
Abstract
Tomato flavor has changed over the course of long-term domestication and intensive breeding. To understand the genetic control of flavor, we report the meta-analysis of genome-wide association studies (GWAS) using 775 tomato accessions and 2,316,117 SNPs from three GWAS panels. We discover 305 significant associations for the contents of sugars, acids, amino acids, and flavor-related volatiles. We demonstrate that fruit citrate and malate contents have been impacted by selection during domestication and improvement, while sugar content has undergone less stringent selection. We suggest that it may be possible to significantly increase volatiles that positively contribute to consumer preferences while reducing unpleasant volatiles, by selection of the relevant allele combinations. Our results provide genetic insights into the influence of human selection on tomato flavor and demonstrate the benefits obtained from meta-analysis.
Similar content being viewed by others


Introduction
The deterioration of tomato flavor has been a source of complaint from consumers for decades1. During long-term domestication and breeding history, flavor has not been a priority, in contrast to yield, disease resistance, and postharvest shelf life1,2. However, flavor is one of the most important traits for improving tomato sensory quality and consumer acceptability3. Flavor is centrally influenced by sugars, acids, amino acids and a diverse set of volatiles4,5,6. Most of these compounds are quantitatively inherited as shown by many QTL studies but only a few QTLs have been positionally cloned7. Genome-wide association studies (GWAS) have detected many significant associated loci for tomato flavor related traits6,8,9,10,11,12 However, reducing a QTL to a causative gene is difficult and only a few candidate genes have been functionally validated7. The underlying genetic control of tomato flavor is still incomplete and remains an important breeding target.
Meta-analysis of genome-wide associations is powerful in dissecting complex human diseases13,14. A recent meta-analysis in cattle stature also demonstrated its power in non-human species15. However, to the best of our knowledge, no GWAS meta-analysis has been reported in major crops, despite the increasing number of GWAS studies in major crops, such as rice. To date, the genomes of over 500 tomato accessions have been fully sequenced6,12,16,17,18,19, making it possible to perform genotype imputation20,21 and subsequent meta-analysis of GWAS using summary data14 to decipher the polygenic architecture of agronomic traits. In this study, we perform a meta-GWAS on 775 tomato accessions and 2,316,117 SNPs and discover 305 significant associations for diverse flavor-related traits. Our results provide genetic insights into tomato flavor.
Results
Meta-analysis
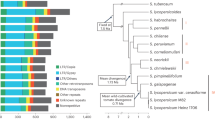
Here we report the first meta-analysis of GWAS in tomato using results of three publicly available GWAS panels: 163 tomato accessions from panel S8, 291 accessions from panel B11, and 402 accessions from panel T6 (Fig. 1). We analyzed a large set of tomato flavor-related quality chemicals, including sugars, organic acids, amino acids, and volatiles measured in each of these panels.

Overview of study design. N, the number of individuals; S.L, S. lycopersicum; S.C, S. lycopersicum var cerasiforme; S.P, S. pimpinellifolium; Genotyping arrays: SOLCAP, Solanaceae Coordinated Agricultural Project; CBSG, Centre of Biosystems Genomics consortium; HWE, Hardy–Weinberg equilibrium; MAF, minor allele frequency; GWAS, genome-wide association study; EMMAX, Efficient Mixed-Model Association eXpedited; DAPC, Discriminant Analysis of Principal Components; eQTL, expression quantitative trait locus; TWAS, transcriptome-wide association study
First, we used IMPUTE2 software22 to increase the genome-wide SNP densities of panel S8 and panel B11, which were genotyped using SNP arrays (Online methods). After quality control (Supplementary Figs. 1–3, Supplementary Tables 1 and 2, Supplementary Data 1–3), a total of 209,152 and 252,414 SNPs was retained for panel S and B, respectively. Imputation greatly increased the density of genomic coverage (Supplementary Fig. 4) and revealed a similar genetic population structure compared with genotyped SNPs for both panels (Supplementary Figs. 5–12 and Supplementary Data 4–5). We used the Efficient Mixed-Model Association eXpedited (EMMAX) software for association tests for panel S and B23, as reported for panel T6 (Online methods, Supplementary Fig. 13). After imputation, we observed a similar or slight statistical increase in terms of the significance and the number of associated loci compared with MLMM24 (Supplementary Figs. 14–44) and no genomic inflation (λ < 1) was detected for most (83.3%) of the traits (Supplementary Data 6). For panel T, which was characterized by 2,040,403 SNPs, the association tests had also been performed using EMMAX6.
By combining the three separate studies, a total of 775 unique tomato accessions were used for the final meta-analysis of 31 flavor-related traits (2 sugars, 2 organic acids, 10 amino acids, and 17 flavor-related volatiles). We performed the meta-analysis with two software: METAL25 using a fixed effect model and METASOFT26 for those SNPs where heterogeneity occurred (I2 > 25) using a random effect model. Manhattan plots and quantile–quantile (Q-Q) plots for all traits are shown in Supplementary Figs. 45–75. Meta-analysis identified a total of 305 significant loci (P < 4 × 10−7 for sugars, acids, and volatiles; P < 2.99 × 10−6 for amino acids), among which 211 were new (Supplementary Data 7). A total of 87 strong effect meta-QTLs were identified with high probability (P < 10−9). Most of these loci passed the suggestive thresholds in at least one panel (Supplementary Figs. 14–75). Among the identified loci, 35 had a moderate to strong heterogeneity (I2 > 25). We generated a local SQLite dataset for tomato (Online methods) and provided the LocusZoom plots for all the genome-wide significant associated loci (Supplementary Figs. 76–123). Among the 305 loci, 24 loci exhibited cis-eQTLs in a previous transcriptome-wide association study12 in fruit tissue (Supplementary Data 7). Among the 211 associated loci, we identified 37 promising candidate genes (7 with significant cis-eQTLs12) with functional annotations related to the pathways of flavor chemicals (Table 1).
We performed a singular enrichment analysis for all associations using agriGO27 (http://bioinfo.cau.edu.cn/agriGO/index.php). Up to 10 biological processes were significantly enriched (P < 0.005) (Supplementary Data 8). All these enriched processes or groups were closely involved in flavor-related metabolites (in terms of sugars, organic acids, amino acids, and volatiles), such as UDP-glycosyltransferase activity, transferase activity, oxidoreductase activity, and carbohydrate metabolic processes.
Previously reported flavor-related loci in the three panels were all strongly associated in the meta-analysis at a higher significance level, such as Lin5 (Solyc09g010080, fructose, P = 6.16 × 10−10; glucose, P = 4.30 × 10−10), TFM6 (Solyc06g072920, malate, P = 2.26 × 10−37), and Phytoene synthase 1 (Solyc03g031860, geranyl acetone, P = 6.73 × 10−26)6,28. In meta-analysis of GWAS, heterogeneity represents the genetic variations observed across combined studies13. In this study, strong heterogeneity occurred even for those loci with major effects, such as Lin5 (fructose, I2 = 95.6, P = 1.05 × 10−10; glucose, I2 = 95.3, P = 5.85 × 10−10). This could be due to population structure, linkage disequilibrium, phenotyping platforms, G × E interactions, etc.13. We then focused on loci in regions showing low LD, where one or a few candidate genes could be identified and regions with medium LD but with candidate genes near the peak SNPs.
