
Abstract
Culture contamination, end-product toxicity, and energy efficient product recovery are long-standing bioprocess challenges. To solve these problems, we propose a high-pressure fermentation strategy, coupled with in situ extraction using the abundant and renewable solvent supercritical carbon dioxide (scCO2), which is also known for its broad microbial lethality. Towards this goal, we report the domestication and engineering of a scCO2-tolerant strain of Bacillus megaterium, previously isolated from formation waters from the McElmo Dome CO2 field, to produce branched alcohols that have potential use as biofuels. After establishing induced-expression under scCO2, isobutanol production from 2-ketoisovalerate is observed with greater than 40% yield with co-produced isopentanol. Finally, we present a process model to compare the energy required for our process to other in situ extraction methods, such as gas stripping, finding scCO2 extraction to be potentially competitive, if not superior.
Similar content being viewed by others


Introduction
Supercritical fluids offer many advantages over conventional solvents due to their gas-like viscosity and diffusivity with liquid-like density and solvation1. Supercritical carbon dioxide (scCO2) is especially promising as a green solvent due to its moderate critical point temperature (31.1 °C) and pressure (7.38 MPa) as well as being non-flammable, non-hazardous, abundant, and inexpensive2. Numerous biocatalytic processes have taken advantage of scCO22,3, including enantiomer specific lipase reactions4, asymmetric alcohol reduction5, and carboxylation of various substrates6. The solvent properties of scCO2 have been exploited for the separation of alcohols, aldehydes, ketones, and acids from aqueous solutions via both in situ and ex situ extraction7. In situ product extraction has been shown to have a dramatic effect on bioproduction titers by alleviating end-product inhibition or toxicity8,9,10,11. scCO2 is especially attractive within biphasic scCO2-aqueous systems12 due to its preferential extraction of alcohols with intermediate-chain length (e.g., 3 to 6 carbons), the result of decreasing water solubility with increasing number of carbons. Conversely, alcohol volatility decreases with chain length, making these molecules more attractive targets for scCO2 extraction compared to gas stripping12.
Despite the potential to couple scCO2 for product removal with microbial synthesis of target compounds, the two have not been combined due to the broad microbial lethality of scCO213, where cellular inactivation is thought to occur through a combination of membrane disruption, cytosolic acidification, enzyme inactivation, and cellular desiccation14. Several early studies have explored processes catalyzed by whole cells under scCO2; however, these were conducted over short time scales ( < 1 day) using biomass grown in the absence of scCO2, and without evidence of cell viability under scCO2. Two results include carboxylation of pyrrole substrate with Bacillus megaterium cells in a biphasic mixture of buffered solution and scCO2 after 1 h15, and reduction of ketones with a high degree of enantiomer specificity using immobilized Geotrichum candidum cells in an aqueous-scCO2 biphasic continuous flow reactor16. Several additional studies have employed scCO2-based extraction of bioproducts, but have not achieved in situ extraction due to inhibited cell growth. Using a semi-continuous process, ethanol was extracted from the spent fermentation broth of yeast17 and Clostridium18 cultures using scCO2. Knutsen et al.19 attempted to produce ethanol from cellobiose in dual-phase aqueous bioreactors containing non-growing Clostridium thermocellum and pressurized scCO2, N2 or ethane headspaces. Significant cellobiose conversion and ethanol production were observed under pressurized nitrogen and ethane, but not under scCO219, indicating process inhibition by CO2.
Bioprospecting is a promising approach to recover organisms capable of active metabolism under high pressures of CO2. Numerous environmental isolates have been characterized with tolerance to high partial pressures of CO220, although rarely are cultures examined above atmospheric pressures (0.1 MPa). Santillan et al.21 found a strain of Lactobacillus casei in a terrestrial CO2-rich spring that can grow in CO2 pressures up to 1 MPa. Recently, Peet et al.22 examined organisms capable of growth at geologic CO2 sequestration sites, isolating several endospore-forming Bacillus species that are tolerant to scCO2 treatment ( > 10 MPa), but with low growth frequency and magnitude. Similarly, growth was reported for laboratory cultures inoculated with deep subsurface sandstone under scCO2, although the identities of microorganisms were not determined23. Motivated by the hypothesis that natural deposits of high-pressure CO2 would harbor microorganisms adapted to actively grow in close contact with it, Freedman et al.24 investigated the microbial diversity at McElmo Dome, a deep subsurface CO2 reservoir that was formed 40–72 million years ago. Laboratory enrichment cultivation of fluids collected from McElmo dome under scCO2 enabled isolation of B. megaterium strain SR7, an endospore-forming, facultative-anaerobe25. We have observed growth of SR7 cultures to a cell density of greater than 107 cells ml–1 after 3 weeks under scCO2 following inoculation as endospores and supplementation with the germination inducer l-alanine25. Endospores of Gram-positive bacteria are known for their resiliency even when exposed directly to scCO222,26, and we have hypothesized that this dormant state enables these bacteria to withstand and adapt to the scCO2 environment prior to outgrowth as vegetative cells. Analysis of fermentation products from SR7 grown under scCO2 indicated the production of lactate, acetate and succinate, demonstrating active metabolism under scCO2 and the potential for microbial production to be coupled with scCO2 extraction25.
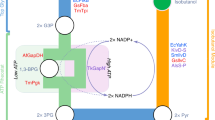
In this work, we seek to develop B. megaterium SR7 as a bioproduction host under scCO2 for useful compounds that have already been established for in situ extraction using scCO2 (Fig. 1), such as branched, intermediate-chain alcohols. We posit that an integrated fermentation-scCO2 extraction process may ultimately, and simultaneously, solve three long-standing challenges in the field by (1) reducing end-product toxicity through extraction9,10,11, (2) mitigating culture contamination under the highly selective conditions of scCO213,22,25, and (3) providing an energy efficient method to recover high-purity products using scCO2 as a sustainable solvent1,12. Branched, intermediate-chain alcohols are not naturally made by SR7, necessitating metabolic engineering to generate them25. Isobutanol is selected due to its importance as a drop-in replacement for gasoline and its favorable fuel characteristics (i.e., high energy density, suitable research octane number, low hygroscopicity). Additionally, isobutanol is an attractive molecule for in situ scCO2 extraction as it is cytotoxic and expected to partition favorably to the scCO2 phase7,12. Isobutanol production requires introduction of two heterologous enzymes, an α-ketoisovalerate decarboxylase and alcohol dehydrogenase, to convert the valine-synthesis intermediate, 2-ketoisovalerate, to the final product with minimal intermediate isobutyraldehyde accumulation27. We conclude our study by analyzing the energy requirements of an integrated fermentation-extraction process, comparing it to alternative in situ extraction technologies such as gas stripping, pervaporation, and adsorption28, in order to address challenges associated with energy efficient product recovery.

Schematic of integrated fermentation and extraction under supercritical CO2. A scCO2-tolerant microbe can be engineered to produce compounds, such as medium-chain alcohols (C4-C5), that may serve as biofuels and may be preferentially extracted from aqueous media into the scCO2 phase. Collection of scCO2 followed by partial de-pressurization will facilitate high-purity biofuel extraction. The presented biphasic scCO2 separation strategy is expected to simultaneously provide a contaminant-free environment for the engineered organism due to the broad microbial lethality of scCO2, and continuously strip microbially produced biofuels to eliminate end-product toxicity. scCO2 is only weakly soluble in the aqueous media phase ( < 1%) and vice versa, resulting in a biphasic system
Results
Heterologous protein expression in aerobically grown SR7
As strain B. megaterium SR7 is an environmental isolate, its genetic modification required optimization and customization of protocols previously developed for laboratory strains and model organisms. A transformation protocol for SR7 was developed by modifying a method based on protoplast-osmotic shock29 (Supplementary Note 1). The xylose-inducible promoter system (pXyl) was selected for use in SR7 due to its simplicity, high expression level, and widespread use in the B. megaterium literature29 (Fig. 2a). Additionally, the IPTG-inducible hyperspank promoter system (pHysp)30 and the p43-growth based promoter (p43) were evaluated, both of which have shown effective protein production in B. subtilis31 (Fig. 2a).

Heterologous protein expression and promoter evaluation in SR7. a Schematic of the promoters tested in SR7. pXyl is a xylose-inducible promoter and contains the xylR gene and xylA promoter from B. megaterium strain DSM 319. pHysp contains the IPTG-inducible hyperspank promoter and lacI, both from plasmid pDR111. The p43 promoter is taken from B. subtilis strain KS438 and is growth-associated. Expected inducers are indicated in parentheses. b Bulk fluorescence measurements of SR7 populations containing gfp-encoding plasmids, with (circles, solid lines) or without (diamonds, dashed lines) inducers. Error bars represent the standard deviation of biological triplicate samples. c Fluorescence populations of SR7 cells expressing gfp at 4 h post induction measured by FACS (Alexa Fluor 488). A GFP positive gate was established using the autofluorescence of SR7 cells containing an empty plasmid control. d Phase-contrast and fluorescence microscopy of SR7 pXyl gfp cells with and without xylose induction. Images were taken 4 h post induction. Source data are provided as a Source Data file
All three promoters were functional in SR7 (Fig. 2b and Supplementary Fig. 1), with pXyl and pHysp promoters resulting in strong GFP expression of 25- and 20-fold greater than uninduced cultures, respectively. The xylose promoter was activated most rapidly, achieving maximal fluorescence 3 h post induction, similar to other B. megaterium strains using this promoter system32. For induced pXyl gfp and pHysp gfp cultures, a large proportion ( > 95%) of the population was found to express GFP (Fig. 2c, d and Supplementary Table 1), which is greater than that observed for other B. megaterium strains using pXyl33. Uninduced pXyl gfp containing cells have many members (40%) that fall into the GFP positive gate, providing evidence that this promoter is leaky when grown aerobically in rich Luria-Bertani (LB) medium (Supplementary Table 1 and Supplementary Fig. 2), in contrast to previous work showing tight regulation of pXyl within B. megaterium strain WH32032. As expected, the growth-associated p43 promoter was activated as the cells entered exponential growth phase (Supplementary Fig. 1e, f), but showed weaker GFP expression, only sixfold greater than the empty plasmid-containing control strain. Inducible production of GFP for the pXyl and pHysp promoters was also observed for cells cultured in semi-defined medium supplemented with xylose and/or glucose, with maximal fluorescence observed for cells containing the pXyl promoter grown in xylose-amended medium (Supplementary Fig. 3). Expression of gfp from the xylose promoter was shown to be lowered when both glucose and xylose sugars are present, likely due to catabolite repression, as observed for other strains of B. megaterium34,35 (Supplementary Fig. 4). Based on the rapidity, strength, and uniformity of induction, the remaining work presented herein utilized the pXyl promoter. Plasmid retention was assessed, as the literature is mixed regarding long-term exogenous plasmid stability in B. megaterium36,37. Plasmid retention ( > 87%) was verified after 74 h of culturing by counting colony forming units (CFUs) on media with and without antibiotic selection as well as confirming the presence of plasmid elements for individual colonies (Supplementary Table 2 and Supplementary Fig. 5).
