
Abstract
Quantifying the genetic correlation between cancers can provide important insights into the mechanisms driving cancer etiology. Using genome-wide association study summary statistics across six cancer types based on a total of 296,215 cases and 301,319 controls of European ancestry, here we estimate the pair-wise genetic correlations between breast, colorectal, head/neck, lung, ovary and prostate cancer, and between cancers and 38 other diseases. We observed statistically significant genetic correlations between lung and head/neck cancer (rg = 0.57, p = 4.6 × 10−8), breast and ovarian cancer (rg = 0.24, p = 7 × 10−5), breast and lung cancer (rg = 0.18, p =1.5 × 10−6) and breast and colorectal cancer (rg = 0.15, p = 1.1 × 10−4). We also found that multiple cancers are genetically correlated with non-cancer traits including smoking, psychiatric diseases and metabolic characteristics. Functional enrichment analysis revealed a significant excess contribution of conserved and regulatory regions to cancer heritability. Our comprehensive analysis of cross-cancer heritability suggests that solid tumors arising across tissues share in part a common germline genetic basis.
Similar content being viewed by others

Introduction
Inherited genetic variation plays an important role in cancer etiology. Large twin studies have demonstrated an excess familial risk for cancer sites including, but not limited to, breast, colorectal, head/neck, lung, ovary, and prostate with heritability estimates ranging between 9% (head/neck) to 57% (prostate)1,2,3. Data from nation-wide and multi-generation registries further show that elevated cancer risks go beyond nuclear families and isolated types, as family history of a specific cancer can increase risk for other cancers4,5,6. Additional evidence for a shared genetic component have been demonstrated by cross-cancer genome-wide association study (GWAS) meta-analyses, which set out to identify genetic variants associated with more than one cancer type. Fehringer et al. studied breast, colorectal, lung, ovarian, and prostate cancer, and identified a novel locus at 1q22 associated with both breast and lung cancer7. Kar et al. focused on three hormone-related cancers (breast, ovarian, and prostate), and identified seven novel susceptibility loci shared by at least two cancers8.
Previous attempts to estimate the genetic correlation across cancers using GWAS data9,10,11,12 have mostly relied on restricted maximum likelihood (REML) implemented in GCTA (genome-wide complex trait analysis)13 and individual-level genotype data. However, these studies have had limited sample sizes, yielding inconclusive results. Sampson et al. quantified genetic correlations across 13 cancers in European ancestry populations and identified four cancer pairs with nominally significant genetic correlations (bladder–lung, testis–kidney, lymphoma–osteosarcoma, and lymphoma–leukemia)9. They did not observe any significant genetic correlations across common solid tumors including cancers of the breast, lung and prostate9. REML becomes computationally challenging for large sample sizes and is sensitive to technical artifacts. LD score regression (LDSC)14,15 overcomes these issues by leveraging the relationship between association statistics and LD patterns across the genome. We recently used cross-trait LDSC to quantify genetic correlations across six cancers based on a subset of the data included here and found moderate correlations between colorectal and pancreatic cancer, as well as between lung and colorectal cancer16. However, the average sample size was only 11,210 cases and 13,961 controls per cancer, resulting in imprecise estimates with wide confidence intervals.
In addition to the development of novel analytical methods tailored to genomic data, several high-quality functional annotations have recently been released into the public domain through large-scale efforts. For example, the ENCODE consortium has built a comprehensive and informative parts list of functional elements in the human genome (http://www.nature.com/encode/#/threads), which allows for the analysis of components of SNP-heritability to unravel the functional architecture of complex traits.
Here, we use summary statistics from the largest-to-date European ancestry GWAS of breast, colorectal, head/neck, lung, ovary, and prostate cancer with an average sample size of 49,369 cases and 50,219 controls per cancer, to quantify genetic correlations between cancers and their subtypes. We also use GWAS summary statistics for 38 non-cancer traits (average N = 113,808 per trait), to quantify the genetic correlations between the six cancers and other diseases. Furthermore, we assessed the proportion of cancer heritability attributable to specific functional categories, with the goal of identifying functional elements that are enriched for SNP-heritability.
Our comprehensive analysis identifies statistically significant genetic correlations between lung and head/neck cancer, breast and ovarian cancer, breast and lung cancer, and breast and colorectal cancer. We also find multiple cancers to be genetically correlated with non-cancer traits including smoking, psychiatric diseases, and metabolic traits. Functional enrichment analysis reveals a significant contribution of conserved and regulatory regions to cancer heritability. Our results suggest that solid tumors arising across tissues share in part a common germline genetic basis.
Results
Heritability estimates across cancers
We first estimated cancer-specific heritability causally explained by common SNPs (\(h_g^2\)) using LDSC (note that this quantity is slightly different from the \(h_g^2\) as defined in Yang et al.17 which estimates the heritability due to genotyped and imputed SNPs) (see Methods). Estimates of \(h_g^2\) on the liability scale ranged from 0.03 (ovarian) to 0.25 (prostate) (Supplementary Table 1). After removing genome-wide significant (p < 5 × 10−8) loci, defined as all SNPs within 500 kb of the most significant SNP in a given region (Supplementary Data 1), we observed an ~50% decrease in SNP-heritability for prostate and breast cancer, and ~20% decrease for lung, ovarian, and colorectal cancer, despite the fact that we were only excluding 1% (colorectal cancer) to 5% (breast cancer) of the genome. In contrast, the SNP-heritability for head/neck cancer was not affected by removing genome-wide significant loci (Fig. 1a). For most of the cancers, the GWAS significant loci for that particular cancer explained most of the heritability. For some cancers, however, significant GWAS loci of other cancers also explained a non-trivial part of its heritability. For example, the significant breast cancer GWAS loci explained 10%, 15%, and 22% heritability of colorectal, ovarian and prostate cancer, respectively; the significant colorectal cancer GWAS loci explained 11% heritability of prostate cancer; the significant lung cancer GWAS loci explained 10% heritability of head/neck cancer; and the significant prostate cancer GWAS loci explained 11 and 15% heritability of breast and ovarian cancer, respectively (Supplementary Table 2). Comparing the liability-scale SNP-heritability to corresponding estimates from twin studies suggests that common SNPs can almost entirely explain the classical heritability of head/neck cancer, whereas for other cancers, only 30–40% of heritability can be explained (Fig. 1b).

Estimates of SNP-heritability (\(h_g^2\)) and cross-cancer heritability (rg) for the six cancer types. SNP-heritability and cross-cancer heritability are calculated based on HapMap3 SNPs using LD score regression (LDSC). a The solid bar represents overall SNP \(h_g^2\) on the liability scale, calculated based on all HapMap3 SNPs. The dark green bar represents \(h_g^2\) calculated based on non-significant SNPs—the remaining SNPs after excluding genome-wide significant hits (p < 5 × 10−8) ± 500 kb. The black bar with density texture indicates proportion of \(h_g^2\) (as reflected by the percentages displayed on top of each bar) that could be explained by top hits ±500 kb surrounded areas. The orange error bars represent 95% confidence intervals. b The solid blue bar represents overall SNP \(h_g^2\) in liability scale (no SNP exclusion), with black error bars indicating 95% confidence intervals. The red short lines correspond to classical estimates of h2 measured in a twin study of Scandinavian countries (Mucci et al.2). c Genetic correlations between cancers. Estimates withstood Bonferroni corrections (p < 0.05/15) are marked with double asterisk (**), and nominal significant results (p < 0.05) are marked with single asterisk (*)
Genetic correlations between cancers
We then estimated the genetic correlation between cancers using cross-trait LDSC (see Methods). After adjusting for the number of tests (p < 0.05/15 = 0.003), we found multiple significant genetic correlations Fig. 1c and Supplementary Table 1), with the strongest result observed for lung and head/neck cancer (rg = 0.57, se = 0.10). In addition, colorectal and lung cancer (rg = 0.28, se = 0.06), breast and ovarian cancer (rg = 0.24, se=0.06), breast and lung cancer (rg = 0.18, se = 0.04), and breast and colorectal cancer (rg = 0.15, se = 0.04) showed statistically significant genetic correlations. We also observed nominally significant genetic correlations (p < 0.05) between lung and ovarian cancer (rg = 0.16, se = 0.08), prostate cancer and head/neck (rg = 0.15, se = 0.08), colorectal (rg = 0.11, se = 0.05), and breast cancer (rg = 0.07, se = 0.03) (Fig. 1c). Some cancer pairs showed minimal correlations with estimates close to 0 (ovarian and prostate: rg = 0.02, se = 0.07; lung and prostate: rg = −0.03, se = 0.04; breast and head/neck: rg = 0.03, se = 0.06). We further calculated the cross-cancer genetic correlation based on data after excluding the GWAS significant regions of each cancer. The estimates were mostly consistent with the results calculated based on all SNPs.