Meta-analysis for sugar content
We looked into six candidate genes that were significantly associated both with fructose and glucose. In addition to Lin5 and SSC11.1, we found four loci from the meta-analysis that were significantly associated both with fructose (Fig. 2a) and glucose content (Fig. 2b). These associations are in strong linkage disequilibrium with four candidate genes: alpha-L-fucosidase 1 (FUCA; chr3: 1,506,106; fructose, P = 3.39 × 10−8; glucose, P = 1.46 × 10−8), fatty acid elongase 3-ketoacyl-CoA synthase (KCS; chr5: 3,403,706, fructose, P = 2.57 × 10−8; chr5: 3,406,424, glucose, P = 1.49 × 10−8), glucosyltransferase (GTF; chr5: 63,485,334; fructose, P = 4.68 × 10−10; glucose, P = 8.36 × 10−10), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; chr10:422,707, fructose, P = 6.27 × 10−10; chr10:332,069, glucose, P = 1.20 × 10−9). Notably, near the region of FUCA (up to ten genes), there are two candidate genes (Solyc03g006870, phosphoglucomutase and Solyc03g006860, fructokinase), which are also promising candidate genes for association with fructose and glucose content. Notably, GTF (P = 7.55 × 10−34) and GAPDH (P = 7.84 × 10−17) also showed significant cis-eQTL in a related transcriptome-wide association study12.

Combinations of fructose and glucose alleles for the improvement of tomato sugar content. Only alleles that were significantly associated both with fructose and glucose were analyzed. a, b Manhattan plot for meta-analysis of genome-wide association analysis of fructose (a) and glucose (b) content. Candidates and previously identified genes were labeled in blue and red, respectively. FUCA, alpha-L-fucosidase 1; KCS, fatty acid elongase 3-ketoacyl-CoA synthase; GTF, glucosyltransferase; GADPH, glyceraldehyde-3-phosphate dehydrogenase. c Allele distribution of fructose/glucose content at positions: chr3:1,506,106, chr5:3,403,706, chr5:63,485,334, chr9:3,477,979, and chr10:422,707 that were both significantly associated with fructose and glucose in S. lycopersicum var cerasiforme (cerasiforme), heirloom + transitional (heir_trans), heir + modern (heir_mod), and the closest wild species S. pimpinellifolium (pimpinellifolium) tomato accessions (see detailed information about groups in online methods). d Comparison of sugar content (fructose + glucose) between different tomato types in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. e Mean (±SE) content of fructose (black) and glucose (brown) at different allele combinations in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. Significant t-test P values are also provided. f Correlation between the number of alternative alleles and sugar content. Fructose, glucose, and the sum of fructose + glucose were colored in brown4, cyan4, and purple. g Comparison of sugar content (fructose + glucose) between all alternative and reference allele combinations at position chr3: 1,506,106, chr5: 3,403,706, chr5: 63,485,334, chr9: 3,477,979, and chr10: 422,707. Center line and limits of box were the mean and interquartile ranges. Error bars represent the maximum and minimum values. Whiskers indicate variability outside the upper and lower quartiles. Significant t-test P values are also provided. Source data of Fig. 2c–g are provided in a Source Data file
Interestingly, all these loci, except Lin5 (which falls in the domestication sweep DW14919), were not associated with any domestication or improvement sweep19. We compared the frequencies of different combinations of alleles of these candidate genes in relation to sugar content in wild, transitional, heirloom and modern accessions (more detailed explanations about group definition in Online Methods). All modern, heirloom, and transitional accessions lost most of the diversity of allele combinations that is present in the wild species group (Fig. 2c). The sugar content of heirloom + transitional (heir_trans) and heirloom + modern (heir_mod) groups were both significantly lower than that of the wild species (Fig. 2d). Fruit sugar content increased gradually as the number of alternative alleles increased (Fig. 2e). We observed significant positive correlations between the number of alternative alleles within allele combinations and sugar content (Fig. 2f). In addition, total sugar content (glucose + fructose) of all alternative allele combinations was significantly higher (P = 0.024) than that of all reference allele combinations (Fig. 2g). Together, these results provide insights into possibilities for tomato sugar improvement.
Meta-analysis for organic acids
The meta-analysis also provided several candidate genes for tomato fruit acid content. A strong association (P = 2.26 × 10−37 was detected for malate at an aluminum-activated malate transporter-like gene on chromosome 6, which has been reported to have a major effect on malate content6,8,11, and was further validated as Al-Activated Malate Transporter 9 (Sl-ALMT9)28. We found a strong significant association for citrate (chr6: 44,955,568, P = 7.46 × 10−27), which was 1.54 kb away from Sl-ALTM9 (Supplementary Fig. 45 and Table 1). We also identified a significant association with another aluminum-activated malate transporter on chromosome 1 (chr1:1,749,084, P = 3.62 × 10−13; Supplementary Fig. 45 and Table 1). The strong linkage with both citrate and malate indicated that Al-Activated Malate Transporter also plays an important role in regulating citrate content in tomato fruit.
Candidate genes directly involved in the biosynthesis of citrate and malate were also identified. For example, we identified an association with citrate on chromosome 7, 150 kb away from a gene coding a citrate synthase (Solyc07g055840, P = 4.70 × 10−12). This candidate gene was also significantly associated with fructose (P = 4.28 × 10−09). For malate content, we found one association on chromosome 12 (chr12: 1,824,226, P = 1.75 × 10−19) close (36 kb) to a gene coding a malic enzyme (Solyc12g008430, four genes away from the peak SNP). We then took six candidate genes to analyze the relationships between different allele combinations and citrate and malate content, respectively (Fig. 3). The six candidate genes for citrate were AIMT (Aluminum-activated malate transporter, chr1: 1,749,084, P = 3.62 × 10−13), GTF (Glycosyl transferase group 1, chr2: 47,904,426, P = 4.30 × 10−13), GS (Glycogen synthase, chr3: 52,998,165, P = 1.84 × 10−15), AIMT (Aluminum-activated malate transporter, chr6: 44,955,568, P = 7.46 × 10−27), CS (Citrate synthase, chr7: 63,601,724, P = 4.70 × 10−12), and Rubisco (Ribulose-1 5-bisphosphate carboxylase/oxygenase activase 1, chr10: 65,378,714, P = 5.35 × 10−09). The six candidate genes for malate were GTF (UDP-glucosyltransferase, chr2: 48,509,791, P = 3.47 × 10−28), PDHB (Pyruvate dehydrogenase E1 component subunit beta, chr4: 2,156,747, P = 4.45 × 10−17), AIMT (Aluminum-activated malate transporter, chr6: 44,999,916, P = 2.26 × 10−37), SS (Sucrose synthase, chr9: 72,364,359, P = 1.34 × 10−15), ME (Malic enzyme, chr12: 1,824,226, P = 1.75 × 10−19), and GAPB (Glyceraldehyde-3-phosphate dehydrogenase B, chr12: 64,816,056, P = 5.99 × 10−16).