Heterologous protein production in SR7 grown under scCO2
The anaerobic functionality of the xylose promoter was next established for SR7 grown under 0.1 MPa CO2 (i.e., atmospheric pressure) as well as a 10 MPa headspace of scCO2. Several challenges were anticipated when moving from aerobic to scCO2-based culturing, including slower and less predictable growth due to stresses associated with scCO2, and reduced maintenance of non-native plasmids through sporulation, germination, and outgrowth. Since the formation of the GFP chromophore is oxygen dependent, the β-galactosidase protein (LacZ) from E. coli was selected as a reporter for anaerobic cultures due to its previous use in B. megaterium32, its oxygen independence, and its easy and sensitive detection via enzymatic assay.
After confirming heterologous lacZ expression (Fig. 3) and plasmid maintenance under 0.1 MPa CO2 (Supplementary Note 2), the functionality of the pXyl promoter was assessed for SR7 pXyl lacZ cells grown under 10 MPa scCO2 at 37 °C in semi-defined medium containing glucose as a carbon source and l-alanine as a germination inducer25. Xylose was added as an inducer at the time of inoculation rather than during early exponential growth since high-pressure cultivation of SR7 in stainless steel columns precludes non-destructive amending of samples during cultivation. After 21 days of incubation under scCO2, LacZ activity was measured in crude lysates derived from columns that displayed growth. Cells grown under 0.1 MPa and 10 MPa CO2 with xylose displayed similar LacZ specific activities of 0.56 and 0.40 U mg–1, respectively (Fig. 3). The 15-fold increase in LacZ expression under 10 MPa relative to uninduced cultures (p = 0.0059) is similar in magnitude to the increase in induced-expression observed under 0.1 MPa (Fig. 3).

β-Galactosidase activity for SR7 cultures grown under 0.1 MPa CO2 and 10 MPa scCO2 at 37 °C. Specific β-galactosidase activity measured in lysates of SR7 cultures that contain an empty pXyl plasmid or pXyl lacZ. Activity was normalized to total protein concentration in the lysate to control for growth differences and differential cell lysis. Errors represent standard deviation of biological replicate cultures (n > 4). For cultures grown under 0.1 MPa CO2, overnight anaerobic cultures grown from spores were subcultured into CO2-evacuated vials to a uniform optical density. Induction with xylose occurred 2 h after subculture, and the time point for LacZ activity measurement was taken 4 h post induction. For cultures grown under 10 MPa scCO2, SR7 pXyl lacZ spores were loaded into stainless steel culture vessels with and without xylose inducer. Cultures were grown for 21 days and microscopy was used to determine which samples showed at least a tenfold increase in cell number prior to measuring LacZ activity. Source data are provided as a Source Data file
Growth of SR7 from endospores under scCO2 has been defined as at least a tenfold increase in cell number and presence of vegetative cells, as determined by direct cell counts using fluorescence microscopy25. SR7 pXyl lacZ cultures amended with xylose grew in 8 of 55 columns (15%) while growth was observed in 5 of 34 unamended columns (15%) (Supplementary Table 3), which together represent a 4.4-fold decrease in growth frequency relative to wild-type SR7 (64%) under the same conditions (i.e., 5 ml culture volume with 3 × 105 spores ml–1 inoculum and 5 ml scCO2 headspace)25. Previous studies have shown bacteria transformed with exogenous plasmids are more susceptible to cell death than wild-type strains, especially under harsh culture conditions38,39, consistent with our results. Improving growth of genetically modified SR7 remains an important target for further optimization.
Alcohol production and optimization in aerobically grown SR7
An isobutanol bioproduction pathway was developed in SR7 due to previous success at achieving high yields and titers of this compound in B. subtilis31,40, and the fact that the isomer n-butanol can be effectively extracted from a biphasic aqueous-scCO2 reactor12. Furthermore, isobutanol generation from 2-ketoisovelerate (αKIV) is expected to require heterologous expression of only one enzyme, α-ketoisovalerate decarboxylase (KivD from L. lactis)27 (Fig. 4a), since the SR7 genome encodes eight predicted alcohol dehydrogenases25. For SR7 grown in LB medium under aerobic conditions, isobutanol production was only observed for cells that contained kivD, with approximately 50% yield on αKIV in cultures that were both induced with xylose and fed the αKIV substrate (Fig. 4b). To increase product yield, the alcohol dehydrogenase ADH6Sc from S. cerevisisae was overexpressed in tandem with KivD, resulting in approximately 70% yield of isobutanol on αKIV for xylose-induced cells (Fig. 4b). In addition to the target isobutanol molecule, isopentanol (Fig. 4c and Supplementary Figs. 6 and 7) and phenylethyl alcohol (PEA) (Supplementary Fig. 8a) production were also observed, with the isopentanol titer being greater than that of isobutanol. Accumulation of these products is not surprising given the known promiscuity of KivD towards other amino acid synthesis pathway intermediates27 (Supplementary Fig. 8b).

Isobutanol and isopentanol production in SR7. a Two-step enzyme pathway to convert the valine intermediate 2-ketoisovalerate (αKIV) to isobutanol using the decarboxylase enzyme KivD from L. lactis and an alcohol dehydrogenase. b Evaluation of isobutanol production at 24 h post induction in SR7 with an empty pXyl plasmid, pXyl kivD, pXyl kivD ADH6Sc, or pXyl kivD yqhDEc, where ADH6 and YqhD are alcohol dehydrogenases from S. cerevisiae and E. coli, respectively. Cultures were grown aerobically in LB medium in the presence and absence of 5 g l–1 xylose as an inducer and 5 mM αKIV as substrate for the pathway. For all aerobic experiments, error bars represent the standard deviation of biological triplicate cultures. c Co-production of isopentanol measured in SR7 samples shown in b. d Accumulation of the isobutyraldehyde intermediate at short culture times, 4 h post induction. Alcohol dehydrogenases ADH6 from S. cerevisiae, AdhA from L. lactis, AdhP from E. coli, YqhD from E. coli, and AdhA from B. megaterium SR7 were evaluated to decrease the buildup of isobutyraldehyde intermediate for aerobic cultures fed 5 mM αKIV and induced with xylose. e Production of isobutanol at 24 h post induction for the cultures shown in d. f Production of isobutanol for SR7 cultures grown under 0.1 MPa CO2 (biological triplicate cultures) and 10 MPa scCO2 at 37 °C. Under scCO2, cultures with at least a tenfold increase in cell number, as enumerated by microscopy, were analyzed for alcohol production, sugar consumption and fermentation product generation. Average isobutanol titers from the aqueous phase of cultures showing at least 1 mM glucose consumption (classified as high activity; Supplementary Fig. 11) are provided. Source data are provided as a Source Data file
Biosynthesis of alcohols entails conversion of aldehyde intermediates (Fig. 4a) that tend to be highly soluble in scCO2, partitioning approximately tenfold more readily into scCO2 than their cognate alcohols7. Thus, any accumulation of aldehyde is hypothesized to result in premature extraction of this compound under scCO2, resulting in lower alcohol production. Under aerobic conditions at short induction times of 4 h, isobutyraldehyde buildup was observed, even for cells that contained the alcohol dehydrogenase ADH6Sc (Fig. 4d). Four additional alcohol dehydrogenases were screened for reduced aldehyde accumulation: three previously shown to be functional within the context of the isobutanol pathway41, and a predicted alcohol dehydrogenase native to SR7 (AdhABm) with high homology to AdhALl from L. lactis. The YqhD enzyme from E. coli, known to be highly effective for producing isobutanol41,42, was identified as the superior variant, with the lowest amount of aldehyde accumulation at short culture times (4 h) (Fig. 4d), over 80% yield of isobutanol from αKIV (Fig. 4e), and maintained capacity for isopentanol production (Fig. 4c).
The optimized two-enzyme isobutanol production pathway was evaluated aerobically in semi-defined medium containing 4 g l–1 glucose and 5 g l–1 xylose (conditions previously shown to support heterologous enzyme production in SR7 under anaerobic conditions). The final isobutanol titer was approximately half of that observed in LB medium (Supplementary Fig. 9a), possibly due to lower heterologous enzyme expression and more αKIV substrate being directed towards biomass in the lower nutrient semi-defined medium. Increasing the glucose concentration to 10 g l–1 in the semi-defined medium increased the titers of all three alcohols, suggesting that cellular resources may be directed towards these products when carbon is provided in greater excess (Supplementary Fig. 9b). When neither glucose nor xylose were supplied, minimal biomass (OD600 < 0.1) or product accumulated (Supplementary Fig. 9c), indicating that neither growth nor alcohol production is due to trace amounts of yeast extract (50 mg l–1). Xylose alone enabled high cell density growth (OD600 > 6.5), and in the case of isopentanol, almost 1 mM production, with a further increase in titer upon adding αKIV substrate (Supplementary Fig. 9c). When combined with xylose, the addition of 100 mM alanine (used under anaerobic CO2 conditions for endospore germination) increased cell growth (OD600 > 15) and more than doubled isobutanol production (Supplementary Fig. 9d), likely due to the deamination of alanine to pyruvate, a precursor for isobutanol via the valine-synthesis pathway and part of central carbon metabolism.