We conducted subtype-specific analysis for breast, lung, ovarian, and prostate cancer (Supplementary Table 1). Estrogen receptor positive (ER+) and negative (ER−) breast cancer showed a genetic correlation of 0.60 (se = 0.03), indicating that the genetic contributions to these two subtypes are in part distinct. The genetic correlation between the two common lung cancer subtypes adenocarcinoma and squamous cell carcinoma was similarly 0.58 (se = 0.10). Further, we observed a significantly larger genetic correlation of lung cancer with ER− (rg = 0.29, se = 0.06) than with ER + breast cancer (rg = 0.13, se = 0.04) (pdifference = 0.002). This also held true for lung squamous cell carcinoma, which showed statistically stronger genetic correlation with ER− (rg = 0.33, se = 0.08) than with ER + breast cancer (rg = 0.11, se = 0.05) (pdifference = 0.0019). We observed no other statistically significant differential genetic correlations across subtypes (all pdifference > 0.1).
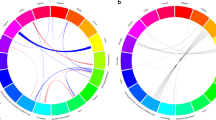
We then estimated local genetic correlations between cancers using ρ-HESS, dividing the genome into 1703 regions (see Methods) (Fig. 2 and Supplementary Fig. 1). We found that although the genome-wide genetic correlation between breast and prostate cancer was modest (rg = 0.07), chr10:123M (10q26.13, p = 1.0 × 10−7) and chr9:20–22 M (9p21, p = 1.0 × 10−6), two previously known pleiotropic regions18, showed significant genetic correlations (rg = −0.00098 and rg = 0.00046). Similarly, although the genome-wide genetic correlation between lung and prostate cancer was negligible (rg = −0.03), two previously identified pleiotropic regions (chr6:30–31 M or 6p21.33, p = 5.7 × 10−7 and chr20:62M or 20q13.33, p = 2.8 × 10−6) exhibited significant local genetic correlations (rg = −0.00060 and rg = 0.00067). Overall, local genetic correlation analysis reinforced shared effects for 44% (31/71) of previously reported pleiotropic cancer regions (Supplementary Data 2). It also identified novel pleiotropic signals. For example, the breast and prostate cancer pleiotropic region at 2q33.1 showed significant local genetic correlation between breast and ovarian cancer (p = 2.3 × 10−6). Additionally, 6p21.32, a region indicated for head/neck and prostate cancer, showed highly significant local genetic correlation for head/neck and lung cancer (p = 8.6 × 10−8).

Local genetic correlation between breast, lung and prostate cancer. The region-specific p-values for the local genetic covariance for breast and prostate cancer are shown in a, and for lung and prostate cancer in b. Each dot presents a specific genomic region. In the QQ plots, red color indicates significance after multiple corrections (p < 0.05/1703 regions compared), and blue color indicates nominal significance (p < 0.05/15 pairs of cancers compared). Manhattan-style plots showing the estimates of local genetic covariance for breast and prostate cancer (c), and for lung and prostate cancer (d). Although breast and prostate cancer only show modest genome-wide genetic correlation, two loci exhibit significant local genetic covariance. Similarly, albeit the negligible overall genetic correlation for lung and prostate cancer, three loci present significant local genetic covariance. In the Manhattan plots, red color indicates even number chromosomes and blue color indicates odd number chromosomes
Genetic correlations between cancer and other traits
Significant genetic correlations (p < 0.05/228 = 0.0002) between the six cancers and 38 non-cancer traits reflected several known associations (Fig. 3 and Supplementary Data 3). We observed a strong genetic correlation between smoking and lung cancer (rg = 0.56, se = 0.06), and similarly for head/neck cancer (rg = 0.47, se = 0.08), both cancers having smoking as its primary risk factor19,20. Educational attainment was negatively genetically correlated with colorectal (rg = −0.17, se = 0.04), head/neck (rg = −0.42, se = 0.07), and lung cancer (rg = −0.39, se=0.04) (all p < 5 × 10−6). Body mass index (BMI) showed a positive genetic correlation with colorectal cancer (rg = 0.15, se = 0.03) and also suggestive but weak negative correlations with prostate (rg = −0.07, se = 0.03) and breast cancer (rg = −0.06, se = 0.03). Lung cancer showed a negative genetic correlation with lung function (rg = −0.15, se = 0.04) and age at natural menopause (rg = −0.25, se = 0.05), and moderate positive genetic correlations with depressive symptoms (rg = 0.25, se=0.06) and waist-to-hip ratio (rg = 0.16, se = 0.04). Breast cancer showed a positive genetic correlation with schizophrenia (rg = 0.14, se = 0.03).

Cross-trait genetic correlation (rg) analysis between cancers and non-cancer traits. The traits were divided into four categories: a Common phenotypes, b Metabolic or cardiovascular related traits, c Psychiatric traits, d Autoimmune inflammatory diseases. Pair-wise genetic correlations withstood Bonferroni corrections (228 tests) are marked with double asterisk (**), with estimates of correlation shown in the cells. Pair-wise genetic correlations with significance at p < 0.01 are marked with a single asterisk (*). The color of cells represents the magnitude of correlation
We did not find evidence of genetic correlations between cancer and several previously suggested risk factors21,22,23 including cardiovascular traits (coronary artery disease, hypertension, and blood pressure) or sleep characteristics (chronotype, duration, and insomnia). Further, we did not observe genetic correlations between cancer and circulating lipids (HDL, LDL, and triglycerides) or type 2 diabetes-related traits except a significant negative correlation between HDL and lung cancer (rg = −0.14, se = 0.04). We observed no significant genetic correlation between breast cancer and age at menarche (rg = −0.03, se = 0.03) or age at natural menopause (rg = −0.01, se = 0.03). We also did not observe notable genetic correlations between cancer and autoimmune inflammatory diseases or height.
Subtype analysis revealed that smoking and educational attainment showed genetic correlations with all lung cancer subtypes (Supplementary Data 3). Educational attainment, forced vital capacity and depressive symptoms showed genetic correlations with ER− but not ER + breast cancer, whilst the observed genetic correlation between schizophrenia and breast cancer was limited to ER + disease, and the genetic correlation between depressive symptoms and lung cancer was observed only for lung squamous cell carcinoma.
We further assessed the support for mediated or pleiotropic causal models for non-cancer traits and cancer using the correlation between trait-specific effect sizes of genome-wide significant SNPs for pairs of phenotypes. We detected four putative directional genetic correlations (defined as p < 0.05 from a likelihood ratio (LR) comparing the best non-causal model to the best causal model) (Fig. 4), where SNPs associated with the non-cancer trait showed correlated effect estimates with cancer but the reverse was not true (circulating HDL concentrations and breast cancer, LRnon-causal vs. causal = 0.04, schizophrenia and breast cancer, LRnon-causal vs. causal = 0.003, age at natural menopause and breast cancer, LRnon-causal vs. causal = 0.04, and lupus and prostate cancer, LRnon-causal vs. causal = 0.0006).