Combinations of citrate and malate alleles for the improvement of tomato organic acid content. a, b Manhattan plot for meta-analysis of genome-wide association analysis of citrate (a) and malate (b) content. AIMT, Aluminum-activated malate transporter; GTF, Glycosyl transferase group 1; GS, Glycogen synthase; AIMT, Aluminum-activated malate transporter; CS, Citrate synthase; Rubisco, Ribulose-1 5-bisphosphate carboxylase/oxygenase activase 1; PDHB, Pyruvate dehydrogenase E1 component subunit beta; SS, Sucrose synthase; ME, Malic enzyme; GAPB, Glyceraldehyde-3-phosphate dehydrogenase B. c Allele distribution of citrate content at positions: chr1:1749084, chr2: 47,904,426, chr3: 52,998,165, chr6: 44,955,568, chr7: 63,601,724, and chr10: 65,378,714 in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. d Allele distribution of malate content at positions: chr2: 48,509,791, chr4: 2,156,747, chr6: 44,999,916, chr9: 72,364,359, chr12: 1,824,226, and chr12: 64,816,056 in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. e Mean (±SE, standard error) content of citrate content at different allele combinations in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. f Correlation between the number of alternative alleles and citrate content. g Mean (±SE) content of malate content at different allele combinations in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. h Correlations between the number of alternative alleles and malate content. Source data of Fig. 3c–h are provided in a Source Data file
Among the selected candidates, GTF on chromosome 2 and AIMT on chromosome 6 were associated with both citrate and malate (Fig. 3a, b). Both GTF and GS are located within improvement sweeps (IS031 and IS044, respectively)19 and domestication sweeps (DS050 and DS175)19 were observed for malate on PDHB and ME. For citrate and malate, the modern tomato accessions presented very different allele combinations than those in wild species and cherry tomatoes (Fig. 3c, d). In comparison, the total number of allele combinations for malate was approximately three times that of citrate. The citrate content was significantly different between some allele combinations (Fig. 3e). With the increase in the total number of alternative alleles in different allele combinations, the citrate content first increased gradually, with a peak at n = 2, and then steadily decreased (Fig. 3f). The malate content also showed a wide range of variation among alleles (Fig. 3g and Supplementary Data 9). We observed a weak but significant (P = 0.02) positive linear correlation (r = 0.16) between the number of alternative alleles and malate content (Fig. 3h).
These results demonstrated that citrate content was more influenced by improvement sweeps while malate was more influenced by domestication sweeps in the long-term breeding history. In addition, citrate has much less allele diversity than malate and a distinct pattern of relationships between the number of alternative alleles and its content.
Meta-analysis for amino acids and volatiles
Many candidate genes associated with amino acid and volatile contents were identified. For example, we found a significant association for serine on chromosome 3 (P = 3.06 × 10−14) (Supplementary Fig. 57 and Table 1), which was only significant in panel B (P = 2.13 × 10−9) (Supplementary Fig. 26). The candidate gene is annotated as a threonine synthase, an enzyme involved in the serine biosynthesis pathway. For proline, we found one associated locus (Solyc03g117770, P = 2.39 × 10−9), which was also reported as a significant eQTL (P = 1.04 × 10−35)12. This gene is a serine incorporator, and directly regulates serine content. One locus corresponding to GDSL esterase/lipase (Solyc12g089350) was also significantly associated with four amino acids (asparagine, GABA, glutamine and threonine). For hexanal, we found the strongest association corresponding to the lipoxygenase gene LoxC (Solyc01g006540, P = 1.45 × 10−10), which encodes an enzyme that is essential for synthesis of C6 and C5 fatty acid-derived volatiles29,30. This candidate gene was also significantly associated with (Z)-3-hexen-1-ol (P = 3.94 × 10−07). For 2-methyl-1-butanol, the strongest association corresponded to a 3-methyl-2-oxobutanoate dehydrogenase gene (Solyc06g059850, P = 5.50 × 10−09), an enzyme associated with branched chain amino acid metabolism.
We then looked at the possibility that significantly increasing the overall intensity of volatiles contributed to consumer liking as well as significantly reducing the overall content of unpleasant volatiles by combining the strongest loci associated with the contents of six volatiles (Fig. 4). The four volatiles positively contributing to liking included geranyl acetone (chr3: 4,328,514, P = 6.73 × 10−26), hexanal (chr1: 1,083,181, P = 1.45 × 10−10), phenylacetaldehyde (chr4: 55,635,636, P = 5.59 × 10−22), and 6-methyl-5-hepten-2-one (chr3: 3,212,583, P = 6.76 × 10−26). The two unpleasant (or negative) volatiles were guaiacol (chr9: 69,299,940, P = 5.90 × 10−18) and methyl salicylate (chr9: 69,293,875, P = 2.34 × 10−19) (Fig. 4a–f). Modern and heirloom + transitional accessions had the lowest allele diversity, especially compared with S. pimpinellifolium and cherry tomato accessions (S. l. cerasiforme). Interestingly, we also found that cherry tomatoes had the greatest diversity of allele combinations and some of them only appeared in this group (Fig. 4g). The highest total content of the four positive volatiles was observed in allele combinations of cherry tomato accessions, which were significantly higher than the allele combinations of all modern tomato accessions (Fig. 4h). In contrast, modern accessions have, on average, a significantly higher content of unpleasant volatiles, compared with the cherry accessions (Fig. 4i). These results revealed the combinations of alleles that have the potential to significantly enhance the total contents of volatiles associated with consumer liking.