Isobutanol production in SR7 under scCO2
After optimizing aldehyde conversion and alcohol production for SR7 under aerobic conditions, functionality of the two-step isobutanol pathway was assessed in semi-defined medium under anaerobic conditions (Fig. 4f). Seed cultures of SR7 pXyl kivD yqhDEc cells passaged under 0.1 MPa CO2 into fresh medium amended with xylose and 5 mM αKIV substrate generated 4.43 mM isobutanol after 48 h of incubation. The final isobutanol titer under anaerobic 0.1 MPa CO2 headspace is greater than the 2 mM isobutanol generated by aerobic cells grown in the same medium conditions. Isopentanol and PEA titers were 1.8 and 0.21 mM, respectively (Supplementary Fig. 10). As expected, no isobutanol or isopentanol was observed for SR7 cultures that contained an empty plasmid (Fig. 4f and Supplementary Fig. 10).
Biofuel production was next evaluated for SR7 cultures grown under supercritical CO2 at 10 MPa and 37 °C. Cultures were grown for 21 days in semi-defined medium from SR7 endospores containing the desired biofuel pathway or empty plasmid, with 4 g l–1 glucose, 5 g l–1 xylose, 5 mM αKIV, and 100 mM alanine added at the time of inoculation. As was found for SR7 pXyl lacZ, the addition of a plasmid resulted in lower frequency growth (27%) as compared to SR7 wild-type (64%)25 (Supplementary Table 3). For cultures that displayed growth, glucose consumption was not correlated with increases in cell number (Supplementary Fig. 11a), possibly due to consumption of yeast extract and l-alanine. Glucose consumption was found to correlate strongly with the production of fermentation products lactate and acetate (R2 = 0.97) (Supplementary Fig 11b), which have previously been shown to accumulate in SR7 cultures grown under scCO225. This finding led to classifying samples that consumed more than 1 mM glucose as having high activity, as all of these samples also produced greater than 1 mM combined lactate and acetate (Supplementary Fig. 11b). Other samples that showed at least tenfold growth, but less than 1 mM glucose consumption were classified as having low activity since they neither consumed sugar substrate nor generated meaningful amounts of fermentation products (Supplementary Fig. 11b).
Isobutanol production was measured from the aqueous phase of scCO2 cultures grown in the presence and absence of xylose (Supplementary Fig. 12). Xylose-containing high activity cultures produced an average of 1.23 mM isobutanol, whereas those without xylose generated approximately half that amount (Fig. 4f and Supplementary Table 4). Isobutanol titers normalized to cell density were comparable under 0.1 MPa CO2 and 10 MPa scCO2, at 2.2 × 10–8 μmol cell–1 and 3.1 × 10–8 μmol cell–1, respectively (Supplementary Table 5). While the high activity, empty vector control cultures consumed up to 1.9 mM αKIV under 10 MPa scCO2, isobutanol was not detected in any of these samples (Supplementary Table 4), an identical outcome to aerobic and 0.1 MPa CO2 cultures. The isobutanol yield from αKIV ranged from 37 to 45% in the aqueous phase for xylose-induced high activity biofuel cultures, 2.8-fold higher than for those that lacked xylose (Supplementary Table 4). Isopentanol generation was also observed for samples grown under 10 MPa scCO2, reaching an average titer of 0.31 mM (Supplementary Table 4), while only trace amounts of PEA were detected under scCO2. The amount of isobutanol produced by low activity cultures was on average 35-fold less than for the high activity cultures (Supplementary Table 4).
Extraction of isobutanol using scCO2
Inspired by our demonstration of microbially generated alcohols under scCO2, we attempted to recover product from SR7 cultures using scCO2. Partitioning of biologically produced isobutanol into the scCO2 headspace was observed for the aforementioned SR7 cultures (Supplementary Fig. 13), albeit at marginal concentrations. Isopentanol was not detected in the headspace samples. Our proof of concept demonstration for isobutanol extraction motivates adoption of two engineering solutions to increase recovery: (1) use of flowing scCO2 to increase mass transfer and (2) controlled de-pressurization to capture scCO2-solubilized alcohol. To evaluate these solutions, we assessed the recovery of isobutanol in the absence of microbial cells using a stirred two-phase reactor that is designed for in situ extraction using scCO2 stripping (Supplementary Fig. 14), as previously done for n-butanol and n-pentanol12. Similar to straight-chained alcohols, the rate of isobutanol extraction from aqueous solutions increases with low scCO2 flow rates, reaching a maximum at a flow rate of 3.2 ml min–1, most likely due to a tradeoff between convective and diffusive mass transfer. Isobutanol extraction rate and efficiency using scCO2 was found to be approximately 25% greater than that for n-butanol (Supplementary Fig. 14). Taken together, these data indicate that scCO2 is effective for rapid extraction of isobutanol from dilute aqueous solutions and identifies scale up of cultures to a similarly designed system as a priority for further development.
Process analysis for extraction using scCO2
The experimental component of this study established growth and heterologous bioproduction in the presence of scCO2 and extraction of isobutanol into the scCO2 phase in a stirred reactor. Next, we constructed a fermentation-extraction process model to assess technological potential using detailed mass and energy balances. The objective of the analysis was calculation of isobutanol recovery energy requirements for comparison with previously reported analyses of butanol recovery using other methods28. In practice, Oudshoorn suggested a benchmark of 5 MJ kg–1 as the maximum energy requirement for butanol recovery28. Unfortunately, previous studies of butanol recovery from dilute fermentation broths ( < 20 g l–1) using a wide range of conventional technologies, such as gas stripping, adsorption, and liquid–liquid extraction, report energy requirements in the range of 10–20 MJ kg–128. By comparison, Tompsett et al.12 estimated the energy demand for the CO2 compression required for scCO2 extraction of dilute n-butanol to be 4.0 MJ kg–1. Since CO2 compression is expected to represent the majority of the energy required by the scCO2 extraction process, these results12 suggest that the energy requirement of scCO2 extraction might compare favorably with alternative technologies — especially since efforts to minimize CO2 compression requirements were not considered. Moreover, this previous study12 did not analyze the performance of an integrated fermentation-extraction process since bioproduction in the presence of scCO2 has never been reported prior to this work. Accordingly, the current study evaluated approaches to reduce CO2 compression requirements, the advantages associated with coupled fermentation-extraction (Supplementary Note 3), and the intrinsic performance of the scCO2 extraction process.
With emphasis on evaluating energy demand for recovering a high-purity product, we developed an Aspen Plus simulation model (Fig. 5a and Supplementary Fig. 15a) to analyze a process consisting of integrated fermentation and extraction using scCO2. Isobutanol extraction performance was simulated using data reported in the current work, literature data on n-butanol extraction12,43,44, and established thermodynamic properties45,46 (Supplementary Note 3). Sensitivity analysis revealed that the most influential parameter in determining isobutanol energy requirement was the CO2:isobutanol feed ratio since it directly relates to the amount of CO2 that must be compressed for extraction (Supplementary Fig. 15b and Supplementary Note 3). Combining our isobutanol extraction data with values reported on n-butanol44, we calculate that 99.73% isobutanol recovery could be achieved at a CO2:isobutanol feed ratio of 3:1 as a base case. To reduce CO2 compression energy demand, product recovery via partial de-pressurization of the CO2 stream was explored, finding > 94% pure isobutanol product could be recovered by partially de-pressurizing the stream exiting the fermenter to between 3.5 and 6.5 MPa45. In this pressure range, liquid–liquid phase splitting occurs, resulting in isobutanol-rich and water-rich liquids45. Partial de-pressurization allows recycling of a high-pressure CO2 stream, further reducing CO2 compression demand and thereby energy consumption (Supplementary Fig. 15b). Lastly, the sensitivity of the process to the ratio of recycled to purged CO2 was investigated using an optimized intermediate pressure of 6.5 MPa (Supplementary Fig. 15b). Increasing the CO2 recycle amount decreased energy requirements nearly linearly, with the best performance observed at the limit of 87% recycle (Supplementary Fig. 15c).

Isobutanol energy recovery requirements for integrated fermentation-extraction under scCO2. a Simplified process schematic of the envisioned scCO2 bioprocess used to develop an Aspen Plus model, including compression of CO2 to scCO2, fermentation/extraction, de-pressurization/biofuel recovery, and CO2 recycle. Further detail is provided in Supplementary Fig. 15a. b Energy requirement for isobutanol recovery as MJ kg−1 for several scenarios. Separate fermentation and scCO2 recovery processes represent the base case (scenario A). Scenario B is coupled fermentation and extraction without use of partial extraction stream de-pressurization or scCO2 recycle. Scenario C de-pressurizes the extraction stream to 6.5 MPa to induce formation of an isobutanol-rich stream, which permits recycling of compressed CO2, set at 50% (by mass). Scenario D again de-pressurizes to 6.5 MPa and recycles the maximum possible amount of CO2 (87%) that results in no accumulation of CO2 in the system. Values for b can be found in Supplementary Table 6. c Comparison of the energy requirements for isobutanol production found for the scCO2 process relative to published literature values for alternative in situ recovery methods28. The scCO2 extraction energy requirement in c is for coupled fermentation and extraction with partial de-pressurization and full recycle (i.e., Scenario D in b). Source data are provided as a Source Data file
Based on these sensitivity analyses, four scenarios were assessed using the process model, with specific emphasis on evaluating the extent to which partial de-pressurization and CO2 recycling could be used to decrease energy requirements of isobutanol extraction (Fig. 5b and Supplementary Table 6). Atmospheric-pressure fermentation followed by scCO2 extraction (scenario A) required an estimated 6.80 MJ kg–1, with compression accounting for approximately 66% of the energy demand. An integrated process, with complete de-pressurization to 0.1 MPa and no CO2 recycling (scenario B), reduced the energy burden to 4.4 MJ kg–1, with most of the improvement stemming from reduced compression requirements. Intermediate de-pressurization to 6.5 MPa combined with 50% CO2 recycle (scenario C)43, further reduces the energy requirement to 2.4 MJ kg–1, again largely due to diminished compression requirements. Finally, placing the CO2 recycle ratio at its maximum value (87%) (scenario D) and de-pressurizing the extraction stream to 6.5 MPa reduces the energy requirement to 1.4 MJ kg–1. As expected from previous arguments, the newly established energy requirement is lower than that previously reported for stand-alone fermentation and scCO2 extraction, and much less than any figure reported for conventional technologies such as gas stripping, liquid–liquid extraction, or adsorption (Fig. 5c). Although the energy requirement estimated here for integrated extraction-fermentation has not yet been demonstrated in practice, the current finding motivates further evaluation, with our model serving as a basis for follow-on techno-economic, emissions, and life cycle assessment studies.