Putative directional relationships between cancers and traits. For each cancer–trait pair identified as candidates to be related in a causal manner, the plots show trait-specific effect sizes (beta coefficients) of the included genetic variants. Gray lines represent the relevant standard errors. a HDL and breast cancer. Trait-specific effect sizes for HDL and breast cancer are shown for SNPs associated with HDL levels (left) and breast cancer (right). b Schizophrenia and breast cancer. Trait-specific effect sizes for schizophrenia and breast cancer are shown for SNPs associated with schizophrenia (left) and breast cancer (right). c Age at natural menopause and breast cancer. Trait-specific effect sizes for age at natural menopause and breast cancer are shown for SNPs associated with age at natural menopause (left) and breast cancer (right). d Lupus and prostate cancer. Trait-specific effect sizes for lupus and prostate cancer are shown for SNPs associated with lupus (left) and prostate cancer (right)
Functional enrichment analysis of cancer heritability
Finally, we partitioned SNP-heritability of each cancer by using 24 genomic functional annotations (the baseline-LD model described in Gazal et al.24) and 220 cell-type-specific histone mark annotations (the cell-type-specific model described in Finucane et al.14). Meta-analysis across the six cancers revealed statistically significant enrichments for multiple functional categories. We observed the highest enrichment for conserved regions (Table 1, Supplementary Table 3) which overlapped with only 2.6% of SNPs but explained 25% of cancer SNP-heritability (9.8-fold enrichment, p = 2.3 × 10−5). Transcription factor binding sites showed the second highest enrichment (4.0-fold, 13% of SNPs explaining 40% of SNP-heritability, p = 1.4 × 10−7). Further, super-enhancers (groups of putative enhancers in close genomic proximity with unusually high levels of mediator binding) showed a significant 2.6-fold enrichment (p = 2.0 × 10−20). Additional enhancers, including regular enhancers (3.2-fold), weak enhancers (3.1-fold) and FANTOM5 enhancers (3.1-fold), presented similar enrichments but were not statistically significant. In addition, multiple histone modifications of epigenetic markers H3K9ac, H3K4me3, and H3K27ac, were all significantly enriched for cancer heritability. Repressed regions exhibited depletion (0.34-fold, p = 1.2 × 10−6). Enrichment analysis of functional categories for each cancer and cancer subtype are shown in Fig. 5 and Supplementary Table 4.

Enrichment p-values of 24 non-cell-type-specific functional categories over six cancer types. The x-axis represents each of the 24 functional categories, y-axis represents log-transformed p-values of enrichment. Annotations with statistical significance after Bonferroni corrections (p < 0.05/24) were plotted in orange, otherwise blue. The horizontal gray dash line indicates p-threshold of 0.05; horizontal red dash line indicates p-threshold of 0.05/24. From top to bottom are six panels representing six cancers: breast cancer, colorectal cancer, head/neck cancer, lung cancer, ovarian cancer, and prostate cancer. TSS transcription start site, UTR untranslated region, TFBS transcription factor binding sites, DHS DNase I hypersensitive sites, DGF digital genomic foot printing, CTCF CCCTC-binding factor
Overall, cell-type-specific analysis of histone marks identified significant enrichments specific to individual cancers (Supplementary Fig. 2). For breast cancer, 3 out of 8 statistically significant tissues were adipose nuclei (H3K4me1, H3K9ac) and breast myoepithelial (H3K4me1) cells. For colorectal cancer, 15 out of the 18 statistically significant enrichments were observed in either colon or rectal tissues (colon/rectal mucosa, duodenum mucosa, small/large intestine, and colon smooth muscle). We observed no significant enrichments for head/neck, lung, and ovarian cancer, but we noted that for both lung (9 out of 10) and ovarian cancer (6 out of 10), the most enriched cell types were immune cells; while in head/neck cancer, 6 out of 10 most highly enriched cell types belonged to CNS (Supplementary Fig. 3, Supplementary Data 4). Cell-type-specific analysis for cancer subtypes are shown in Supplementary Data 5. Comparing cell-type-specific enrichment for cancers to the additional 38 non-cancer traits revealed notably differential clustering patterns (Supplementary Fig. 4). Breast, colorectal, and prostate cancer showed enrichment mostly for adipose and epithelial tissues, in contrast to autoimmune diseases (enriched for immune/hematopoietic cells) or psychiatric disorders (enriched for brain tissues).
Discussion
We performed a comprehensive analysis quantifying the heritability and genetic correlation of six cancers, leveraging summary statistics from the largest cancer GWAS conducted to date. Our study demonstrates shared genetic components across multiple cancer types. These results contrast with a prior study conducted by Sampson et al. which reported an overall negligible genetic correlation among common solid tumors9. Our results are, however, in line with a recent study,16 which analyzed a subset of the data included here, and identified a significant genetic correlation between lung and colorectal cancer.
Our data support, and for the first time quantify, the strong genetic correlation (rg = 0.57) between lung and head/neck cancer, two cancers linked to tobacco use20,25. We also for the first time observed a significant genetic correlation between breast and ovarian cancer (rg = 0.24), two cancers that are known to share rare genetic factors including BRCA1/2 mutations, and environmental exposures associated with endogenous and exogenous hormone exposures26. Prostate cancer is also considered as hormone-dependent and associated with BRCA1/2 mutations, but interestingly, we only observed a nominally significant and modest (rg = 0.07) genetic correlation between breast and prostate cancer, while ovarian and prostate cancer showed no genetic correlation (rg = 0.02, se = 0.07).
Our large sample sizes allowed us to conduct well-powered analyses for cancer subtypes. While head/neck cancer showed negligible genetic correlation with overall (rg = 0.03, se = 0.06) and ER + breast cancer (rg = −0.02, se = 0.07), it showed a stronger genetic correlation with ER− breast cancer (rg = 0.21, se = 0.09). Similarly, lung cancer showed a statistically more pronounced genetic correlation with ER− (rg = 0.29, se = 0.06) than ER + breast cancer (rg = 0.13, se = 0.04). A recent pooled analysis of smoking and breast cancer risk demonstrated a smoking-related increased risk for ER + but not for ER− breast cancer27, and thus it is unlikely that the stronger genetic correlation between ER− subtype and lung and head/neck cancer is due to smoking behavior. Perhaps surprisingly, despite literature suggesting substantial similarities between ER− breast cancer and serous ovarian cancer in particular28, we did not observe statistically significant different genetic correlations between ER− or ER + breast cancer and serous ovarian cancer (rg = 0.17, se = 0.08 vs. rg = 0.11, se = 0.06). This suggests that rare high penetrance variants may play a more important role in driving the similarities behind ER− breast cancer and serous ovarian cancer than common genetic variation.
Heritability analysis confirms that common cancers have a polygenic component that involves a large number of variants. Although susceptibility variants identified at genome-wide significance explain an appreciable fraction of the heritability for some cancers, we estimate that the majority of the polygenic effect is attributable to other, yet undiscovered variants, presumably with effects that are too weak to have been identified with current sample sizes. We found the genetic component that could be attributed to genome-wide significant loci varied greatly from ~0% for head/neck cancer to ~50% for breast and prostate cancer. These results reflect in part the strong correlation between number of GWAS-identified loci and sample size, as we had more than twice as many breast and prostate cancer samples compared to the other cancers. One corollary is that larger GWAS are likely to identify new susceptibility loci that could help our understanding of disease development, improve prediction power of genetic risk scores and hence contribute to screening and personalized risk prediction29.