Combinations of six volatile alleles for the improvement of tomato volatile content. a–f Manhattan plot for meta-analysis of genome-wide association analysis of geranyl acetone (a), guaiacol (b), hexanal (c), methyl salicylate (d), phenylacetaldehyde (e), and 6-methyl-5-hepten-2-one (f) content. g Allele distribution of six volatiles content at positions: chr3: 4,328,514 (geranyl acetone), chr9: 69,299,940 (guaiacol), chr1: 1,083,181 (hexanal), chr9: 69,293,875 (methyl salicylate), chr4: 55,635,636 (phenylacetaldehyde), and chr3: 3,212,583 (6-methyl-5-hepten-2-one) in cerasiforme, heir_trans, heir_mod, and pimpinellifolium tomato accessions. h, i Mean (±SE, standard error) content of total content of the four positive volatiles (geranyl acetone, hexanal, phenylacetaldehyde and 6-methyl-5-hepten-2-one) (h) and two unpleasant volatiles (lower panel, guaiacol and methyl salicylate) (i) at different allele combinations in cerasiforme, heir_trans, heir_mod and pimpinellifolium tomato accessions. Source data of Fig. 4g–i are provided in a Source Data file
Discussion
With the development of next-generation sequencing technology, GWAS has become a classical genetic approach to identify QTLs and causal genes in crops31. We herein demonstrate the potential of meta-analysis of GWAS following the detailed protocols first proposed in human genetics32,33, which can be easily applied in other crops. Meta-analysis of GWAS is used when pooling raw data of separate panels (mega-analysis) is not possible. It has been shown both theoretically and numerically that meta-analysis is statistically as efficient as mega-analysis34,35. Even when possible, it is thus not necessary to re-analyze the raw data to perform meta-analysis. Only summary data (beta, standard error and p-values of associations at each SNP) from each panel is needed and should be provided with each GWAS result. For mega-analysis, genotypes and phenotypes from all panels should be first combined and then analyzed, which requires proper management of phenotypic structure (data coming from different studies with different plant growth conditions, different harvesting and sampling procedures, different metabolic analysis protocols etc.) and genotypic structure (such as population structure and kinship). Compared to mega-analysis, meta-analysis can assess the heterogeneity (consistency) of studies, which can be caused by many factors, such as phenotypic structure, genetic structure, linkage disequilibrium, imputation accuracies or G × E interactions13,34.
Flavor remains a major breeding challenge in tomato1,6. Here, we used imputation-driven meta-analysis of genome-wide association studies to greatly increase the number of SNPs linked to chemicals associated with flavor. Among the 305 significantly associated loci, 41% of the SNPs had a low frequency (MAF < 0.1). Very low-frequency (0.01 < MAF < 0.05) SNPs were also detected (3 significant associated loci) (Supplementary Fig. 124). These results demonstrated that a sufficiently large sample size is needed to uncover these low-frequency and less common variants and to account for missing heritability36,37,38. Although hundreds of tomato genome sequences have been published6,12,16,17,18,19, a high sequence depth reference panel is needed, such as the 1000 Genomes Project39 in humans or the 1135 Arabidopsis genomes40 in Arabidopsis, to perform genotype imputation20,21, heritability estimation36,41,42,43 and meta-analysis13,14 with higher accuracy. Also, an imputation server could greatly enhance the integration of genetic resources44.
In this study, we identified 37 promising candidate genes with functional annotations consistent with their involvement in biosynthesis of flavor chemicals. With the advancement of genome editing technologies, their functional analysis could greatly promote our knowledge of the genetic architecture of tomato flavor, provide fully linked markers for breeding and ensure consumer satisfaction45,46,47,48. It is also possible now to introduce desirable traits into wild stress-tolerant tomato accessions by genome editing49,50. However, tomato flavor can only be significantly improved when multiple genes are modified.
Many consumers are more attracted by small and medium size tomatoes with superior taste51, as higher sugar content is usually associated with smaller fruit size6. In the meta-analysis, we found that modern cultivars have lost the majority of high-sugar alleles that were present in transitional, cherry tomato varieties and wild species. All these loci did not seem to have been influenced by any domestication or improvement sweeps, with the exception of Lin5, but some were loosely linked to fruit weight QTLs due to large LD in tomato. These results reflect the fact that sugar content has not been a breeding priority, in contrast to fruit size, yield, biotic, and abiotic resistances1,6. Strong positive correlations between the number of alternative alleles and sugar content provide clues on how to select higher sugar content tomato cultivars. However, sugar content can only be significantly improved when almost all the alternative alleles are selected, and will probably be accompanied by reduced fruit size6 except if precise recombination or genetic modifications limits the linkage drag effect.
Malate and citrate are the main organic acids in most ripe fruits52. In tomato, citrate has a stronger impact on consumer preferences. In this study, candidate genes potentially impacting both citrate and malate contents were identified. We also demonstrated that citrate has been more influenced by improvement sweeps and malate by domestication sweeps. These results show that citrate was probably selected for improving tomato flavor.
Flavor-related volatiles are strongly influenced by the environment53,54. Nevertheless this meta-analysis illustrates that it should be possible to significantly enhance the content of favorable aromas via replacement of undesirable alleles. However, unlike sugars, the undesirable alleles should be carefully chosen6. Cherry tomato varieties have been introduced to the market since the 1990s. Their genomes are an admixture of those of big-fruited tomatoes and S. pimpinellifolium species19,55 and may still contain a large number of favorable alleles. Thus they may serve as the most promising allele reservoir for breeding of high-flavor tomatoes.
In conclusion, we performed the first meta-analysis of genome-wide association analyses in a major vegetable and identified numerous loci involved in tomato flavor that were not identified in the three independent studies. A strong positive correlation between allele combinations and sugar content provides clues for breeding for higher sugar content. Modern cultivars have lost most of the allelic diversity for sugars, acids, and volatiles that is present within the species. Significant improvements should be achieved by replacing undesirable alleles. Taken together, our meta-analysis provides genetic insights into the genetic control of tomato flavor and gives a roadmap for flavor improvement.
Methods
Three GWAS panels
The meta-GWASs approach is based on three different GWAS panels already published and genotyped using different technologies. Our approach consisted in imputing SNP data for panels S8 and B11 from a reference panel, then conducting separate GWAS using the same mixed linear model (MLM) as described in6 and collecting the summary statistics to run a meta-GWAS.
Panel S consists of 163 accessions8, including 28S. lycopersicum (large tomato), 119S. lycopersicum var cerasiforme (cherry tomato), and 16S. pimpinellifolium (closest wild species). This panel was genotyped using the Solanaceae Coordinated Agricultural Project (SOLCAP) genotyping array56,57, generating 5995 high quality SNPs. The minimal success genotyping rate per accession was fixed at 90%. The minor allele frequency of SNPs ranged from 0.037 to 0.45. Tomato accessions in Panel S were grown in Avignon, France, following a randomized complete block design, in a greenhouse during the summers of 2007 and 20088,58.
Panel B consists of 300 accessions with 62S. pimpinellifolium, 48S. lycopersicum, and 190S. l. cerasiforme accessions11. This panel was genotyped both with the SOLCAP56,57 and CBSG arrays59. After quality control, 9013 SNPs (minor allele frequency, MAF > 0.1) and 291 accessions were kept. Accessions in Panel B were grown in Agadir, Morocco, France, under passive greenhouse irrigated conditions in 2011 and 201211. Each trial followed a randomized complete block design, with three and two blocks, in 2011 and 2012, respectively.
Panel T consists of 402 tomato accessions from two separate panels6. Panel T was genotyped by whole genome resequencing technology, generating a number of 2,014,488 SNPs passing quality control (MAF > 0.05, missing rate < 10%). This panel includes five tomato types, including modern (51), transitional (50), cherry (27), heirloom (243), and wild species (27)6.