Discussion
In this work, we report the production of alcohol biofuels under supercritical carbon dioxide. Using an environmentally isolated strain (Bacillus megaterium SR7) native to a deep subsurface CO2 reservoir, a transformation protocol was developed and heterologous protein expressed from three promoters. Furthermore, inducible β-galactosidase production was observed for cells grown under scCO2. SR7 was engineered to produce isobutanol from αKIV through a two-step metabolic pathway that simultaneously produced isopentanol. Both isobutanol and isopentanol have potential as drop-in biofuels or fuel additives, with comparable energy densities (29 MJ l–1 and 28 MJ l–1, respectively) and research octane numbers (98 and 105)47. Similar to isobutanol, isopentanol is predicted to be highly miscible in scCO212. For cultures grown under scCO2 that showed high activity, isobutanol titers were greater than 1 mM, with 42% average yield on consumed αKIV substrate. Importantly, while total titers were reduced at 10 MPa, specific titers under scCO2 were similar to those observed under 0.1 MPa conditions, at 2.2 × 10−8 μmol cell–1 and 3.1 × 10−8 μmol cell–1, respectively. Recovery of microbially generated isobutanol using scCO2 was demonstrated, but limited, due to both the configuration of the high-pressure culturing vessels and the low titers achieved. Separation of dilute isobutanol from aqueous media using scCO2 is possible using a two-phase extractor-fermenter, as previously observed for straight-chain alcohols12. Lastly, process analysis shows that using scCO2 to extract biofuels is energetically feasible and comparable, if not better, than other in situ extraction techniques if sufficiently high product titers can be achieved. As such, this work serves as a starting point for bioproduction under scCO2 with numerous avenues for continued optimization.
Further development of the genetic toolbox for SR7 will enable future metabolic engineering of SR7 for enhanced biochemical production. Isobutanol pathway optimization was briefly explored by screening five alcohol dehydrogenases and selecting the variant that resulted in highest substrate conversion ( > 80%) and minimal aldehyde accumulation at short incubation times. Additionally, isopentanol and phenylethyl alcohol production were observed through the simple addition of a decarboxylase, possibly due to upregulation of the leucine and phenylalanine synthesis pathways in SR7. Other strains of B. megaterium are known to accumulate the amino acids lysine48 and possibly valine49; however, evidence for leucine and phenylalanine accumulation was not previously observed49. The leucine biosynthesis pathway may become a starting point for future engineering efforts to increase isopentanol titers or make additional bioproducts due to the already presumably high flux of the pathway50. To ensure that coupled fermentation-extraction technology is utilized to its full potential, initial screening of subsequent compounds to be made by SR7 will be determined based on their preferential partitioning into scCO2 and toxicity to bioproduction hosts.
Taking advantage of scCO2 for properties beyond its well-established extractive12 and sterilizing capabilities13 would confer additional utility to this technology. For example, scCO2 has been used for the depolymerization of lignocellulosic biomass to release fermentable sugars51. Combining the two technologies could provide an inexpensive substrate for SR7 growth and bioproduction. Carboxylation reactions may also be possible in SR7, directly using CO2 or HCO3– found in solution for bioproduction52,53. Even with the low solubility of scCO2 in water54, the approximate HCO3– concentration of 1.1–1.3 mM (based on Henry’s law and the reaction of CO2(aq) with water) is advantageous relative to other studies that use air as a CO2 source for carbon fixation through heterologous overexpression of a carboxylase55,56. SR7 contains several predicted carboxylases, including pyruvate carboxylase, two PEP carboxylases and a biotin-dependent acetyl-CoA carboxylase, each of which could serve as pathways to route HCO3– into central carbon metabolism25. Additionally, the SR7 genome encodes two predicted carbonic anhydrases for converting CO2(aq) to HCO3–. Lastly, Bacillus megaterium strains are known for their ability to secrete proteins57, conferring further biotechnological utility to this host for ex vivo reactions.
One of the limitations to using scCO2 for bioprocessing is the slow and infrequent growth observed for SR7, especially when bearing an exogenous plasmid. Significant improvements were previously made for scCO2 culturing using SR7 through media optimization and recognition of endospore germination as a key step for acclimation of this strain to conditions associated with scCO2 exposure25. Future work will be conducted to better understand the factors limiting growth of SR7 under scCO2 and to select for strains of SR7 that demonstrate more robust and rapid outgrowth into vegetative cells. Enhanced understanding and substantial improvement of SR7 growth will similarly inform economic, environmental, and energy analysis of the integrated process, due to the inter-relationships between growth, productivity, titer, scCO2 volumes and extraction efficiency, energy requirements, and ultimately cost. Numerous studies have shown the ability to improve the tolerance of microbes to harsh environments, including through the use of adaptive laboratory evolution58. Many of these studies, however, found that increased tolerance of strains does not necessarily correlate with enhanced specific titers when used for bioproduction59,60,61. For SR7, the fact that specific titer is comparable under atmospheric and scCO2 conditions suggests that scCO2 does not impair specific productivity and that improvements in titer observed under 1 atm CO2 will positively translate into higher titers under scCO2. Using the plasmid bearing strains in this work, future selection of SR7 mutants that have a higher growth capacity despite the burden associated with plasmid maintenance and expression may also be possible62. Additionally, genomic integration could alleviate plasmid-maintenance burden for SR7 strains, and recently developed Cas9-based genomic modification strategies for B. subtilis strains63,64 provide a starting point for integrating and removing desired and competing pathways, respectively, in SR7.
The isolation, characterization and genetic engineering of an scCO2-tolerant strain of B. megaterium establishes exciting biotechnological opportunities for in vivo high-pressure CO2 bioprocessing coupled with in situ product extraction. While low-frequency growth from Bacillus spp. spores22 and enzymatic catalysis15 have previously been shown under scCO2, this work demonstrates combined heterologous enzyme expression and bioproduct synthesis under these harsh conditions. Proven scCO2 stripping of biofuels and favorable energy requirements of scCO2-based bioprocessing lend credibility to the adoption of this process for commercial applications, motivating continued work to improve titers, yields, and productivities. Achieving such improvements would enable demonstration of the integrated extraction-fermentation process at a larger scale, facilitating collection of data needed to form the basis for full techno-economic and life cycle analyses, and more detailed comparisons to other extraction strategies and renewable energy sources.
Methods
Plasmid construction
For plasmid construction (Supplementary Table 7), restriction enzymes, T4 DNA ligase, and Q5 High-Fidelity PCR MasterMix were purchased from NEB and used according to manufacturer recommendations. The pXyl gfp plasmid is based on the pRBBm34 shuttle vector65 (purchased from Addgene), which contains a xylose-inducible promoter, the xylose repressor (xylR) under control of its native B. megaterium promoter, ampicillin resistance marker (Ampr) for selection in E. coli, tetracycline resistance marker (Tetr) for selection in B. megaterium, and two origins of replication. The gene encoding SuperFolder GFP was PCR amplified from the pTRC99a gfpSF (lab stock) template using the oligonucleotides SpeI gfp for and gfp SphI rev (Supplementary Table 8). The resulting PCR product and pRBBm34 plasmid were digested with SpeI and SphI, and resulting fragments ligated to create pXyl gfp. The pXyl empty plasmid was generated by adding the DNA sequence 5ʹ-atttaatgttgatgaaagctggtctggtgtcaaaaataatga-3ʹ between the SpeI and SphI restriction enzyme sites. Ligated plasmid products were used to transform DH5α and plated on LB agar containing 100 μg ml–1 carbenicillin (Sigma). Plasmids were purified (Zippy plasmid miniprep kit, Zymo research) prior to verification by DNA sequencing (Genewiz) using appropriate sequencing oligonucleotide primers (Supplementary Table 8).
The xylose repressor and promoter of pRBBm34 were replaced with a hyperspank promoter and lacI using circular polymerase extension cloning (CPEC). The pRBBm34 plasmid was PCR linearized with two sets of primers to remove xylR and the xylose promoter: pRBBm34 for/bla rev and bla for/pRBBm34 rev. The hyperspank promoter and lacI were PCR amplified from pDR11166 (gift from Alan Grossman, MIT Department of Biology) using primers pRBBm34-pHysp for and lacI-pRBBm34. A standard CPEC protocol67 was used to assemble the three PCR products into the pHysp empty plasmid. Gfp was PCR amplified from pXyl gfp using primers SalI RBS gfp for and gfp NheI rev. The gfp PCR product and pHysp empty were digested with SalI and NheI, and the fragments were assembled to make pHysp gfp. To generate the p43 empty and p43 gfp plasmids, the pXyl empty and pXyl gfp plasmid (respectively) were PCR linearized with oligonucleotide sets RBS-start for/bla rev and bla for/pRBBm34 rev. The p43 promoter was PCR amplified from Bacillus subtilis strain KS438 using primers pRBBm34-p43 for and p43-RBS-start rev. The two p43 plasmids were created by assembling the three PCR fragments using CPEC.
