–heteroatom σ-bonds. Synthesis 44, 3401–3420 (2012).
Huang, X., Klimczyk, S. & Maulide, N. Charge-accelerated sulfonium [3,3]-sigmatropic rearrangements. Synthesis 44, 175–183 (2012).
Castro, A. M. M. Claisen rearrangement over the past nine decades. Chem. Rev. 104, 2939–3002 (2004).
Nasveschuk, C. G. & Rovis, T. The [1, 3] O-to-C rearrangement: opportunities for stereoselective synthesis. Org. Biomol. Chem. 6, 240–254 (2008).
Meek, S. J. & Harrity, J. P. A. O→C rearrangements: a powerful strategy for the synthesis of functionalised carbocycles. Tetrahedron 63, 3081–3092 (2007).
Nasveschuk, C. G. & Rovis, T. Stereoselective Lewis acid mediated [1,3] ring contraction of 2,5-dihydrooxepins as a route to polysubstituted cyclopentenes. Angew. Chem. Int. Ed. 44, 3264–3267 (2005).
Shiina, I. & Nagasue, H. [1,3] Sigmatropic rearrangement of ketene silyl acetals derived from benzylα-substituted propanoates. Tetrahedron Lett. 43, 5837–5840 (2002).
Wu, H., Zi, W., Li, G., Lu, H. & Toste, F. D. Gold(I)-catalyzed desymmetrization of 1,4-dienes by an enantioselective tandem alkoxylation/Claisen rearrangement. Angew. Chem. Int. Ed. 54, 8529–8532 (2015).
Dubé, P. & Toste, F. D. Synthesis of indenyl ethers by gold(I)-catalyzed intramolecular carboalkoxylation of alkynes. J. Am. Chem. Soc. 128, 12062–12063 (2006).
Kim, C. et al. Formal alkyne aza-Prins cyclization: gold(I)-catalyzed cycloisomerization of mixed N,O-acetals generated from homopropargylic amines to highly substituted piperidines. J. Am. Chem. Soc. 131, 14660–14661 (2009).
Kim, H. & Rhee, Y. H. Stereodefined N,O-acetals: Pd-catalyzed synthesis from homopropargylic amines and utility in the flexible synthesis of 2,6-substituted piperidines. J. Am. Chem. Soc. 134, 4011–4014 (2012).
Kim, J., Jeong, W. & Rhee, Y. H. Flexible tetrahydropyran synthesis from homopropargylic alcohols using sequential Pd–Au catalysis. Org. Lett. 19, 242–245 (2017).
Jaimes, M. C. B., Weingand, V., Rominger, F. & Hashmi, A. S. K. From ynamides to highly substituted benzo[b]furans: gold(I)-catalyzed 5-endo-dig-cyclization/rearrangement of alkylic oxonium intermediates. Chem. Eur. J. 19, 12504–12511 (2013).
Wang, C.-D., Hsieh, Y.-F. & Liu, R.-S. Gold-catalyzed carboalkoxylations of 2-ethynylbenzyl ethers to form 1-and 2-indanones chemoselectively: effects of ligands and solvents. Adv. Synth. Catal. 356, 144–152 (2014).
Adcock, H. V., Langer, T. & Davies, P. W. 1,2-N-migration in a gold-catalysed synthesis of functionalised indenes by the 1,1-carboalkoxylation of ynamides. Chem. Eur. J. 20, 7262–7266 (2014).
Zhang, J., Liao, Z., Chen, L., Jiang, H. & Zhu, S. Catalytic [1,3] O-to-C rearrangement: rapid access to bridged bicyclic systems. Chem. Eur. J. 24, 6927–6931 (2018).
Zi, W. & Toste, F. D. Gold(I)-catalyzed enantioselective carboalkoxylation of alkynes. J. Am. Chem. Soc. 135, 12600–12603 (2013).
Zhou, B. et al. Yttrium-catalyzed intramolecular hydroalkoxylation/Claisen rearrangement sequence: efficient synthesis of medium-sized lactams. Angew. Chem. Int. Ed. 56, 4015–4019 (2017).
Evano, G., Theunissen, C. & Lecomte, M. Ynamides: powerful and versatile reagents for chemical synthesis. Aldrichim Acta 48, 59–70 (2015).
Wang, X.-N. et al. Ynamides in ring forming transformations. Acc. Chem. Res. 47, 560–578 (2014).
DeKorver, K. A. et al. Ynamides: a modern functional group for the new millennium. Chem. Rev. 110, 5064–5106 (2010).
Evano, G., Coste, A. & Jouvin, K. Ynamides: versatile tools in organic synthesis. Angew. Chem. Int. Ed. 49, 2840–2859 (2010).
Li, L. et al. Metal-free alkene carbooxygenation following tandem intramolecular alkoxylation/Claisen rearrangement: stereocontrolled access to bridged [4.2.1] lactones. Chem. Sci. 10, 3123–3129 (2019).
Shen, W.-B. et al. Divergent synthesis of N-heterocycles via controllable cyclization of azido-diynes catalyzed by copper and gold. Nat. Commun. 8, 1748 (2017).
Shen, W.-B. et al. Highly site selective formal [5+2] and [4+2] annulations of isoxazoles with heterosubstituted alkynes by platinum catalysis: rapid access to functionalized 1,3-oxazepines and 2,5-dihydropyridines. Angew. Chem. Int. Ed. 56, 605–609 (2017).
Li, L. et al. Reversal of regioselectivity in catalytic arene-ynamide cyclization: direct synthesis of valuable azepino[4,5-b]indoles and β-carbolines and DFT calculations. ACS Catal. 7, 4004–4010 (2017).
Shu, C. et al. Generation of α-imino gold carbenes through gold-catalyzed intermolecular reaction of azides with ynamides. J. Am. Chem. Soc. 137, 9567–9570 (2015).