Among the genetic correlations between cancer and non-cancer traits, we observed positive correlations for psychiatric disorders (depressive symptoms, schizophrenia) with lung and breast cancer, where findings from epidemiological studies have been suggestive but inconclusive. It has been proposed that the linkage between psychiatric traits and cancers are more likely to be mediated through cancer-associated risk phenotypes such as smoking, excessive alcohol consumption in depressed populations30, and reduced fertility patterns (e.g., nulliparous) in psychiatric populations31. Detailed analyses considering confounding traits like reproductive history and smoking are needed to make inference about the mechanisms involved. GWAS have identified pleiotropic regions influencing both lung cancer and nicotine dependence, such as 15q25.132,33. In line with those results, we identified a strong genetic correlation between smoking and both lung (rg = 0.56) and head/neck cancer (rg = 0.47). It remains unclear whether this genetic correlation is completely explained by the direct influence of smoking or if the shared genetic component affects the traits through separate pathways. Interestingly, a genetic correlation (rg = 0.35, se = 0.14) between lung and bladder cancer, another smoking-associated cancer, has been identified previously9. Due to the small numbers of GWAS-identified smoking-associated SNPs, we were unable to assess a directional correlation between smoking and cancer, but we expect such analyses to become feasible as additional smoking-related SNPs are identified. We found modest positive, yet significant genetic correlations between adiposity-related measures (as reflected by waist-to-hip ratio, circulating HDL levels and BMI) and both colorectal and lung cancer, but negative genetic correlations between BMI and prostate and breast cancer, consistent with previous reported findings34 and reinforce the complex dynamics between obesity and cancer where multiple factors including age, smoking, endogenous hormones and reproductive status play a role.
We did not observe genetic correlations between breast cancer and age at menarche or age at natural menopause. These null observations were largely driven by ER + breast cancer (ER + : rg = 0.006, se = 0.03 vs. ER−: rg = −0.09, se = 0.04 for age at menarche. ER + : rg = 0.0005, se = 0.04 vs. ER−: rg = −0.10, se = 0.05 for age at natural menopause), and were unexpected given that both factors play pivotal roles in breast cancer etiology35 and previous Mendelian randomization (MR) analyses have identified a link36,37. An important difference between genetic correlation and MR analyses is that the latter only considers genome-wide significant SNPs while the former incorporates the entire genome. It is possible that a relatively small overlap in strongly associated SNPs can result in significant MR results despite low evidence of an overall genetic correlation. Indeed, the directional genetic correlations we observed for age at natural menopause, schizophrenia, and HDL with breast cancer, and for lupus with prostate cancer, highlight again that although an overall genetic correlation may be negligible, there can still be genetic links between traits. It is important to note that we cannot rule out unmeasured confounding, including the possibility that these genetic variants affect an intermediate phenotype that is pleiotropic for both target traits. Given the observational nature of our data, these putative causal directions should be interpreted with caution.
Pan-cancer tumor-based studies have demonstrated that different cancers are sometimes driven by similar somatic functional events such as specific copy number abnormalities and mutations38,39. Our enrichment results of germline genetic across functional annotation data shed new light on the biological mechanisms leading to cancer development. The more pronounced enrichment identified for conserved regions compared with coding regions provides evidence for the biological importance of the former, which has been shown to be true for multiple traits14,40. Even though the biochemical function of many conserved regions remains uncharacterized, transcribed ultra-conserved regions have been found to be frequently located at fragile sites. Compared to normal cells, cancer cells have a unique spectrum of transcribed ultra-conservative regions, suggesting that variation in expression of these regions are involved in the malignant process41,42. These results bridge the link between germline and somatic genetics in cancer development, which was also observed in a recent breast cancer GWAS that has demonstrated a strong overlap between target genes for GWAS hits and somatic driver genes in breast tumors43. We also found a four-fold enrichment for transcription factor binding sites and a three-fold enrichment for super-enhancers, consistent with prior observations that breast cancer GWAS loci fall in enhancer regions involved in distal regulation of target genes43. Cell-type-specific analysis of histone marks demonstrated the importance of tissue specificity, primarily for colorectal and breast cancer. Further, our results suggest that immune cells are important for ovarian and lung cancer whilst CNS is important to head/neck cancer. Unfortunately, we did not have data on prostate-specific tissues, but we note that tissue-specific enrichment of prostate cancer heritability for epigenetic markers has been observed previously10. We note that generation of rich functional annotation is ongoing and we expect to include additional tissue-specific functional elements in our future work.
Our study has several strengths. We were able to robustly quantify pair-wise genetic correlations between multiple cancers using the largest available cancer GWAS, comprising almost 600,000 samples across six major cancers and subtypes. We were also able to systematically assess the genetic correlations between cancer and 38 non-cancer traits. Notwithstanding the large sample sizes, several limitations need to be acknowledged. We did not have the sample sizes required to assess relevant cancer subgroups including oropharyngeal cancer, clear cell, mucinous and endometrioid ovarian cancer, or lung cancer among never smokers (each with ~2000 cases). In addition, we did not have access to GWAS summary statistics for pre- vs. post-menopausal breast cancer. We were not able to consider all cancer risk factors when selecting non-cancer traits, since some of the well-established risk factors such as infection were either not available, showed no evidence of heritability or were not based on adequate sample sizes for robust analyses. SNP-heritability varies with minor allele frequency, linkage disequilibrium, and genotype certainty; we note that approaches to estimate heritability leveraging GWAS data are constantly evolving. We also note that estimate variability needs to be taken into account when comparing the SNP-heritability with the classical twin-heritability, in particular for cancers with small sample sizes such as head/neck cancer (SNP-heritability varied between 5–14% and twin-heritability varied between 0–60%, although both point estimates were 9%). Further, our data were based on GWAS meta-analysis from multiple individual GWAS across European ancestry populations from Europe, Australia and the US. Intra-European ancestry differences are likely to be a source of bias. However, since we limited our analysis to SNPs with MAF > 1% and HapMap3 SNPs (which have proven to be well imputed across European ancestry populations), we believe that any population structure across cancers will have minimal effect on our results. Finally, as more non-European and multi-ethnic GWAS data become available, it is important to examine trans-ethnic genetic correlation in cancer.
In conclusion, results from our comprehensive analysis of heritability and genetic correlations across six cancer types indicate that solid tumors arising from different tissues share common germline genetic influences. Our results also demonstrate evidence for common genetic risk sharing between cancers and smoking, psychiatric, and metabolic traits. In addition, functional components of the genome, particularly conserved and regulatory regions, are significant contributors to cancer heritability across multiple cancer types. Our results provide a basis and direction for future cross-cancer studies aiming to further explore the biological mechanisms underlying cancer development.
Methods
Studies and quality control
We used summary statistics from six cancer GWASs based on a total of 597,534 participants of European ancestry. Cancer-specific sample sizes were: breast cancer: 122,977 cases/105,974 controls; colorectal cancer: 36,948/30,864; head/neck cancer (oral and oropharyngeal cancers): 5452/5984; lung cancer: 29,266/56,450; ovarian cancer: 22,406/40,941; prostate cancer: 79,166/61,106. These data were generated through the joint efforts of multiple consortia. Details on study characteristics and subjects contributed to each cancer-specific GWAS summary dataset have been described elsewhere43,44,45,46,47,48,49. SNPs were imputed to the 1000 Genomes Project reference panel (1KGP) using a standardized protocol for all cancer types18. We included autosomal SNPs with a minor allele frequency (MAF) larger than 1% and present in HapMap3 (NSNPs = ~1 million) because those SNPs are usually well imputed in most studies (note that excluding sex chromosomes could reduce the overall heritability estimates). A brief overview of the quality control in each cancer dataset are presented in Supplementary Table 5. For some of the cancers, we further obtained summary statistics data on subtypes (ER + and ER− breast cancer; lung adenocarcinoma, and squamous cell carcinoma; serous invasive ovarian cancer and advanced stage prostate cancer, defined as metastatic disease or Gleason score ≥ 8 or PSA > 100 or prostate cancer death). Sample sizes and more details shown in Supplementary Table 1.
We additionally assembled European ancestry GWAS summary statistics from 38 traits, which spanned a wide range of phenotypes including anthropometric (e.g., height and body mass index (BMI)), psychiatric disorder (e.g., depressive symptoms and schizophrenia), and autoimmune disease (e.g., rheumatoid arthritis and celiac disease) (Supplementary Table 6). We calculated trait-specific SNP-heritability and restricted our analysis to traits with a heritable component (Supplementary Table 7)14. We removed the major histocompatibility complex (MHC) region from all analysis because of its unusual LD and genetic architecture.