Phenotypes
A total of 31 flavor-related quality traits in tomato were analyzed for meta-analysis, including two sugars (fructose and glucose), two organic acids (citrate and malate), 10 amino acids, and 17 flavor-related volatiles. The 10 amino acids were asparagine, aspartate, GABA, glutamine, lysine, methionine, phenylalanine, proline, serine, and threonine. The 17 volatiles were (E)-2-heptenal (E2HEP), (E)-2-hexenal (E2HEX), (E)-2-pentenal (E2PEN), (E,E)-2,4-decadienal (EE24D), (Z)-3-hexen-1-ol (Z3H1X), (Z)-3-hexenal (Z3HEX), 1-octen-3-one (X1O3ON), 1-penten-3-one (X1P3ON), 2-methyl-1-butanol (X2M1BU), 3-methyl-1-butanol (X3M1BU), 6-methyl-5-hepten-2-one (X6MHON), beta-ionone (BIONO), geranylacetone (GRACE), guaiacol (GUAIA), hexanal (XEXAN), phenylacetaldehyde (PHEAC), and methylsalicylate (METHY).
Sugars and organic acids were measured in all three panels. Amino acids were measured both in panel S and B, while flavor-related volatiles were measured both in panel B and T. Briefly, fructose and glucose in panel S were measured using the micro-method. Citrate and malate were measured by gas chromatography-mass spectrometry (GC-MS)8. Data distribution was tested using the Shapiro–Wilk test and data with a non-normal distribution were Log10 transformed. In panel B, these metabolites were measured within the Product Metabolism and Analytical Sciences Endogenous Metabolite Profiling Platform at Syngenta Jealott’s Hill International Research Center, Bracknell, UK. Fructose and glucose were analyzed by high pH ion-exchange chromatography. Citrate and malate were analyzed using electrospray ionization-liquid chromatography (ESI-LC-MS/MS). Fructose and malate were transformed using the Boxcox method. Citrate was transformed using the Log10 method. In panel T, citrate and malate were measured using the citrate and malate analysis kits (R-Biopharm, Marshall, MI), according to the manufacturer’s instructions60. Measurements of amino acids and volatiles in panel S was measured using GC-MS by comparing with a database of authentic standards. Small organic acids and amino acids in panel B were analyzed using electrospray ionization-liquid chromatography (ESI-LC-MS/MS).Volatiles in panel T were first captured by headspace solid phase micro extraction (HS-SPME) coupled GC-MS.
Reference panel for SNP imputation
A reference panel was selected from the 360 re-sequenced tomato accessions19 to perform SNP imputation in panels S and B. Among this panel, only accessions with genome coverage ≥90% and mean sequencing depth ≥4.0 were kept. Wild tomato species were also removed, generating a total reference set of 221 accessions genotyped with 3,809,156 SNPs (Supplementary Table 1).
Recombination map
A high-density recombination map is required for imputation and computing genomic partitions. However, the available tomato genetic maps EXPIM 2012 and EXPEN 201257 have a limited genomic coverage (~3500 mapped SNPs). In order to use a much denser genetic map, we developed a Python script to infer the corresponding genetic positions of the 3,809,156 SNPs in the reference panel. Before calculating the recombination rate, we first compared the physical vs genetic distribution patterns for each chromosome (Supplementary Fig. 1). Comparing with EXPIM 2012, this newly built genetic map had the same distribution pattern (Supplementary Fig. 1). This comparison indicated the inferred genetic positions were accurate and were then used for estimating the recombination rate, as required for imputation. Minor adjustments were also done for some SNPs in order to follow an overall increasing positional order. Extreme recombination rate values were also removed (>2000 cM/Mb).
Genotype imputation
One unphased reference panel from IMPUTE2 (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html#home)22 was adopted for imputation of panel S and B independently. The 221 filtered sequenced accessions passing quality control were used as the reference panel. The newly built recombination map was used instead of EXPIM 2012. The whole genome was then divided into genomic intervals of 5 Mb for imputation and the effective size of population (Ne) was set at 2000.
Quality control
After imputation, the minimum MAF for panel S and B was set at 0.037 and 0.021, respectively, according to the formula: [Number of chromosomes/(2 × Number of individuals)]61. After combining all the imputed data, basic statistic summaries were obtained in QCTOOL v2 (http://www.well.ox.ac.uk/~gav/qctool_v2/) with the following command:./qctool -g GWAS.gen -snp-stats. We then filtered all imputed SNPs with Hardy-Weinberg equilibrium (HWE) ≥ 0.000001, MAF ≥ 0.037 (0.021 for panel B), missing rate ≤ 0.10 and missing call rate ≤ 0.10. After these primary control steps, a total of 224,097 and 327,436 SNPs were retained for panel S and B, respectively.
In order to determine the optimal threshold of imputation quality (Info criteria), we compared the imputed and sequenced genotype data of the nine overlapping accessions in panel S that have been genotyped by SNP arrays and whole-genome sequencing. If the maximum of the three probabilities at a locus was higher than 0.9, we treated it as a certainty. This was done by converting the imputed data to ped/map format via GTOOL (http://www.well.ox.ac.uk/~cfreeman/software/gwas/gtool.html). We then compared the imputed and genotyped values of the nine accessions (Supplementary Fig. 2). Total numbers of corrected SNPs at different MAF and Info thresholds were obtained to validate the optimal threshold of MAF and Info. The average value of Info was 0.882 (with no filtering of MAF). With the increase of Info, the number of correctly genotyped SNPs increased from less than 200 to about 50,000 for panel S (Supplementary Fig. 2a, Supplementary Table 2). On average, 51.45% of the SNPs have been correctly imputed for all Info values. There was no significant difference between the numbers of corrected imputed SNPs for different Info values of the three tomato groups (Supplementary Fig. 2b). The majority of imputed SNPs had a MAF value ranging from 0.037 to 0.25, with a mean value of 0.172 ± 0.103 (with no filtering of Info). The percentage of successfully genotyped SNPs averaged at 57.3% and a higher percentage of corrected imputed SNPs decreased gradually with the increase of MAF (Supplementary Fig. 2c). Similarly, no significant difference was found between the numbers of corrected imputed SNPs for different MAF values of three tomato genetic groups (Supplementary Fig. 2d). Details of the number and percentage of corrected imputed SNPs at different MAF bins among the nine accessions are listed in Supplementary Data 1. We than compared the relationship between MAF and Info. The average value of Info was 0.912 for all values of MAF (Supplementary Fig. 2e). We found that the lowest mean value of Info (0.622) was observed on less common SNPs (0.037 < MAF < 0.05) (Supplementary Fig. 2e, Supplementary Data 2). However, this value is still higher than the proper imputation quality threshold (0.4) in common quality control of meta-analysis of genome-wide association studies33. So, we decided to set the Info threshold at 0.60 as the threshold of high imputation quality.