Li, L. et al. Zinc-catalyzed alkyne oxidation/C−H functionalization: highly site-selective synthesis of versatile isoquinolones and β-carbolines. Angew. Chem. Int. Ed. 54, 8245–8249 (2015).
Chen, J., Goforth, S. K., McKeown, B. A. & Gunnoe, T. B. Brønsted acid-catalysed intramolecular hydroamination of unactivated alkenes: metal triflates as an in situ source of triflic acid. Dalton Trans. 46, 2884–2891 (2017).
Schmidt, R. K., Müther, K., Mück-Lichtenfeld, C., Grimme, S. & Oestreich, M. Silylium ion-catalyzed challenging Diels–Alder reactions: the danger of hidden proton catalysis with strong Lewis acids. J. Am. Chem. Soc. 134, 4421–4428 (2012).
Dang, T. T., Boeck, F. & Hintermann, L. Hidden Brønsted acid catalysis: pathways of accidental or deliberate generation of triflic acid from metal triflates. J. Org. Chem. 76, 9353–9361 (2011).
Merad, J., Lalli, C., Bernadat, G., Maury, J. & Masson, G. Enantioselective Brønsted acid catalysis as a tool for the synthesis of natural products and pharmaceuticals. Chem. Eur. J. 24, 3925–3943 (2018).
Parmar, D., Sugiono, E., Raja, S. & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 114, 9047–9153 (2014).
Zhu, S.-F. & Zhou, Q.-L. Privileged Chiral Ligands and Catalysts (ed Zhou, Q.-L.) chapter 4, p 137 (Wiley-VCH: Weinheim, 2011).
Xie, J.-H. & Zhou, Q.-L. Chiral diphosphine and monodentate phosphorus ligands on a spiro scaffold for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res. 41, 581–593 (2008).
Xie, J.-H. & Zhou, Q.-L. Magical chiral spiro ligands. Acta Chim. Sin. 72, 778–797 (2014).
Hong, X. et al. Mechanism and selectivity of N-triflylphosphoramide catalyzed (3+ +2) cycloaddition between hydrazones and alkenes. J. Am. Chem. Soc. 136, 13769–13780 (2014).
Dehli, J. R. & Gotor, V. Parallel kinetic resolution of racemic mixtures: a new strategy for the preparation of enantiopure compounds? Chem. Soc. Rev. 31, 365–370 (2002).
Eames, J. Parallel kinetic resolutions. Angew. Chem. Int. Ed. 39, 885–888 (2000).
Becke, A. D. Density-functional thermochemistry. III. The role of exact change. J. Chem. Phys. 98, 5648–5652 (1993).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Agrafiotis, D. K. & Rzepa, H. S. Dihydrogen transfer reactions. An SCF-MO study of the relative energies of the concerted and stepwise pathways. J. Chem. Soc. Chem. Commun. 902–904 https://doi.org/10.1039/C39870000902. (1987).
Tantillo, D. J. Recent excursions to the borderlands between the realms of concerted and stepwise: carbocation cascades in natural products biosynthesis. J. Phys. Org. Chem. 21, 561–570 (2008).
Hess, B. A. Jr. & Smentek, L. The concerted nature of the cyclization of squalene oxide to the Protosterol cation. Angew. Chem. Int. Ed. 52, 11029–11033 (2013).
Pham, H. V. & Houk, K. N. Diels−alder reactions of allene with benzene and butadiene: Concerted, stepwise, and ambimodal transition states. J. Org. Chem. 79, 8968–8976 (2014).
Mackey, J. L., Yang, Z.-Y. & Houk, K. N. Dynamically concerted and stepwise trajectories of the cope rearrangement of 1,5-hexadiene. Chem. Phys. Lett. 683, 253–257 (2017).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21772161, 21622204, 21625204, 21702109, and 21890722), the President Research Funds from Xiamen University (20720180036), NFFTBS (No. J1310024), PCSIRT, Science and Technology Cooperation Program of Xiamen (3502Z20183015), the Fundamental Research Funds for the Central Universities (Nankai University: Nos. 63191515, 63191523, 63191321), and the Natural Science Foundation of Tianjin City (18JCYBJC21400). We also thank Professor Dr. Xianming Deng from Xiamen University (School of Life Sciences) for assistance with biological tests and Professor Dr. Nanfeng Zheng from Xiamen University (College of Chemistry and Chemical Engineering) for assistance with X-ray crystallographic analysis.
Author information
Authors and Affiliations
Contributions
B.Z., Y.-Q.Z., M.-Y.Y., Y.-B.C., and Y.L. performed experiments. K.Z. and Q.P. performed DFT calculations. Q.P., S.-F.Z., and Q.-L.Z. revised the paper. L.-W.Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, B., Zhang, YQ., Zhang, K. et al. Stereoselective synthesis of medium lactams enabled by metal-free hydroalkoxylation/stereospecific [1,3]-rearrangement. Nat Commun 10, 3234 (2019). https://doi.org/10.1038/s41467-019-11245-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11245-2
This article is cited by
-
Enantioselective functionalization of unactivated C(sp3)–H bonds through copper-catalyzed diyne cyclization by kinetic resolution
Nature Communications (2024)
-
Copper-catalyzed enantioselective desymmetrizing C(sp2)–H functionalization of azide-ynamides via α-imino copper carbenes
Science China Chemistry (2024)
-
Modular enantioselective access to β-amino amides by Brønsted acid-catalysed multicomponent reactions
Nature Chemistry (2023)
-
Chiral Brønsted acid-catalyzed asymmetric intermolecular [4 + 2] annulation of ynamides with para-quinone methides
Science China Chemistry (2023)
-
Photoinduced ynamide structural reshuffling and functionalization
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