Estimation of SNP-heritability and genetic correlation
We estimated the SNP-heritability due to genotyped and imputed SNPs (\(h_g^2\), the proportion of phenotypic variance causally explained by common SNPs) of each cancer using LDSC15. Briefly, this method is based on the relationship between LD score and χ2-statistics:
where \(E\left[ {\chi _j^2} \right]\) denotes the expected χ2-statistics for the association between the outcome and SNP j, Nj is the study sample size available for SNP j, M is the total numbers of variants and lj denotes the LD score of SNP j defined as \(l_j = \mathop {\sum }\limits_k r^2\left( {j,k} \right)\) (k denotes other variants within the LD region). Note that the quantity estimated by LDSC is the causal heritability of common SNPs, which is different from the SNP-heritability as defined in Yang et al.17. To estimate \(h_g^2\) attributable to undiscovered loci, we identified SNPs that were associated with a given cancer at genome-wide significance (p < 5 × 10−8) and removed all SNPs within (+/−) 500,000 base-pairs of those loci prior to calculation (number of regions (+/− 500 kb) for each cancer that reach the 5 × 10−8 threshold and measures of effect size are shown in Supplementary Data 1). We also converted the SNP-heritability from observed scale to liability scale by incorporating sample prevalence (P) and population prevalence (F) of each cancer:
We subsequently calculated the genome-wide genetic correlations (rg) between different cancers, and between cancers and non-cancer traits, using an algorithm14:
where βj and γj are the effect sizes of SNP j on traits 1 and 2, rg is the genetic covariance, M is number of SNPs, N1 and N2 are the sample sizes for trait 1 and 2, Ns is the number of overlapping samples, r is the phenotypic correlation in overlapping samples and lj is the LD score defined as above. For genetic correlation between 6 cancers, the significance level is 0.05/15 = 0.003; for genetic correlation between 6 cancers and 38 traits, the significance level is 0.05/(6 × 38) = 0.0002.
Overall genetic correlations as estimated by LDSC are based on aggregated information across all variants in the genome. It is possible that even though two traits show negligible overall genetic correlation, there are specific regions in the genome that contribute to both traits. We therefore examined local genetic correlations between cancer pairs using ρ-HESS50, an algorithm which partitions the whole genome into 1703 regions based on LD-pattern of European populations and quantifies correlation between pairs of traits due to genetic variation restricted to these genomic regions. Local genetic correlation was considered statistically significant if p < 0.05/1,703 = 2.9 × 10−5. In particular, we assessed the local genetic correlations for previously reported pleiotropic regions18,51 known to harbor SNPs affecting multiple cancers.
Directional genetic correlation analysis
In addition to the genetic correlation analysis, which reflects overall genetic overlaps, we also attempted to identify directions of potential genetic correlations using a subset of SNPs as proposed by Pickrell et al.52. The method adopts the following assumption: if a trait X influences trait Y, then SNPs influencing X should also influence Y, and the SNP-specific effect sizes for the two traits should be correlated. Further, since Y does not influence X, but could be influenced by mechanisms independent of X, genetic variants that influence Y do not necessarily influence X. Based on this assumption, the method proposes two causal models and two non-causal models; and calculates the relative likelihood ratio (LR) of the best non-causal model compared to the best causal model. We determined significant SNPs for each given cancer or trait in two independent ways, (1) LD pruned SNPs: we selected genome-wide significant (p < 5 × 10−8) SNPs and pruned on LD-pattern in the European populations in Phase1 of 1KGP; (2) posterior probability of association (PPA) SNPs: we used a method implemented in fgwas53, which splits the genome into independent blocks based on LD patterns in 1KGP and estimates the prior probability that any block contains an association. The model outputs posterior probability that the region contains a variant that influences the trait. We selected the lead SNP from each of the regions with a PPA of at least 0.9. We scanned through all pairs of cancers and traits to identify directional correlations. Only pairs of traits with evidence of directional correlations (LR comparing the best non-causal model over the best causal model < 0.05) and without evidence of heteroscedasticity (pleiotropic effects)54 were reported as relatively more likely to exhibit mediated causation.
Functional partitioning of SNP-heritability
To assess the importance of specific functional annotations in SNP-heritability across cancers, we partitioned the cancer-specific heritability using stratified-LDSC14. This method partitions SNPs into functional categories and calculates category-specific enrichments based on the assumption that a category of SNPs is enriched for heritability if SNPs with high LD to that category have higher χ2 statistics than SNPs with low LD to that category. The analysis was performed using two models14,24.
-
1.
A full baseline-LD model including 24 publicly available annotations that are not specific to any cell type. When performing this model, we adjusted for MAF via MAF-stratified quantile-normalized LD score, and other LD-related annotations such as predicted allele age and recombination rate, as implemented by Gazal et al.24. Briefly, the 24 annotations included coding, 3′UTR and 5′UTR, promoter and intronic regions, obtained from UCSC Genome Browser and post-processed by Gusev et al.55; the histone marks mono-methylation (H3K4me1) and tri-methylation of histone H3 at lysine 4 (H3K4me3), acetylation of histone H3 at lysine 9 (H3K9ac) processed by Trynka et al.56,57,58 and two versions of acetylation of histone H3 at lysine 27 (H3K27ac, one version processed by Hnisz et al.59, another used by the Psychiatric Genomics Consortium (PGC)60); open chromatin, as reflected by DNase I hypersensitivity sites (DHSs and fetal DHSs)55, obtained as a combination of ENCODE and Roadmap Epigenomics data, processed by Trynka et al.58; combined chromHMM and Segway predictions obtained from Hoffman et al.61, which make use of many annotations to produce a single partition of the genome into seven underlying chromatin states (The CCCTC-binding factor (CTCF), promoter-flanking, transcribed, transcription start site (TSS), strong enhancer, weak enhancer categories, and the repressed category); regions that are conserved in mammals, obtained from Lindblad-Toh et al.40 and post-processed by Ward and Kellis62; super-enhancers, which are large clusters of highly active enhancers, obtained from Hnisz et al.59; FANTOM5 enhancers with balanced bi-directional capped transcripts identified using cap analysis of gene expression in the FANTOM5 panel of samples, obtained from Andersson et al.63; digital genomic footprint (DGF) and transcription factor binding site (TFBS) annotations obtained from ENCODE and post-processed by Gusev et al.55
-
2.
In addition to the baseline-LD model, we also performed analyses using 220 cell-type-specific annotations for the four histone marks H3K4me1, H3K4me3, H3K9ac, and H3K27ac. Each cell-type-specific annotation corresponds to a histone mark in a single cell type (for example, H3K27ac in CD19 immune cells), and there were 220 such annotations in total. We further divided these 220 cell-type-specific annotations into 10 groups (adrenal and pancreas, central nervous system (CNS), cardiovascular, connective and bone, gastrointestinal, immune and hematopoietic, kidney, liver, skeletal muscle, and other) by taking a union of the cell-type-specific annotations within each group (for example, SNPs with any of the four histone modifications in any hematopoietic and immune cells were considered as one big category). When generating the cell-type-specific models, we added annotations individually to the baseline model, creating 220 separate models.
We performed a random-effects meta-analysis of the proportion of heritability over six cancers for each functional category. We set significance thresholds for individual annotations at p < 0.05/24 for baseline model and at p < 0.05/220 for cell-type-specific annotation.