After filtering with imputation quality threshold (Info) ≥ 0.60, total of 209,152 and 252,414 SNPs were retained for panel S and B, respectively. The mean Info value at different MAF values for panel S and B were 0.929 and 0.922, respectively (Supplementary Data 3). The lowest mean value of Info at different MAF value was 0.810 and 0.783, respectively (Supplementary Fig. 2f, Supplementary Fig. 3). These SNPs offered a much denser genomic coverage for both panel S and B (35-fold and 28-fold, respectively) (Supplementary Fig. 4). Only some large genomic gaps still remained where there were few genotyped SNPs over a long genomic region (Supplementary Fig. 4). These results indicated that all the retained SNPs had a high imputation quality and were used for further analyses.
Linkage disequilibrium analysis
For population structure and kinship analyses, only independent SNPs (r2 < 0.2) were used. This was done in PLINK (https://www.cog-genomics.org/plink2) with: --indep-pairwise 50 5 0.2 (windows, step, r2) –maf 0.05, generating a total of 3,602 and 4,294 independent SNPs for panel S and B, respectively.
Principal component analysis
In order to compare the genetic structure revealed before and after imputation, we performed a principal component analysis (PCA) for panels S and B, using all genotyped SNPs and independent imputed SNPs (r2 < 0.2) in PLINK: --pca. Principal component analysis showed that genotype imputation did not lead to significant differences in genetic group composition and pairwise individual distances, for all three accession classes of panel S (S.C., S.L., S.P.) (Supplementary Fig. 5a–c). For the first principal component (PC1), there were strong positive correlations (0.93, 0.82, and 0.93 for S.C, S.L., and S.P. respectively) between genotyped and imputed SNPs (only imputed SNPs) (Supplementary Fig. 5d). By combining genotyped and imputed SNPs together (hereafter called ‘All’ dataset), a similar strong positive correlation (0.94, 0.82, and 0.94 for S.C, S.L., and S.P. respectively) was also found (Supplementary Fig. 5e). Correlation between imputed and all SNPs was also strong for all tomato classes (Supplementary Fig. 5f). For the panel B, a previous study revealed a population structure composed of six groups62. After imputation, we found they had a similar distribution pattern (Supplementary Fig. 6). PC1 between genotyped SNPs and all (genotyped and imputed) SNPs had a strong positive correlation (higher than 0.7 for all six groups) (Supplementary Fig. 6c). In contrast, the second principal component (PC2) had strong negative correlations for all six groups (lower than −0.6 for all six groups) (Supplementary Fig. 6d).
Population structure
In a previous study, the population structure of panel S was evaluated by Structure v2.3.463 (https://web.stanford.edu/group/pritchardlab/structure_software/release_versions/v2.3.4/html/structure.html). So we first compared the structure following the same parameters, with 1 × 106 burn-in period and 5 × 106 MCMC steps. Based on the Evanno method63, the optimal number of ancestral populations was two. Only minor population assignment differences were found for both subpopulations, compared with structure from genotyped SNPs (Supplementary Fig. 7).
We further used discriminant analysis of principal components (DAPC)64 (http://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf) using the independent 3,602 and 4,294 SNPs (r2 < 0.2) to infer the optimal population structure for panels S and B. This method partitioned the variance within and among groups without assumptions on LD or Hardy–Weinberg equilibrium65, which has shown a better performance in clustering individuals11. The optimal number of clusters was determined by Bayesian Information Criteria (BIC) with a minor increase or decrease. All PCs and all discriminant functions were retained to find the optimal number of clusters. In the following DAPC analyses, all discriminant functions and the first 50 PCs were retained in order to achieve 80% of cumulative variance for both panel S and B.
For panel S, the optimal number of clusters was six (Supplementary Fig. 8) and DAPC revealed a clear structure of all the accessions (Supplementary Fig. 9). For panel B, the optimal number of cluster was six, which was the same as that revealed by using genotyped SNPs (Supplementary Fig. 10). Membership of each cluster was also quite similar (Supplementary Fig. 11), compared with that of genotyped SNPs (Supplementary Fig. 12). Detailed information of the membership of each cluster revealed by all independent SNPs for panels S and B is listed in Supplementary Data 4 and Supplementary Data 5, respectively. These results indicated that imputation did not cause significant differences in the genetic structure for both panels S and B. For panel T, the optimal number of clusters was five from DAPC with the first 20 PCs retained and a cross validation run of 100 times6.
Genome-wide association analysis
Though SNPTEST v2.5.4 (https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html#introduction) can use the imputed data from IMPUTE2 to detect associations directly, it cannot however handle too many cofactors in the model. For accessions from each panel used in this study, there is strong genetic structure. We first took one trait (malate) in panel S as an example to choose the optimal association software to perform the association tests.
In order to add kinship as a cofactor in SNPTEST, we performed a principal component analysis of the kinship calculated in SPAGeDi (http://ebe.ulb.ac.be/ebe/SPAGeDi.html) and structure in Structure v2.3.4. We then added the first 20 PCs as cofactors in the frequentist association test model in SNPTEST. In the next step, we used EMMAX (http://genetics.cs.ucla.edu/emmax/index.html) with the BN kinship matrix and DAPC results to conduct association analyses. For BN kinship calculation, the default command was used: emmax-kin -v -h -d 10. A uniform threshold (P = 1/n, n is the effective number of independent SNPs) was used as the genome-wide significance threshold for all three panels. The effective number of independent SNPs was calculated in Genetic type 1 Error Calculator (GEC)66 (http://grass.cgs.hku.hk/gec/download.php). The suggestive p-value for the 224,097 SNPs of panel S was 9.63 × 10−5 and the significant p-value was 4.82 × 10−6. For the 327,436 SNPs of panel B, the suggestive and significant p-value was 5.99 × 10−5 and 2.99 × 10−6, respectively.
After comparing the association results for malate of panel S, we found the strongest p-value in SNPTEST was still quite low, compared with other approaches (Supplementary Fig. 13). Results from MLMM (https://github.com/Gregor-Mendel-Institute/MultLocMixMod) and EMMAX were quite similar. So, in the following analyses, we only used SNPTEST to compute summary statistics, not for finding associations. For MLMM, this model adds the marker as co-factor using a window of 10. If too many markers are in full LD, the genetic variance calculation may be biased24. So, we used EMMAX for association analyses for all traits with the BN kinship matrix and DAPC results as covariance.
Meta-analysis
A total of 788 tomato accessions and 2,316,117 SNPs from three GWAS panels were used for the final meta-analysis. Since each panel was stratified and a small number of individuals overlapped between panels (38 between panel B and S, 18 between panel S and T, 17 between panel B and T), genomic inflation factor (λ) was corrected before meta-analysis using GenABEL61 (http://www.genabel.org/packages/GenABEL) in R. Genomic inflation can be caused by population structure, cryptic relatedness, genotyping errors, sample size, LD, trait heritability, number of causal variants and other technical artefacts67. Though no adjustment is necessary when λ is lower or equal to one, we still corrected the standard errors of beta coefficients by applying the formula SE × \(\sqrt \lambda\) in general for each individual studies to get the chi-squares to its optimal values68.