All the remaining plasmids were made using standard restriction enzyme digest and ligation construction; the pairs of restriction enzymes used are indicated in the oligonucleotide primer names. The gene encoding β-galactosidase from E. coli was PCR amplified from the plasmid pKVS45 lacZ_LVA (lab stock) using primers SpeI lacZEc for and lacZEcSphI rev. The ketoisovalerate decarboxylase gene from L. lactis was amplified from pCOLA (Fjoh-2967) (kivD)68 with primers SpeI kivDLl for and kivDLlBamHI SphI rev for the construction of pXyl kivDLl. The ADH6 gene from Saccromyces cerevisiae was amplified from pACYC (car sfp) (ADH6Sc)68 using primers BamHI RBS ADH6Sc for and ADH6ScSphI rev to make the plasmid pXyl kivDLlADH6Sc. For all the alcohol dehydrogenases studied, the RBS 5ʹ-agggggaaa-3ʹ was used. The adhALl gene was amplified from the L. lactis subsp. lactis (ATCC 19435D) genome (primers: BamHI adhALl for and adhALlSphI rev), adhPEc and YqhDEc from the E. coli MG1655 genome (primers: BamHI adhPEc for and adhPEcSphI rev; BamHI yqhDEc for and yqhDEcSphI rev, respectively), and adhABm from the B. megaterium SR7 geneome (primers: BamHI adhABm for and adhABmSphI rev). All alcohol dehydrogenases were ligated between the BamHI and SphI sites in pXyl kivDLl.
Transformation of SR7 and verification
Protoplast transformation of SR7 was adapted from an established method by Biedendieck et al.29. All buffers described in this protocol were identical to those in Biedendieck et al.29 and were made with chemicals from Millipore-Sigma unless otherwise described. An overnight culture of SR7 was grown from the inoculation of 50 ml of LB medium (BD Difico) with a single SR7 colony, and was incubated at 37 °C and 250 r.p.m. SR7 was subcultured at a dilution of 1:100 in fresh LB medium and grown at 37 °C and 250 r.p.m. until an OD600 reading of 1.0 was reached (~3 h) (Implen NanoPhotometer). Cells were collected by centrifugation at 4000 × g for 10 min and the cell pellet was retained. The cell pellet was resuspended in 5 ml 1x SMMP buffer (2.32 g l-1 L-malic acid (Alfa Aesar), 1.6 g l-1 NaOH, 4.06 g l-1 MgCl2•6H20, 171.16 g l-1 sucrose, 17.5 g l-1 antibiotic medium number 3 (AB3) (BD Difico)). Lysozyme (Sigma) was added to a concentration of 30 μg ml–1, which was tenfold lower than used in previous methods29. Protoplast formation was performed at 37 °C for 10 min at 70 r.p.m. and was visualized by microscopy.
Protoplasts were collected by centrifugation at 1300 r.p.m. for 10 min, followed by a wash with 5 ml 1x SMMP. Protoplasts were resuspended gently in 5 ml 1x SMMP and were aliquoted into 0.5 ml fractions. Each sample was transformed with 3 μg freshly prepared plasmid DNA (Zippy plasmid miniprep kit, Zymo research) and 1.5 ml of a 20% PEG 8000 (Promega) solution in 1x SMM (2.32 g l-1 L-malic acid (Alfa Aesar), 1.6 g l-1 NaOH, 4.06 g l-1 MgCl2•6H20, 171.16 g l-1 sucrose). Samples were incubated with gentle mixing for 2 min, followed by addition of 5 ml 1x SMMP and centrifugation to collect the transformed protoplasts. The protoplasts were resuspended in 0.5 ml 1x SMMP and incubated at 37 °C for 1.5 h at 100 r.p.m. to recover. Transformed protoplasts were plated in a soft agar overlay29 on dried and pre-warmed LB agar plates containing 5 μg ml–1 tetracycline (Calibiochem). Additionally, protoplasts were plated in agar overlays on LB agar without antibiotic to determine viability. Plates were incubated for two days at 30 °C.
Confirmation of SR7 transformation was carried out using several techniques including colony PCR, streaking colonies on fresh LB agar plates containing 5 μg ml–1 tetracycline, sequencing isolated plasmid DNA, and back transforming plasmid DNA into E. coli. For colony PCR, colonies were picked and boiled for 10 min to serve as template for the PCR reaction using OneTaq Quick-Load Master Mix (NEB) and appropriate sequencing primers (Supplementary Table 8). Plasmid DNA was isolated from SR7 using a standard plasmid isolation protocol and materials (Zippy plasmid miniprep kit, Zymo research), except 10 μg ml–1 lysozyme was added for the initial resuspension step with a 1 h incubation at 37 °C to promote cell lysis. Standard Sanger sequencing of isolated plasmids was used to verify identity (Genewiz). Purified plasmids were also backtransformed into E. coli DH5α, which were subsequently plated on LB agar containing 100 μg ml–1 carbenicillin. Plasmid stability was assessed by isolating the backtransformed plasmid DNA and restriction enzyme digestion with EcoRI (NEB). For a positive control, purified pXyl gfp directly from E. coli DH5α was also digested with EcoRI. Lastly, SR7 species and strain identity was confirmed through preparation of genomic DNA (Wizard genomic DNA purification kit, Promega) and sequencing of the PCR amplified 16s rRNA and xylE genes, respectively. The 16s fragment was PCR amplified using Q5 High-Fidelity MasterMix (NEB) and the 27F and 1492R primers (Supplementary Table 8); the xylE gene was amplified using xylEBm seq for and xylEBm seq rev primers.
Aerobic SR7 culturing
SR7 was cultured aerobically in complex LB medium and a semi-defined M9-based medium. The semi-defined medium consisted of 1x M9 salts (Sigma) (7 g l–1 Na2HPO4•7H2O, 3 g l–1 KH2PO4, 0.5 g l–1 NaCl, 1 g l–1 NH4Cl), 2 mM MgSO4, 0.04 mM MgCl2, 0.05 g l–1 yeast extract (BD Difico), 1x trace metals supplement69 (5 mg l–1 Na2(EDTA), 0.2 mg l–1 NiSO4•6H2O, 0.5 mg l–1 CoCl2•6H2O, 0.1 mg l–1 H2FeO3, 1 mg l–1 FeSO4•7H2O, 0.1 mg l–1 H3BO3, 1 mg l–1 ZnCl2, 0.1 mg l–1 NaMoO4•2H2O, 0.4 mg l–1 AlCl3•6H2O, 1 mg l–1 MnCl2•4H2O, 0.3 mg l–1 Na2NO4•2H2O, 0.2 mg l–1 CaCl2), and desired concentration of glucose or xylose. For SR7 strains that contained a plasmid, the medium was amended with 5 μg ml–1 tetracycline.
Overnight cultures were started in desired medium by picking colonies from freshly grown LB agar plates with 5 μg ml–1 tetracycline; different colonies were selected for experiments performed in biological triplicate. Overnight cultures (5 ml) were incubated for approximately 16 h at 37 °C and 250 r.p.m.. For gfp production, SR7 was subcultured into 1 ml of fresh medium using 20 μl of an overnight culture into a 48-well flower-plate (m2p labs). Plates were sealed with porous adhesive film for culture plates (VWR) to reduce evaporation, and cultures were grown in a BioLector (m2p labs) at 1200 r.p.m., 37 °C, and 80% humidity. The BioLector continuously monitored (15 min cycle) OD and GFP fluorescence. OD measurements from the BioLector were converted to OD600 measurements with a standard pathlength of 1 cm by generating a standard curve using exponentially grown SR7 cells and comparing a dilution series on the BioLector and Implen spectrophotometer. Protein induction occurred 3 h after subculturing (OD600 ~0.4) by adding xylose to a final concentration of 5 g l–1 for strains that contain the pXyl plasmid, and IPTG to 1 mM for strains containing the pHysp plasmid.
For alcohol production experiments, identical medium and starter culture procedures were used. Overnight cultures were subcultured to an OD600 of 0.05 into 3 ml fresh medium in a 50 ml screw-capped tube (Pyrex) to avoid loss of volatile compounds. Protein induction occurred when the OD600 of the culture reached approximately 0.4 by adding xylose to a final concentration of 5 g l–1 for strains that contain the pXyl plasmid. At the time of induction, αKIV was added to a final concentration of 5 mM. Alcohol production and isobutyraldehyde intermediate accumulation was measured 24 h and 4 h post induction, respectively. Cultures were centrifuged at 21,000 × g for 5 min to obtain cell-free supernatants. Alcohols and isobutyraldehyde were extracted from the cell-free supernatant using a 1:1 ratio of GC-grade ethyl acetate (Sigma), vortexing vigorously for 5 min, centrifuging at 21,000 × g for 5 min, and recovering the ethyl acetate fraction.
Plasmid maintenance
For plasmid maintenance studies, a 25 ml culture of SR7 pXyl gfp was grown in LB medium with 5 μg ml–1 tetracycline in a 250 ml baffled flask. Cultures were inoculated with 250 μl of an overnight culture and were subsequently incubated at 37 °C and 250 r.p.m. Protein induction occurred at 3 h post subculture using 5 g l–1 xylose. For each time point, an OD600 reading was recorded, and cells were diluted to a uniform OD600 in a black-lined 96-well plate (Costar) using LB medium prior to reading GFP fluorescence on a plate reader (Tecan infinite f200 pro, ex: 485/20 nm, em: 535/25 nm). To confirm plasmid maintenance at 74 h post induction, dilutions of cells were made in 1x M9 salts and were plated on LB agar with and without 5 μg ml–1 tetracycline to determine colony forming units. Colony PCR was performed using primers RepU seq for and RepU seq rev to amplify the RepU fragment, and primers pXyl seq for and gfp seq rev to amplify gfp (Supplementary Table 8). A negative control PCR reaction did not contain DNA template and a positive control used 1 ng μl–1 purified pXyl gfp.