Data availability
The datasets generated during and/or analyzed during the current study are available from the authors on request. Breast cancer: summary results for all variants are available at http://bcac.ccge.medschl.cam.ac.uk/. Requests for further data should be made through the Data Access Coordination Committee (http://bcac.ccge.medschl.cam.ac.uk/). Ovarian cancer: summary results are available from the Ovarian Cancer Association Consortium (OCAC) (http://ocac.ccge.medschl.cam.ac.uk/). Requests for further data can be made to the Data Access Coordination Committee (http://cimba.ccge.medschl.cam.ac.uk/). Prostate cancer: summary results are publicly available at the PRACTICAL website (http://practical.icr.ac.uk/blog/). Lung cancer: genotype data for lung cancer are available at the database of Genotypes and Phenotypes (dbGaP) under accession phs001273.v1.p1. Readers interested in obtaining a copy of the original data can do so by completing the proposal request form at http://oncoarray.dartmouth.edu/. Head/neck cancer: genotype data for the oral and pharyngeal OncoArray study have been deposited at the database of Genotypes and Phenotypes (dbGaP) under accession phs001202.v1.p1. Colorectal cancer: genotype data have been deposited at the database of Genotypes and Phenotypes (dbGaP) under accession number phs001415.v1.p1 and phs001078.v1.p1.
Change history
23 September 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Lichtenstein, P. et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 343, 78–85 (2000).
Mucci, L. A. et al. Familial risk and heritability of cancer among twins in nordic countries. JAMA 315, 68 (2016).
Polderman, T. J. C. et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015).
Amundadottir, L. T. et al. Cancer as a complex phenotype: pattern of cancer distribution within and beyond the nuclear family. PLoS Med. 1, e65 (2004).
Yu, H., Frank, C., Sundquist, J., Hemminki, A. & Hemminki, K. Common cancers share familial susceptibility: implications for cancer genetics and counselling. J. Med. Genet. 54, 248–253 (2017).
Frank, C., Sundquist, J., Yu, H., Hemminki, A. & Hemminki, K. Concordant and discordant familial cancer: familial risks, proportions and population impact. Int. J. Cancer 140, 1510–1516 (2017).
Fehringer, G. et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 76, 5103–5114 (2016).
Kar, S. P. et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 6, 1052–1067 (2016).
Sampson, J. N. et al. Analysis of heritability and shared heritability based on genome-wide association studies for thirteen cancer types. J. Natl. Cancer Inst. 107, djv279 (2015).
Gusev, A. et al. Atlas of prostate cancer heritability in European and African-American men pinpoints tissue-specific regulation. Nat. Commun. 7, 10979 (2016).
Jiao, S. et al. Estimating the heritability of colorectal cancer. Hum. Mol. Genet. 23, 3898–3905 (2014).
Lu, Y. et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum. Mol. Genet. 23, 6112–6118 (2014).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Lindström, S. et al. Quantifying the genetic correlation between multiple cancer types. Cancer Epidemiol. Biomark. Prev. 26, 1427–1435 (2017).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Amos, C. I. et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 26, 126–135 (2017).
SMOKING and health. Joint report of the Study Group on Smoking and Health. Science 125, 1129–1133 (1957).
Shaw, R. & Beasley, N. Aetiology and risk factors for head and neck cancer: United Kingdom National Multidisciplinary Guidelines. J. Laryngol. Otol. 130, S9–S12 (2016).
Koene, R. J., Prizment, A. E., Blaes, A. & Konety, S. H. Shared risk factors in cardiovascular disease and cancer. Circulation 133, 1104–1114 (2016).
Thompson, C. L. et al. Short duration of sleep increases risk of colorectal adenoma. Cancer 117, 841–847 (2011).
Sigurdardottir, L. G. et al. Sleep disruption among older men and risk of prostate cancer. Cancer Epidemiol. Prev. Biomark. 22, 872–879 (2013).
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017).
Field, R. W. & Withers, B. L. Occupational and environmental causes of lung cancer. Clin. Chest Med. 33, 681–703 (2012).
Hulka, B. S. Epidemiologic analysis of breast and gynecologic cancers. Prog. Clin. Biol. Res. 396, 17–29 (1997).
Gaudet, M. M. et al. Pooled analysis of active cigarette smoking and invasive breast cancer risk in 14 cohort studies. Int. J. Epidemiol. 46, 881–893 (2017).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Maas, P. et al. Breast cancer risk from modifiable and nonmodifiable risk factors among white women in the United States. JAMA Oncol 2, 1295–1302 (2016).
Nakaya, N. et al. Personality traits and cancer risk and survival based on finnish and swedish registry data. Am. J. Epidemiol. 172, 377–385 (2010).
Oksbjerg Dalton, S., Munk Laursen, T., Mellemkjaer, L., Johansen, C. & Mortensen, P. B. Schizophrenia and the risk for breast cancer. Schizophr. Res. 62, 89–92 (2003).
Hung, R. J. et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637 (2008).
Amos, C. I. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40, 616–622 (2008).
Gao, C. et al. Mendelian randomization study of adiposity-related traits and risk of breast, ovarian, prostate, lung and colorectal cancer. Int. J. Epidemiol. 45, 896–908 (2016).
Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 13, 1141–1151 (2012).
Day, F. R. et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 47, 1294–1303 (2015).
Day, F. R. et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Genet. 49, 834–841 (2017).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Ciriello, G. et al. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133 (2013).
Lindblad-Toh, K. et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011).
Calin, G. A. et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229 (2007).
Peng, J. C., Shen, J. & Ran, Z. H. Transcribed ultraconserved region in human cancers. RNA Biol. 10, 1771–1777 (2013).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Milne, R. L. et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 49, 1767–1778 (2017).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132 (2017).
Schmit, S. L. et al. Novel Common Genetic Susceptibility Loci for Colorectal Cancer. J. Natl. Cancer Inst. 111, djy099 (2019).
Phelan, C. M. et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 49, 680–691 (2017).
Lesseur, C. et al. Genome-wide association analyses identify new susceptibility loci for oral cavity and pharyngeal cancer. Nat. Genet. 48, 1544–1550 (2016).
Schumacher, F. R. et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 50, 928–936 (2018).
Shi, H., Mancuso, N., Spendlove, S. & Pasaniuc, B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am. J. Hum. Genet. 101, 737–751 (2017).
Sakoda, L. C., Jorgenson, E. & Witte, J. S. Turning of COGS moves forward findings for hormonally mediated cancers. Nat. Genet. 45, 345–348 (2013).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Pickrell, J. K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94, 559–573 (2014).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Gusev, A. et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 95, 535–552 (2014).
Roadmap Epigenomics Consortium. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Trynka, G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 45, 124–130 (2013).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Hoffman, M. M. et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 41, 827–841 (2013).