METAL25 (fixed-effect model) (https://genome.sph.umich.edu/wiki/METAL_Documentation) and METASOFT26 (random-effect model) (http://genetics.cs.ucla.edu/meta/) are two most commonly used meta-analysis software13. Meta-analysis was first performed using the inverse variance-weighted fixed-effect model in METAL25. The genome-wide significant p-value for meta-analysis was set as 4.0 × 10−7, except for SNPs that only appeared between panel S and B (the significant p-value was set at 2.99 × 10−6). For those SNPs where heterogeneity occurs (I2 > 25, indicating moderate heterogeneity), we used the Han and Eskin random-effects model (RE2) in METASOFT26. This model assumes no heterogeneity under the null hypothesis and offers greater power under heterogeneity, compared with conventional random-effect models26.
Local SQLite database for LocusZoom
In order to obtain a regional zoom plot of the candidate SNPs in LocusZoom69 (https://genome.sph.umich.edu/wiki/LocusZoom_Standalone), a local SQLite database of tomato was required. We thus created a custom SQLite database in LocusZoom with the following steps. SNP positions in the 221 accessions of the reference panel were inserted by: dbmeister.py --db my_database.db --snp_pos my_snp_pos_file. For the gene information, we first downloaded the gene annotation file from Solgenomics (ftp://ftp.solgenomics.net/genomes/Solanum_lycopersicum/annotation/ITAG2.4_release/). We then converted it to genePred file format by gff3ToGenePred (http://hgdownload.cse.ucsc.edu/admin/exe/). Gene names were replaced with short codes instead of providing full names to avoid long names and overlapping. We then inserted the gene information by the following command line: dbmeister.py --db my_database.db --refflat my_refflat_file. For the recombination file, we used the recombination map previously inferred and inserted the data into our database by: dbmeister.py --db my_database.db --snp_set my_snpset_file. We used the 221 reference panel to calculate the linkage disequilibrium (LD) in PLINK by the following parameter: --ld-snp my.snp --ld-window-kb 100000 --ld-window 1000 --r2 --ld-window-r2 0 (windows, step, r2).
LD in candidate gene regions
In order to define the window size of the candidate genes, we first calculated the LD around the significant associated SNP with the window size of 5 Mb in PLINK with the following command line: --ld-window-kb 500000 --ld-window 1000 --r2 --ld-window-r2 0 (windows, step, r2). We then chose LD higher than 0.5 as the threshold of LD decay for the candidate gene region sizes. Within the regions, we chose the candidate genes based on both the distance of the peak SNP as well as the closest genes with known functions related to the trait. If no gene fell in the candidate regions, we provided the closest gene. We further crosschecked the candidate gene expression patterns using the Tomato Expression Atlas70 (http://tea.solgenomics.net/expression_viewer/input).
Group re-definition of panel T
The relationship between allele combinations and flavor-related metabolites (sugars, organic acids and volatiles) was only based on panel T. For the accessions in panel T, they were previously defined as five clusters, namely S. lycopersicum var cerasiforme, heirloom, transitional, modern and the closest wild species S. pimpinellifolium tomato accessions6. However, there were up to 11 accessions with duplicated individual IDs (Supplementary Data 10) and we cross-checked these duplicated lines and only kept one. In addition, some accessions in the group of heirloom, modern and transitional were labeled inappropriately based on the DAPC analysis. In order to correct for this, we generated the principal component analysis (PCA) based on independent SNPs (LD = 0.1) (Supplementary Fig. 125). Based on PCA, some heirloom accessions are mixed with modern accessions and were labeled as heir_mod (heirloom and modern). For the remaining heirloom accessions, they were combined with transitional accessions and labeled as heir_trans (heirloom and transitional) (Supplementary Fig. 126). The accessions of panel T were thus re-defined as four clusters, namely S. lycopersicum var cerasiforme, (cerasiforme, 26 members), heirloom and modern (heir_mod, 196 members), heirloom and transitional (heir_trans, 138 members), and S. pimpinellifolium (27 members) (Supplementary Data 10–11). These redefined groups were then used for allelic combination analyses. Statistical tests were only performed for those allele combinations with at least two observations (either labeled with letters or with p-values).
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. All new meta-analysis data associated with the paper are available in a repository [https://doi.org/10.15454/TWFDYW]. The source data underlying Figs. 2c–g, 3c–h, and 4g–i and Supplementary Figs. 5a–f, 6a–d, and 124–126 are provided as a Source Data file. Additional datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Klee, H. J. & Tieman, D. M. The genetics of fruit flavour preferences. Nat. Rev. Genet. 19, 347–356 (2018).
Tieman, D. et al. The chemical interactions underlying tomato flavor preferences. Curr. Biol. 22, 1035–1039 (2012).
Causse, M. et al. Consumer preferences for fresh tomato at the European scale: a common segmentation on taste and firmness. J. Food Sci. 75, S531–S541 (2010).
Baldwin, E. A., Scott, J. W., Shewmaker, C. K. & Schuch, W. Flavor trivia and tomato aroma: biochemistry and possible mechanisms for control of important aroma components. HortScience 35, 1013–1022 (2000).
Goff, S. A. & Klee, H. J. Plant volatile compounds: sensory cues for health and nutritional value? Science 311, 815–819 (2006).
Tieman, D. et al. A chemical genetic roadmap to improved tomato flavor. Science 355, 391–394 (2017).
Rothan, C., Diouf, I. & Causse, M. Trait discovery and editing in tomato. Plant J. 97, 73–90 (2019).
Sauvage, C. et al. Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 165, 1120–1132 (2014).
Zhang, J. et al. Genome-wide association mapping for tomato volatiles positively contributing to tomato flavor. Front. Plant Sci. 6, 1042 (2015).
Zhao, J. et al. Association mapping of main tomato fruit sugars and organic acids. Front. Plant Sci. 7, 1–11 (2016).
Bauchet, G. et al. Identification of major loci and genomic regions controlling acid and volatile content in tomato fruit: implications for flavor improvement. New Phytol. 215, 624–641 (2017).
Zhu, G. et al. Rewiring of the fruit metabolome in tomato breeding. Cell 172, 249–261.e12 (2018).
Evangelou, E. & Ioannidis, J. P. A. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 14, 379–389 (2013).
Pasaniuc, B. & Price, A. L. Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet. 18, 117–127 (2017).
Bouwman, A. C. et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 50, 362–367 (2018).
Sato, S. et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Aflitos, S. et al. Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 80, 136–148 (2014).
Bolger, A. et al. The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet. 46, 1034–1038 (2014).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226 (2014).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2010).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS. Genet. 5, e1000529 (2009).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Segura, V. et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830 (2012).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Tian, T. et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129 (2017).