FACS and microscopy
Samples for fluorescence activated cell sorting (FACS) were collected 4 h post induction for cells (or 7 h total culturing time for p43-containing cells) grown aerobically in LB medium as described above. Samples were prepared for FACS by diluting 5 μl of culture into 200 μl 1x M9 salts in a 96-well plate. FACS measurements were performed on a FACS Canto (BD) equipped with a high-throughput sampler, and GFP fluorescence was recorded from 488 nm emission laser coupled with the 530/30 nm filter channel. Populations of wild-type SR7 and SR7 with pXyl grown aerobically in LB medium were used to establish forward and side scatter gates to select events corresponding to the morphology of SR7. Fluorescence data was collected for 10,000 cell events for each sample. FACS data was analyzed using FlowJo software and a GFP positive gate was established for fluorescence values above all the negative control samples where SR7 contained an empty plasmid.
Samples for microscopy of SR7 expressing gfp were also collected 4 h post induction for cultures grown aerobically in LB medium. Each sample was prepared as a wet-mount on a glass slide below a cover slip that had been prepared with poly-l-lysine (Sigma) to enable cells to adhere. Cells were visualized at 1000x magnification using an epifluorescence microscope (Zeiss Axioscope 2). Cell morphology was observed using phase-contrast microscopy and GFP fluorescence was visualized using a 480/30 nm excitation and 535/40 emission FITC filter set with a 505 nm long pass mirror and an X-Cite Series 200 fluorescence source. Images were captured on a Nikon D100 camera using the NKRemote live-imaging software.
SR7 endospore preparation
Overnight cultures of SR7 grown in LB medium aerobically were diluted 1:50 in modified G medium (2 g l–1 yeast extract, 2 g l–1 (NH4)2SO4, 0.025 g l–1 CaCl2•2H2O, 0.5 g l–1 K2HPO4, 0.2 g l–1 MgSO4•7H2O, 0.05 g l–1 MnSO4•4H2O, 0.005 g l–1 ZnSO4•7H2O, 0.005 g l–1 CuSO4•5H2O, 0.0005 g l–1 FeSO4•7H2O, pH 7.1). For SR7 cells that contain a plasmid, both the vegetative cell growth medium and sporulation medium were supplemented with 5 μg ml–1 tetracycline. Sporulation was induced by incubating cutures in modified G medium for 72 h at 37 °C. Endospores were collected by centrifugation at 4000 × g and 4 °C for 15 min, and sporulation medium was removed by washing 5–10 times with cold wash buffer (0.058 g l–1 NaH2PO4•H2O, 0.155 g l–1 Na2HPO4•7H2O, 0.01% (vol/vol) Tween 20. Spore preparations were treated at 80 °C for 10 min to kill vegetative cells, and colony forming units were determined pre- and post-heat inactivation. Spore preparations were stored in wash buffer at 4 °C until use22.
SR7 growth under 0.1 MPa CO2 and 10 MPa scCO2
Prior to anaerobic culturing, SR7 strains were prepared as endospores22. For cultures under 0.1 MPa CO2 or 10 MPa scCO2, a semi-defined M9-based medium consisting of 1x M9 salts, 2 mM MgSO4, 0.04 mM MgCl2, 0.05 g l–1 yeast extract, 0.1x trace metals supplement, and 4 g l–1 glucose was degassed with CO2 (Air gas) prior to use. Additionally, the medium was supplemented with 0.25 g l–1 of reducing agent Na2S and 1 mg l–1 of the redox indicator resazurin. All anaerobic culture manipulation occurred in an anaerobic chamber (Coy labs) under 0.1 MPa CO2.
For low-pressure anaerobic experiments, SR7 cultures were grown under 0.1 MPa CO2 headspace in 10 ml of CO2-degassed semi-defined medium in 100 ml serum vials with clamped rubber stoppers. Seed cultures were started by inoculating the degassed media in serum vials with 100 μl of previously prepared endospores; for SR7 samples that contain a plasmid, 0.5 μg ml–1 tetracycline was added. Cultures were grown for approximately 24 h at 37 °C and 250 r.p.m. Anaerobically grown vegetative cells were passaged into 10 ml of fresh medium to an OD600 of 0.05. Cultures were incubated at 37 °C and 250 r.p.m., and protein expression was induced using a final concentration of 5 g l–1 xylose 2 h post subculture. For β-galactosidase production experiments, a 1 ml sample was taken 24 h after induction and centrifuged at 21,000 × g for 5 min, and cell pellets were retained and stored at –20 °C until analysis. For biofuel generation protocols, 5 mM αKIV was also added at the time of induction. Alcohols were collected 48 h post induction by centrifuging samples at 21,000 × g for 5 min and extracting the alcohols using ethyl acetate as was done for the aerobic samples.
For 10 MPa scCO2 cultures, M9 semi-defined medium was amended with 100 mM l-alanine (Sigma) to promote germination under pressure25. Samples were inoculated with endospores to a final concentration of 3 × 105 spores ml–1. Xylose was added to a final concentration of 5 g l–1 for cultures to be induced, and αKIV to 5 mM for those to produce biofuels. As described in previous work22, high-pressure culturing vessels were constructed of ¾ inch 316 stainless steel tubing for 10 ml total capacity, and fitted with quarter turn plug valves (Swagelok or Hylok). Each column was loaded with 5 ml of medium containing appropriate amendments and endospores, prior to pressurization of the 5 ml remaining headspace to 10 MPa with CO2 over a 1 h period. Cultures loaded in steel vessels were grown at 37 °C and shaking at 250 r.p.m. for 21 days prior to de-pressurization. The density of the scCO2 phase under these conditions is 0.68 g ml–170. The pressure of each column was verified to ensure supercritical conditions were maintained throughout the experiment. 1 ml samples of cells were stained with Syto9 (Invitrogen) and filtered on 0.22 μm polycarbonate filters (Whatman) for cell visualization and enumeration by fluorescence microscopy25. The remaining cell culture was centrifuged at 21,000 × g for 5 min. For β-galactosidase production experiments, the cell pellets were retained and stored at –20 °C until analysis. For alcohol generation experiments, alcohols were extracted from the cell-free supernatant using ethyl acetate and an identical protocol to what was used for cultures grown under aerobic and 0.1 MPa CO2 conditions.
β-Galactosidase activity assays
Cells from 0.1 MPa CO2 and 10 MPa scCO2 cultures were lysed by addition of 100 μl of Bacterial Protein Extraction Reagent (B-PER) (Thermo Scientific) to the previously collected cell pellets and vortexing for 30 min at room temperature. Soluble cellular protein was collected by centrifuging the lysed cell pellets for 20 min at 18,500 × g and 4 °C. Total protein content for each sample was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific) according to the manufacture’s instructions, and measuring the resulting absorbance at 562 nm (Beckman DU600). To determine protein concentration, samples were compared to a standard curve generated using 0.05–1.0 mg ml–1 of bovine serum albumin (BSA) and the same protocol. B-PER negative control samples were also analyzed and subtracted from the total protein content of the lysed samples.
To assay for β-galactosidase activity, lysed culture supernatant was added to assay buffer (0.1 M sodium phosphate buffer pH 8, 10 mM KCl, 1 mM MgSO4) to a final volume of 800 μl in a plastic cuvette (VWR). Samples were equilibrated at room temperature prior to the addition of 200 μl of 4 mg ml–1 freshly prepared, β-galactosidase substrate o-nitrophenyl-β-D-galactoside (ONPG) (Sigma). Absorbance at 420 nm was collected every 15 s for 10 min using a Beckman DU600 spectrophotometer. The volume of lysed culture was adjusted to ensure the concentration of ONPG was not limiting during the assay. The initial rate of absorbance change was calculated for each sample and converted to μmol min–1 using 2130 M–1 cm–1 as the extinction coefficient for ONPG. Rates were normalized by total protein added to each reaction to generate a specific activity per unit total protein for each sample.
Compound quantification
Glucose, xylose, αKIV, and fermentation products (acetate, lactate, formate) were detected using an HPLC (Agilent 1200) with an Aminex HPX-87H anion exchange column (Bio-Rad). Cell-free supernatants were loaded onto the column with an inlet and outlet temperature of 35 °C and flow rate of 0.6 ml min–1 under isocratic conditions with 5 mM sulfuric acid. Compound concentrations were determined by integrating the refractive index detection chromatogram and comparison to a standard curve generated with known concentrations of each commercially available standard (Sigma). Purified standards were also used to determine retention times: glucose—9 min, xylose—9.7 min, αKIV—10.2 min, lactate— 13.0 min, formate—14.3 min, and acetate—15.6 min.
Alcohols and isobutyraldehyde were detected using a gas chromatography system (Agilent 7890B GC) equipped with a VF-wax column (Agilent 30 m × 0.25 mm × 0.50 μm), flame-ionization detector (FID) and mass spectrometer (MS). All compounds were extracted into ethyl acetate from cell-free supernatants prior to analysis. The temperature program for the GC oven was 80 °C hold for 3 min, 5 °C min–1 ramp to 210 °C, and 210 °C hold for 3 min. Compound concentrations were determined by integrating the FID chromatogram and comparison to a standard curve generated with known concentrations of each commercially available standard (Sigma). To generate a standard curve using purified compounds, mixtures of compounds were extracted from LB or M9-based semi-defined medium using ethyl acetate, identical to the procedure for microbially generated compounds. Purified standards were also used to determine retention times, and identification of microbially produced compounds was confirmed using the MS fingerprint. The retention times for the molecules in this study are: isobutyraldehyde —4.2 min, isobutanol—7.2 min, isopentanol—8.9 min, and phenylethyl alcohol (PEA)—27.2 min. Yield of isobutanol on αKIV was calculated by dividing the moles of isobutanol produced by the moles of αKIV substrate consumed, noting a 1:1 molar relationship of aKIV conversion to isobutanol (Fig. 4a and Supplementary Table 4).
To measure microbially generated compounds found in the scCO2 headspace of high-pressure stainless steel columns, the headspace was de-pressurized directly into chilled ethyl acetate. The 316 stainless steel tubing and valves in a pressurization manifold22 enabled the simultaneous de-pressurization of 12 samples. Compounds present in the ethyl acetate were analyzed by GC, as was performed for other extracted molecules. Prior to extraction, steel tubing was flushed with ethyl acetate to eliminate cross contamination.
Isobutanol was also extracted from a custom built extractor-fermenter12 using scCO2. A 1% w/v isobutanol (Sigma) solution was prepared in deionized water and extraction was carried out at scCO2 flow rates of 1.3, 3.2, 5.4, and 9.0 ml min–1. The scCO2 was de-pressurized through chilled methanol to collect the isobutanol, which was quantified by GC-FID12. Recovery rate was determined by fitting the natural log of the fraction recovered as a function of time12.
scCO2 fermentation-extraction process model
Aspen Plus (AspenTech) was used to simulate the isobutanol extraction process (full process: Supplementary Fig. 15a, simplified process schematic: Fig. 5a). Preliminary calculations and comparison with published data45,46 suggested use of the Soave-Redlich-Kwong equation of state to model phase equilibrium in all cases except for scCO2 extraction of isobutanol, for which our own measurements on isobutanol were used in concert with literature data on n-butanol separation performance43,44. Thermodynamic properties were modeled using the Lee-Kessler-Plöcker equation of state, as recommended by Aspen for supercritical fluids. The process feeds were CO2 at 0.1 MPa and a stream of aqueous glucose (media and substrate). The water feed (P1) was pumped to 10 MPa in a single stage. Consistent with typical engineering practice, CO2 pressurization to 10 MPa was completed in four stages (C1-4), with each stage accomplishing a threefold pressure increase. In all cases, the thermodynamic efficiency of the pumps/compressors was 85% and their mechanical efficiency was 35%. Heat exchangers (HE1-3) were used to maintain CO2 temperature at 40 °C after compression and for feeding the fermenter. The fermenter was simulated as consisting of a continuously fed stirred tank reactor and a separator (V2) configured in series. The reactor was modeled as achieving a steady state isobutanol concentration of 20 g l–1, consistent with reported titers of isobutanol27. The entire contents of the reactor were fed to the separator (V2), a counter-current isobutanol extraction column44, which separated the feed into water-rich and CO2-rich streams. The isobutanol composition of the exit streams were set to that reported by Laitinen and Kaunisto44 and modified using our performance data for isobutanol (i.e., 99.73% butanol recovery) and the exiting water and CO2 streams were assumed to be at mutual saturation, using the data reported by King et al.54 for binary mixtures. Downstream of the fermenter, a three-phase separator (V3) was used to decrease the pressure to permit the separation of isobutanol-rich, water-rich, and CO2-rich streams, as suggested by de Filippi and Moses43. Pressures tested in the three-phase separator ranged from 0.1 to 6.5 MPa, the pressure at which an isobutanol-rich phase first appears. The CO2-rich stream exiting the three-phase separator was either recycled or purged, with the ratio between these streams set by the splitter (V5). The CO2 recycle ratio was defined by the relative mass flow rates of the CO2 streams exiting the splitter; e.g., 50% CO2 recycle implies that 50% of the CO2 entering the splitter is recycled and 50% is off gassed. The isobutanol-rich phase was further de-pressurized to 0.1 MPa (V4), if necessary, to recover high-purity isobutanol liquid. Finally, the water exiting the three-phase separator was purged, though it could be recycled in an industrial process.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this Article.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request. A Reporting Summary for this Article is available as a Supplementary Information file. The source data underlying Figs. 2, 3, 4, and 5, Supplementary Figs. 1, 3–5, 8–11, 14, and 15, Supplementary Tables 1–6 are provided as a Source Data file.
References
Knez, Ž. et al. Industrial applications of supercritical fluids: A review. Energy 77, 235–243 (2014).
Darani, K. K. & Mozafari, M. R. Supercritical fluids technology in bioprocess industries: A review. J. Biochem Tech. 2, 144–152 (2009).
Matsuda, T. Recent progress in biocatalysis using supercritical carbon dioxide. J. Biosci. Bioeng. 115, 233–241 (2013).
Salgın, U., Salgın, S. & Takaç, S. The enantioselective hydrolysis of racemic naproxen methyl ester in supercritical CO2 using Candida rugosa lipase. J. Supercrit. Fluid 43, 310–316 (2007).
Matsuda, T., Harada, T. & Nakamura, K. Alcohol dehydrogenase is active in supercritical carbon dioxide. Chem. Commun. 0, 1367–1368 (2000).
Matsuda, T., Watanabe, K., Harada, T. & Nakamura, K. Enzymatic reactions in supercritical CO2: carboxylation, asymmetric reduction and esterification. Catal. Today 96, 103–111 (2004).
Timko, M. T., Nicholson, B. F., Steinfeld, J. I., Smith, K. A. & Tester, J. W. Partition coefficients of organic solutes between supercritical carbon dioxide and water: Experimental measurements and empirical correlations. J. Chem. Eng. Data 49, 768–778 (2004).
Dunlop, M. J. Engineering microbes for tolerance to next-generation biofuels. Biotechnol. Biofuels 4, 32 (2011).
Van Hecke, W., Kaur, G. & De Wever, H. Advances in in-situ product recovery (ISPR) in whole cell biotechnology during the last decade. Biotechnol. Adv. 32, 1245–1255 (2014).
Chen, C. T. & Liao, J. C. Frontiers in microbial 1-butanol and isobutanol production. FEMS Microbiol. Lett. 363, fnw020 (2016).
Nielsen, D. R. & Prather, K. J. In situ product recovery of n-butanol using polymeric resins. Biotechnol. Bioeng. 102, 811–821 (2009).
Tompsett, G. A. et al. Extraction rate and energy efficiency of supercritical carbon dioxide recovery of higher alcohols from dilute aqueous solution. Energy Technol. 6, 683–693 (2018).
Zhang, J. et al. Sterilization using high-pressure carbon dioxide. J. Supercrit. Fluid 38, 354–372 (2006).
Spilimbergo, S. & Bertucco, A. Non-thermal bacterial inactivation with dense CO2. Biotechnol. Bioeng. 84, 627–638 (2003).
Matsuda, T. et al. Conversion of pyrrole to pyrrole-2-carboxylate by cells of Bacillus megaterium in supercritical CO2. Chem. Commun. 0, 2194–2195 (2001).
Matsuda, T., Marukado, R., Mukouyama, M., Harada, T. & Nakamura, K. Asymmetric reduction of ketones by Geotrichum candidum: immobilization and application to reactions using supercritical carbon dioxide. Tetrahedron-Asymmetr. 19, 2272–2275 (2008).
Guvenc, A., Mehmetoglu, U. & Calimli, A. Supercritical CO2 extraction of ethanol from fermentation broth in a semicontinuous system. J. Supercrit. Fluid 13, 325–329 (1998).
Qureshi, N. & Eller, F. Recovery of butanol from Clostridium beijerinckii P260 fermentation broth by supercritical CO2 extraction. J. Chem. Technol. Biot. 93, 1206-1212 (2018).
Knutson, B. L. et al. Effect of pressurized solvents on ethanol production by the thermophilic bacterium Clostridium thermocellum. J. Supercrit. Fluid 16, 149–156 (1999).
Ueda, K. et al. Isolation of bacteria whose growth is dependent on high levels of CO2 and implications of their potential diversity. Appl. Environ. Microbiol. 74, 4535–4538 (2008).
Santillan, E. F., Shanahan, T., Omelon, C., Major, J. & Bennett, P. Isolation and characterization of a CO2-tolerant Lactobacillus strain from Crystal Geyser, Utah, USA Front. Earth Sci. 3, 41, 1–11, (2015).
Peet, K. C. et al. Microbial growth under supercritical CO2. Appl. Environ. Microbiol. 81, 2881–2892 (2015).
Szecsody, J. E. et al. Laboratory study of the influence of scCO2 injection on metals migration, precipitation, and microbial growth. Int J. Greenh. Gas. Cont. 47, 71–85 (2016).
Freedman, A. J. E., Tan, B. & Thompson, J. R. Microbial potential for carbon and nutrient cycling in a geogenic supercritical carbon dioxide reservoir. Environ. Microbiol. 19, 2228–2245 (2017).
Freedman, A. J. et al. Isolation, development, and genomic analysis of Bacillus megaterium SR7 for growth and metabolite production under supercritical carbon dioxide. Front. Microbiol. 9, 2152, 1–21 (2018).
Setlow, P. Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J. Appl. Microbiol 101, 514–525 (2006).
Atsumi, S., Hanai, T. & Liao, J. C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451, 86–89 (2008).
Oudshoorn, A. Recovery of Bio-based Butanol. Doctoral Thesis, TU Delft (2012).
Biedendieck, R. et al. Systems biology of recombinant protein production using Bacillus megaterium. Methods Enzymol. 500, 165–195 (2011).
Vavrova, L., Muchova, K. & Barak, I. Comparison of different Bacillus subtilis expression systems. Res. Microbiol. 161, 791–797 (2010).
Li, S., Wen, J. & Jia, X. Engineering Bacillus subtilis for isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression. Appl. Microbiol. Biot. 91, 577–589 (2011).
Rygus, T. & Hillen, W. Inducible high-level expression of heterologous genes in Bacillus megaterium using the regulatory elements of the xylose-utilization operon. Appl. Microbiol. Biot. 35, 594–599 (1991).