Wang, B. et al. An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. Plant Cell 29, 2249–2268 (2017).
Chen, G. et al. Identification of a specific isoform of tomato lipoxygenase (TomloxC) involved in the generation of fatty acid-derived flavor compounds. Plant Physiol. 136, 2641–2651 (2004).
Shen, J. et al. A 13-lipoxygenase, TomloxC, is essential for synthesis of C5 flavour volatiles in tomato. J. Exp. Bot. 65, 419–428 (2014).
Liu, H. J. & Yan, J. Crop genome-wide association study: a harvest of biological relevance. Plant J. 97, 8–18 (2019).
Turner, S. et al. Current Protocols in Human Genetics Chapter 1, Unit 1. 19 (NIH Public Access, Hoboken, New Jersey, USA, 2011).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Panagiotou, O. A., Willer, C. J., Hirschhorn, J. N. & Ioannidis, J. P. A. The power of meta-analysis in genome-wide association studies. Annu. Rev. Genom. Hum. Genet. 14, 441–465 (2013).
Lin, D. & Zeng, D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet. Epidemiol. 34, 60–66 (2010).
Yang, J., Zeng, J., Goddard, M. E., Wray, N. R. & Visscher, P. M. Concepts, estimation and interpretation of SNP-based heritability. Nat. Genet. 49, 1304–1310 (2017).
Gibson, G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 (2012).
Marouli, E. et al. Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190 (2017).
Gibbs, R. A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Alonso-Blanco, C. et al. 1,135 Genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166, 481–491 (2016).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Eichler, E. E. et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450 (2010).
Speed, D., Cai, N., Johnson, M. R., Nejentsev, S. & Balding, D. J. Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992 (2017).
Wang, D. R. et al. An imputation platform to enhance integration of rice genetic resources. Nat. Commun. 9, 3519 (2018).
Gao, C. The future of CRISPR technologies in agriculture. Nat. Rev. Mol. Cell Biol. 19, 275–276 (2018).
Rodríguez-Leal, D., Lemmon, Z. H., Man, J., Bartlett, M. E. & Lippman, Z. B. Engineering quantitative trait variation for crop improvement by genome editing. Cell 171, 470–480.e8 (2017).
Huang, S., Weigel, D., Beachy, R. N. & Li, J. A proposed regulatory framework for genome-edited crops. Nat. Genet. 48, 109–111 (2016).
Yin, K., Gao, C. & Qiu, J.-L. Progress and prospects in plant genome editing. Nat. Plants 3, 17107 (2017).
Zsögön, A. et al. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 36, 1211–1216 (2018).
Gao, C. et al. Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 36, 1160–1163 (2018).
Oltman, A. E., Jervis, S. M. & Drake, M. A. Consumer attitudes and preferences for fresh market tomatoes. J. Food Sci. 79, S2091–S2097 (2014).
Etienne, A., Génard, M., Lobit, P., Mbeguié-A-Mbéguié, D. & Bugaud, C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 64, 1451–1469 (2013).
Cebolla-Cornejo, J. et al. Evaluation of genotype and environment effects on taste and aroma flavor components of Spanish fresh tomato varieties. J. Agric. Food Chem. 59, 2440–2450 (2011).
Karppinen, K., Zoratti, L., Nguyenquynh, N., Häggman, H. & Jaakola, L. On the developmental and environmental regulation of secondary metabolism in Vaccinium spp. berries. Front. Plant Sci. 7, 655 (2016).
Blanca, J. et al. Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genom. 16, 257 (2015).
Hamilton, J. P. et al. Single nucleotide polymorphism discovery in cultivated tomato via sequencing by synthesis. Plant Genome J. 5, 17 (2012).
Sim, S. C. et al. Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS One 7, e40563 (2012).
Xu, J. et al. Phenotypic diversity and association mapping for fruit quality traits in cultivated tomato and related species. Theor. Appl. Genet. 126, 567–581 (2013).
Víquez-Zamora, M. et al. Tomato breeding in the genomics era: insights from a SNP array. BMC Genom. 14, 354 (2013).
Tieman, D. M. et al. Identification of loci affecting flavour volatile emissions in tomato fruits. J. Exp. Bot. 57, 887–896 (2006).
Aulchenko, Y. S., Ripke, S., Isaacs, A. & van Duijn, C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007).
Bauchet, G. et al. Use of modern tomato breeding germplasm for deciphering the genetic control of agronomical traits by Genome Wide Association Study. Theor. Appl. Genet. 130, 875–889 (2017).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Jombart, T. et al. Package ‘ adegenet’. Bioinforma. Appl. Note 24, 1403–1405 (2008).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Li, M. X., Yeung, J. M. Y., Cherny, S. S. & Sham, P. C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756 (2012).
Yang, J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011).
de Bakker, P. I. W. et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 17, 122–128 (2008).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 27, 2336–2337 (2011).
Fernandez-Pozo, N. et al. The tomato expression atlas. Bioinformatics 33, 2397–2398 (2017).
Acknowledgements
J-T.Z. was funded by a Chinese Scholarship Council (CSC) scholarship. We thank Guangtao Zhu from Huang’s group in helping by providing the original GWAS results of panel T and discussions about the results. We thank Qi Wu from the University of Cambridge for detailed theoretical explanations about linkage disequilibrium and population genetics. We thank David Francis from Ohio State University for the positive discussions and cross-checking the misclassification of the accessions in panel T. We thank Rebecca Stevens for the English language editing.
Author information
Authors and Affiliations
Contributions
Study design/conception: M.C., J-T.Z., C.S.; supervision: C.S, M.C.; data collection and analysis: J-T.Z., F.B., J-H.Z., D.L., G.B., S.H., D.M.T., H.J.K.; data interpretation: J-T.Z., F.B., C.S., M.C., D.M.T., H.J.K.; first draft of the manuscript: J-T.Z.; critical revisions of the manuscript: all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal Peer Review Information: Nature Communications thanks Yun Li, and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Sauvage, C., Zhao, J. et al. Meta-analysis of genome-wide association studies provides insights into genetic control of tomato flavor. Nat Commun 10, 1534 (2019). https://doi.org/10.1038/s41467-019-09462-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-09462-w
This article is cited by
-
Biofortification of Triticum species: a stepping stone to combat malnutrition
BMC Plant Biology (2024)
-
Transcriptomes of developing fruit of cultivated and wild tomato species
Molecular Horticulture (2023)
-
Bayesian estimation of multi-allele QTLs for agricultural traits in tomato using recombinant inbred lines derived from two F1 hybrid cultivars
Euphytica (2023)
-
Identification and characterization of RNA polymerase II (RNAP) C-Terminal domain phosphatase-like 3 (SlCPL3) in tomato under biotic stress
Molecular Biology Reports (2023)
-
Plasticity of maternal environment-dependent expression-QTLs of tomato seeds
Theoretical and Applied Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