Bunk, B. et al. A short story about a big magic bug. Bioeng. Bugs 1, 85–91 (2010).
Rygus, T. & Hillen, W. Catabolite repression of the xyl operon in Bacillus megaterium. J. Bacteriol. 174, 3049–3055 (1992).
Schmiedel, D. & Hillen, W. Contributions of XylR, CcpA, and cre to diauxic growth of Bacillus megaterium and to xylose isomerase expression in the presence of glucose and xylose. Mol. Gen. Genet. 250, 259–266 (1996).
Vary, P. S. Prime time for Bacillus megaterium. Microbiology 140(Pt 5), 1001–1013 (1994).
Munch, K. M. et al. Polar fixation of plasmids during recombinant protein production in Bacillus megaterium results in population heterogeneity. Appl. Environ. Microbiol. 81, 5976–5986 (2015).
Silva, F., Queiroz, J. A. & Domingues, F. C. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 30, 691–708 (2012).
Diaz Ricci, J. C. & Hernandez, M. E. Plasmid effects on Escherichia coli metabolism. Crit. Rev. Biotechnol. 20, 79–108 (2000).
Li, S., Huang, D., Li, Y., Wen, J. & Jia, X. Rational improvement of the engineered isobutanol-producing Bacillus subtilis by elementary mode analysis. Microb. Cell Fact. 11, 101 (2012).
Atsumi, S. et al. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl. Microbiol. Biot. 85, 651–657 (2010).
Miao, R., Liu, X., Englund, E., Lindberg, P. & Lindblad, P. Isobutanol production in Synechocystis PCC 6803 using heterologous and endogenous alcohol dehydrogenases. Metab. Eng. Commun. 5, 45–53 (2017).
de Filippi, R. P. & Moses, J. M. Extraction of organics from aqueous solutions using critical-fluid carbon dioxide. Biotech. Bioeng. Symp. 12, 205–219 (1982).
Laitinen, A. & Kaunisto, J. Supercritical fluid extraction of 1-butanol from aqueous solutions. J. Supercrit. Fluid 15, 245–252 (1999).
Panagiotopoulos, A. Z. & Reid, R. C. Multiphase high pressure equilibria in ternary aqueous systems. Fluid Phase Equilibr. 29, 525–534 (1986).
Chen, H. -I., Chang, H. -Y. & Chen, P. -H. High-pressure phase equilibria of carbon dioxide+1-butanol, and carbon dioxide+water+1-butanol systems. J. Chem. Eng. Data 47, 776–780 (2002).
Wallner, T., Ickes, A. & Lawyer, K. Analytical Assessment of C2-C8 Alcohols as Spark-Ignition Engine Fuels. In Proc. of the FISITA2012 Automotive Congress, LectureNotes in Electrical Engineering (Springer, Beijing, China, 2013).
Ekwealor, I. & A N Obeta, J. Studies on lysine production by Bacillus megaterium. Afr. J. Biotechnol. 4, 633–638 (2005).
Korneli, C., Bolten, C. J., Godard, T., Franco-Lara, E. & Wittmann, C. Debottlenecking recombinant protein production in Bacillus megaterium under large-scale conditions--targeted precursor feeding designed from metabolomics. Biotechnol. Bioeng. 109, 1538–1550 (2012).
Marcheschi, R. J. et al. A synthetic recursive “+1” pathway for carbon chain elongation. ACS Chem. Biol. 7, 689–697 (2012).
Luterbacher, J. S., Tester, J. W. & Walker, L. P. High-solids biphasic CO2-H2O pretreatment of lignocellulosic biomass. Biotechnol. Bioeng. 107, 451–460 (2010).
Erb, T. J. Carboxylases in natural and synthetic microbial pathways. Appl. Environ. Microbiol. 77, 8466–8477 (2011).
Cotton, C. A., Edlich-Muth, C. & Bar-Even, A. Reinforcing carbon fixation: CO2 reduction replacing and supporting carboxylation. Curr. Opin. Biotechnol. 49, 49–56 (2017).
King, M. B., Mubarak, A., Kim, J. D. & Bott, T. R. The mutual solubilities of water with supercritical and liquid carbon dioxides. J. Supercrit. Fluid 5, 296–302 (1992).
Erb, T. J. & Zarzycki, J. Biochemical and synthetic biology approaches to improve photosynthetic CO2-fixation. Curr. Opin. Chem. Biol. 34, 72–79 (2016).
Butti, S. K. & Mohan, S. V. Autotrophic biorefinery: dawn of the gaseous carbon feedstock. FEMS Microbiol. Lett. 364, https://doi.org/10.1093/femsle/fnx166 (2017).
Stammen, S. et al. High-yield intra- and extracellular protein production using Bacillus megaterium. Appl. Environ. Microbiol 76, 4037–4046 (2010).
Nguyen, A. T. T. Directed Evolution and Characterization of an Escherichia coli Mutant Displaying Improved Growth at High Hydrostatic Pressure. Masters thesis, University of California, San Diego (2013).
Atsumi, S. et al. Evolution, genomic analysis, and reconstruction of isobutanol tolerance in Escherichia coli. Mol. Syst. Biol. 6, 449, 1–11 (2010).
Wang, X., Khushk, I., Xiao, Y., Gao, Q. & Bao, J. Tolerance improvement of Corynebacterium glutamicum on lignocellulose derived inhibitors by adaptive evolution. Appl. Microbiol Biot. 102, 377–388 (2018).
Demeke, M. M. et al. Development of a D-xylose fermenting and inhibitor tolerant industrial Saccharomyces cerevisiae strain with high performance in lignocellulose hydrolysates using metabolic and evolutionary engineering. Biotechnol. Biofuels 6, 89 (2013).
Marietou, A., Nguyen, A. T. T., Allen, E. E. & Bartlett, D. H. Adaptive laboratory evolution of Escherichia coli K-12 MG1655 for growth at high hydrostatic pressure. Front. Microbiol. 5, 749 (2015).
Altenbuchner, J. Editing of the Bacillus subtilis Genome by the CRISPR-Cas9 System. Appl. Environ. Microbiol. 82, 5421–5427 (2016).
Zhang, K., Duan, X. & Wu, J. Multigene disruption in undomesticated Bacillus subtilis ATCC 6051a using the CRISPR/Cas9 system. Sci. Rep. 6, 27943 (2016).
Biedendieck, R. et al. A sucrose-inducible promoter system for the intra- and extracellular protein production in Bacillus megaterium. J. Biotechnol. 132, 426–430 (2007).
Ben-Yehuda, S. et al. Defining a centromere-like element in Bacillus subtilis by Identifying the binding sites for the chromosome-anchoring protein RacA. Mol. Cell 17, 773–782 (2005).
Quan, J. & Tian, J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS ONE 4, e6441 (2009).
Sheppard, M. J., Kunjapur, A. M., Wenck, S. J. & Prather, K. L. J. Retro-biosynthetic screening of a modular pathway design achieves selective route for microbial synthesis of 4-methyl-pentanol. Nat. Commun. 5, 5031 (2014).
Boone, D. R., Johnson, R. L. & Liu, Y. Diffusion of the interspecies electron carriers H2 and formate in methanogenic ecosystems and its implications in the measurement of K m for H2 or formate uptake. Appl. Environ. Microbiol. 55, 1735–1741 (1989).
Span, R. & Wagner, W. A new equation of state for carbon dioxide covering the fluid region from the triple-point temperature to 1100 K at pressures up to 800 MPa. J. Phys. Chem. Ref. Data 25, 1509–1596 (1996).
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0012555. pDR111 was generously provided by Dr. Alan Grossman (MIT Department of Biology). FACS analysis was performed at the Swanson Biotechnology Center Flow Cytometry Facility (MIT Koch Institute) funded by the Cancer Center Support Grant P30-CA14051 from the NCI. We thank the Chisholm and Polz lab (MIT Department of Civil and Environmental Engineering) for access to their microscope. L.A.J.’s contribution was funded by a Summer Undergraduate Research Fellowship (WPI). S.K.M. and A.J.A.’s contributions were performed as part of their major qualifying project (MQP). The authors also wish to acknowledge members of the Prather, Thompson and Timko labs for helpful discussions on this work.
Author information
Authors and Affiliations
Contributions
J.T.B., A.J.E.F., J.R.T. and K.L.J.P. designed biological experiments. J.T.B. completed aerobic experiments, and A.J.E.F. anaerobic biological experiments under 0.1 MPa CO2 and 10 MPa scCO2. G.A.T performed isobutanol extraction using scCO2. G.A.T., S.K.M., A.K.A., L.A.J., B.C. and M.T.T. built the Aspen Plus model and completed analysis. All authors analyzed data, and wrote, reviewed, and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.T.B, A.J.E.F., M.T.T., J.R.T. and K.L.J.P declare a pending patent application for an invention related to this work. The remaining authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks Tomoko Matsuda and the other anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boock, J.T., Freedman, A.J.E., Tompsett, G.A. et al. Engineered microbial biofuel production and recovery under supercritical carbon dioxide. Nat Commun 10, 587 (2019). https://doi.org/10.1038/s41467-019-08486-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-08486-6
This article is cited by
-
Toward bioproduction of oxo chemicals from C1 feedstocks using isobutyraldehyde as an example
Biotechnology for Biofuels and Bioproducts (2022)
-
Supercritical CO2 applications in microfluidic systems
Microfluidics and Nanofluidics (2022)
-
Bioprospecting of microbial strains for biofuel production: metabolic engineering, applications, and challenges
Biotechnology for Biofuels (2021)
-
The “beauty in the beast”—the multiple uses of Priestia megaterium in biotechnology
Applied Microbiology and Biotechnology (2021)
-
Recent advances in the microbial production of isopentanol (3-Methyl-1-butanol)
World Journal of Microbiology and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