Ward, L. D. & Kellis, M. Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science 337, 1675–1678 (2012).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Acknowledgements
The authors in this manuscript were working on behalf of BCAC, CCFR, CIMBA, CORECT, GECCO, OCAC, PRACTICAL, CRUK, BPC3, CAPS, PEGASUS, TRICL-ILCCO, ABCTB, APCB, BCFR, CONSIT TEAM, EMBRACE, GC-HBOC, GEMO, HEBON, kConFab/AOCS Mod SQuaD, and SWE-BRCA. The breast cancer genome-wide association analyses: BCAC is funded by Cancer Research UK [C1287/A16563, C1287/A10118], the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST, respectively), and by the European Community’s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation, or writing of the report. Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344) and, the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSR-SIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978), and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065, and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The DRIVE Consortium was funded by U19 CA148065. The Australian Breast Cancer Family Study (ABCFS) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia), and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow. The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363]. The Australian Breast Cancer Tissue Bank (ABCTB) is generously supported by the National Health and Medical Research Council of Australia, The Cancer Institute NSW and the National Breast Cancer Foundation. The ACP study is funded by the Breast Cancer Research Trust, UK. The AHS study is supported by the intramural research program of the National Institutes of Health, the National Cancer Institute (grant number Z01-CP010119), and the National Institute of Environmental Health Sciences (grant number Z01-ES049030). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). The BCEES was funded by the National Health and Medical Research Council, Australia and the Cancer Council Western Australia and acknowledges funding from the National Breast Cancer Foundation (JS). For the BCFR-NY, BCFR-PA, and BCFR-UT this work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. For BIGGS, ES is supported by NIHR Comprehensive Biomedical Research Centre, Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London, United Kingdom. IT is supported by the Oxford Biomedical Research Centre. BOCS is supported by funds from Cancer Research UK (C8620/A8372/A15106) and the Institute of Cancer Research (UK). BOCS acknowledges NHS funding to the Royal Marsden/Institute of Cancer Research NIHR Specialist Cancer Biomedical Research Centre. The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). The CAMA study was funded by Consejo Nacional de Ciencia y Tecnología (CONACyT) (SALUD-2002-C01-7462). Sample collection and processing was funded in part by grants from the National Cancer Institute (NCI R01CA120120 and K24CA169004). CBCS is funded by the Canadian Cancer Society (grant # 313404) and the Canadian Institutes of Health Research. CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The CNIO-BCS was supported by the Instituto de Salud Carlos III, the Red Temática de Investigación Cooperativa en Cáncer and grants from the Asociación Española Contra el Cáncer and the Fondo de Investigación Sanitario (PI11/00923 and PI12/00070). COLBCCC is supported by the German Cancer Research Center (DKFZ), Heidelberg, Germany. D.T. was in part supported by a postdoctoral fellowship from the Alexander von Humboldt Foundation. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The CTS was initially supported by the California Breast Cancer Act of 1993 and the California Breast Cancer Research Fund (contract 97-10500) and is currently funded through the National Institutes of Health (R01 CA77398, UM1 CA164917, and U01 CA199277). Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885. H.A.C eceives support from the Lon V Smith Foundation (LVS39420). The University of Westminster curates the DietCompLyf database funded by Against Breast Cancer Registered Charity No. 1121258 and the NCRN. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). FHRISK is funded from NIHR grant PGfAR 0707-10031. The GC-HBOC (German Consortium of Hereditary Breast and Ovarian Cancer) is supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE - Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 713-241202, 14505/2470, and 14575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0, and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The GEPARSIXTO study was conducted by the German Breast Group GmbH. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). GLACIER was supported by Breast Cancer Now, CRUK and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research, by the Lower Saxonian Cancer Society, and by the Rudolf-Bartling Foundation. The HEBCS was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society, and the Sigrid Juselius Foundation. The HERPACC was supported by MEXT Kakenhi (No. 170150181 and 26253041) from the Ministry of Education, Science, Sports, Culture and Technology of Japan, by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from Ministry Health, Labour and Welfare of Japan, by Health and Labour Sciences Research Grants for Research on Applying Health Technology from Ministry Health, Labour and Welfare of Japan, by National Cancer Center Research and Development Fund, and “Practical Research for Innovative Cancer Control (15ck0106177h0001)” from Japan Agency for Medical Research and development, AMED, and Cancer Bio Bank Aichi. The HMBCS was supported by a grant from the Friends of Hannover Medical School and by the Rudolf Bartling Foundation. The HUBCS was supported by a grant from the German Federal Ministry of Research and Education (RUS08/017), and by the Russian Foundation for Basic Research and the Federal Agency for Scientific Organizations for support the Bioresource collections and RFBR grants 14-04-97088, 17-29-06014, and 17-44-020498. ICICLE was supported by Breast Cancer Now, CRUK, and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (A.L.F.) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (E.V.O.) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command [DAMD17-01-1-0729], Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). G.C.-T. and P.W. are supported by the NHMRC. RB was a Cancer Institute NSW Clinical Research Fellow. The KOHBRA study was partially supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), and the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (HI16C1127; 1020350; 1420190). LAABC is supported by grants (1RB-0287, 3PB-0102, 5PB-0018, 10PB-0098) from the California Breast Cancer Research Program. Incident breast cancer cases were collected by the USC Cancer Surveillance Program (CSP) which is supported under subcontract by the California Department of Health. The CSP is also part of the National Cancer Institute’s Division of Cancer Prevention and Control Surveillance, Epidemiology, and End Results Program, under contract number N01CN25403. L.M.B.C. is supported by the ‘Stichting tegen Kanker’. D.L. is supported by the FWO. The MABCS study is funded by the Research Centre for Genetic Engineering and Biotechnology “Georgi D. Efremov” and supported by the German Academic Exchange Program, DAAD. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC) and by funds from the Italian citizens who allocated the 5/1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects “5 × 1000”). The MCBCS was supported by the NIH grants CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation. MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057 and 396414, and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. The MEC was support by NIH grants CA63464, CA54281, CA098758, CA132839, and CA164973. The MISS study is supported by funding from ERC-2011-294576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH grants CA97396, CA128931, CA116201, CA140286, and CA177150. MSKCC is supported by grants from the Breast Cancer Research Foundation and Robert and Kate Niehaus Clinical Cancer Genetics Initiative. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program – grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade – grant # PSR-SIIRI-701. MYBRCA is funded by research grants from the Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Malaysia. MYMAMMO is supported by research grants from Yayasan Sime Darby LPGA Tournament and Malaysian Ministry of Higher Education (RP046B-15HTM). The NBCS has been supported by the Research Council of Norway grant 193387/V50 (to A.-L. Børresen-Dale and V.N. Kristensen) and grant 193387/H10 (to A.-L. Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39346 to A.-L. Børresen-Dale and 27208 to V.N. Kristensen) and the Norwegian Cancer Society (to A.-L. Børresen-Dale and 419616 - 71248 - PR-2006-0282 to V.N. Kristensen). It has received funding from the K.G. Jebsen Centre for Breast Cancer Research (2012-2015). The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The Northern California Breast Cancer Family Registry (NC-BCFR) and Ontario Familial Breast Cancer Registry (OFBCR) were supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The Carolina Breast Cancer Study was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, and U01 CA179715), and the North Carolina University Cancer Research Fund. The NGOBCS was supported by Grants-in-Aid for the Third Term Comprehensive Ten-Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan, and for Scientific Research on Priority Areas, 17015049 and for Scientific Research on Innovative Areas, 221S0001, from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The NHS was supported by NIH grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH grants UM1 CA176726 and U19 CA148065. The OBCS was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland (grant number 250083, 122715 and Center of Excellence grant number 251314), the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation, and the special Governmental EVO funds for Oulu University Hospital-based research activities. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997-1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956, and Breast Cancer Campaign 2010PR62, 2013PR044. PROCAS is funded from NIHR grant PGfAR 0707-10031. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCGS was supported primarily by NIH grants R01CA64277, R01CA148667, UMCA182910, and R37CA70867. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The scientific development and funding of this project were, in part, supported by the Genetic Associations and Mechanisms in Oncology (GAME-ON) Network U19 CA148065. The SBCS was supported by Sheffield Experimental Cancer Medicine Centre and Breast Cancer Now Tissue Bank. The SCCS is supported by a grant from the National Institutes of Health (R01 CA092447). Data on SCCS cancer cases used in this publication were provided by the Alabama Statewide Cancer Registry; Kentucky Cancer Registry, Lexington, KY; Tennessee Department of Health, Office of Cancer Surveillance; Florida Cancer Data System; North Carolina Central Cancer Registry, North Carolina Division of Public Health; Georgia Comprehensive Cancer Registry; Louisiana Tumor Registry; Mississippi Cancer Registry; South Carolina Central Cancer Registry; Virginia Department of Health, Virginia Cancer Registry; Arkansas Department of Health, Cancer Registry, 4815 W. Markham, Little Rock, AR 72205. The Arkansas Central Cancer Registry is fully funded by a grant from National Program of Cancer Registries, Centers for Disease Control and Prevention (CDC). Data on SCCS cancer cases from Mississippi were collected by the Mississippi Cancer Registry which participates in the National Program of Cancer Registries (NPCR) of the Centers for Disease Control and Prevention (CDC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the CDC or the Mississippi Cancer Registry. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. SEBCS was supported by the BRL (Basic Research Laboratory) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2012-0000347). SGBCC is funded by the NUS start-up Grant, National University Cancer Institute Singapore (NCIS) Centre Grant and the NMRC Clinician Scientist Award. Additional controls were recruited by the Singapore Consortium of Cohort Studies-Multi-ethnic cohort (SCCS-MEC), which was funded by the Biomedical Research Council, grant number: 05/1/21/19/425. The Sister Study (SISTER) is supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES049033). The Two Sister Study (2SISTER) was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES102245), and, also by a grant from Susan G. Komen for the Cure, grant FAS0703856. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation. The SZBCS was supported by Grant PBZ_KBN_122/P05/2004. The TBCS was funded by The National Cancer Institute, Thailand. The TNBCC was supported by a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation. The TWBCS is supported by the Taiwan Biobank project of the Institute of Biomedical Sciences, Academia Sinica, Taiwan. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledges NHS funding to the NIHR Biomedical Research Centre. The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The US3SS study was supported by Massachusetts (K.M.E., R01CA47305), Wisconsin (P.A.N., R01 CA47147) and New Hampshire (L.T.-E., R01CA69664) centers, and Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The USRT Study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The WAABCS study was supported by grants from the National Cancer Institute of the National Institutes of Health (R01 CA89085 and P50 CA125183 and the D43 TW009112 grant), Susan G. Komen (SAC110026), the Dr. Ralph and Marian Falk Medical Research Trust, and the Avon Foundation for Women. The WHI program is funded by the National Heart, Lung, and Blood Institute, the US National Institutes of Health and the US Department of Health and Human Services (HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C). This work was also funded by NCI U19 CA148065-01. D.G.E. is supported by the all Manchester NIHR Biomedical research center Manchester (IS-BRC-1215-20007). HUNBOCS, Hungarian Breast and Ovarian Cancer Study was supported by Hungarian Research Grant KTIA-OTKA CK-80745, NKFI_OTKA K-112228. C.I. received support from the Nontherapeutic Subject Registry Shared Resource at Georgetown University (NIH/NCI P30-CA-51008) and the Jess and Mildred Fisher Center for Hereditary Cancer and Clinical Genomics Research. K.M. is supported by CRUK C18281/A19169. City of Hope Clinical Cancer Community Research Network and the Hereditary Cancer Research Registry, supported in part by Award Number RC4CA153828 (PI: J Weitzel) from the National Cancer Institute and the office of the Directory, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The colorectal cancer genome-wide association analyses: Colorectal Transdisciplinary Study (CORECT): The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CORECT Consortium, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the CORECT Consortium. We are incredibly grateful for the contributions of Dr. Brian Henderson and Dr. Roger Green over the course of this study and acknowledge them in memoriam. We are also grateful for support from Daniel and Maryann Fong. ColoCare: we thank the many investigators and staff who made this research possible in ColoCare Seattle and ColoCare Heidelberg. ColoCare was initiated and developed at the Fred Hutchinson Cancer Research Center by Drs. Ulrich and Grady. CCFR: the Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff, and financial support from the U.S. National Cancer Institute, without which this important registry would not exist. Galeon: GALEON wishes to thank the Department of Surgery of University Hospital of Santiago (CHUS), Sara Miranda Ponte, Carmen M Redondo, and the staff of the Department of Pathology and Biobank of CHUS, Instituto de Investigación Sanitaria de Santiago (IDIS), Instituto de Investigación Sanitaria Galicia Sur (IISGS), SERGAS, Vigo, Spain, and Programa Grupos Emergentes, Cancer Genetics Unit, CHUVI Vigo Hospital, Instituto de Salud Carlos III, Spain. MCCS: this study was made possible by the contribution of many people, including the original investigators and the diligent team who recruited participants and continue to work on follow-up. We would also like to express our gratitude to the many thousands of Melbourne residents who took part in the study and provided blood samples. SEARCH: We acknowledge the contributions of Mitul Shah, Val Rhenius, Sue Irvine, Craig Luccarini, Patricia Harrington, Don Conroy, Rebecca Mayes, and Caroline Baynes. The Swedish low-risk colorectal cancer study: we thank Berith Wejderot and the Swedish low-risk colorectal cancer study group. Genetics & Epidemiology of Colorectal Cancer Consortium (GECCO): we thank all those at the GECCO Coordinating Center for helping bring together the data and people that made this project possible. ASTERISK: we are very grateful to Dr. Bruno Buecher without whom this project would not have existed. We also thank all those who agreed to participate in this study, including the patients and the healthy control persons, as well as all the physicians, technicians and students. DACHS: we thank all participants and cooperating clinicians, and Ute Handte-Daub, Renate Hettler-Jensen, Utz Benscheid, Muhabbet Celik, and Ursula Eilber for excellent technical assistance. HPFS, NHS and PHS: we acknowledge Patrice Soule and Hardeep Ranu of the Dana-Farber Harvard Cancer Center High-Throughput Polymorphism Core who assisted in the genotyping for NHS, HPFS, and PHS under the supervision of Dr. Immaculata Devivo and Dr. David Hunter, Qin (Carolyn) Guo, and Lixue Zhu who assisted in programming for NHS and HPFS and Haiyan Zhang who assisted in programming for the PHS. We thank the participants and staff of the Nurses’ Health Study and the Health Professionals Follow-Up Study, for their valuable contributions as well as the following state cancer registries for their help: A.L., A.Z., A.R., C.A., C.O., C.T., D.E., F.L., G.A., I.D., I.L., I.N., I.A., K.Y., L.A., M.E., M.D., M.A., M.I., N.E., N.H., N.J., N.Y., N.C., N.D., O.H., O.K., O.R., P.A., R.I., S.C., T.N., T.X., V.A., W.A., W.Y. In addition, this study was approved by the Connecticut Department of Public Health (DPH) Human Investigations Committee. Certain data used in this publication were obtained from the DPH. We assume full responsibility for analyses and interpretation of these data. PLCO: we thank Drs. Christine Berg and Philip Prorok, Division of Cancer Prevention, National Cancer Institute, the Screening Center investigators and staff or the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial, Mr. Tom Riley and staff, Information Management Services Inc., Ms. Barbara O’Brien and staff, Westat Inc. and Drs. Bill Kopp, Wen Shao and staff, SAIC-Frederick. Most importantly, we acknowledge the study participants for their contributions for making this study possible. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by NCI. PMH: we thank the study participants and staff of the Hormones and Colon Cancer study. WHI: we thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at https://cleo.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short20List.pdf. CORECT: The CORECT Study was supported by the National Cancer Institute, National Institutes of Health (NCI/NIH), U.S. Department of Health and Human Services (grant numbers U19 CA148107, R01 CA81488, P30 CA014089, R01 CA197350; P01 CA196569; and R01 CA201407) and National Institutes of Environmental Health Sciences, National Institutes of Health (grant number T32 ES013678). The ATBC Study was supported by the US Public Health Service contracts (N01-CN-45165, N01-RC-45035, N01-RC-37004, and HHSN261201000006C) from the National Cancer Institute. The Cancer Prevention Study-II Nutrition Cohort is funded by the American Cancer Society. ColoCare: This work was supported by the National Institutes of Health (grant numbers R01 CA189184, U01 CA206110, 2P30CA015704-40 (Gilliland)), the Matthias Lackas-Foundation, the German Consortium for Translational Cancer Research, and the EU TRANSCAN initiative. Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): funding for GECCO was provided by the National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (grant numbers U01 CA137088, R01 CA059045, and U01 CA164930). This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704. The Colon Cancer Family Registry (CFR) Illumina GWAS was supported by funding from the National Cancer Institute, National Institutes of Health (grant numbers U01 CA122839, R01 CA143247). The Colon CFR/CORECT Affymetrix Axiom GWAS and OncoArray GWAS were supported by funding from National Cancer Institute, National Institutes of Health (grant number U19 CA148107 to S.G.). The Colon CFR participant recruitment and collection of data and biospecimens used in this study were supported by the National Cancer Institute, National Institutes of Health (grant number UM1 CA167551) and through cooperative agreements with the following Colon CFR centers: Australasian Colorectal Cancer Family Registry (NCI/NIH grant numbers U01 CA074778 and U01/U24 CA097735), USC Consortium Colorectal Cancer Family Registry (NCI/NIH grant numbers U01/U24 CA074799), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (NCI/NIH grant number U01/U24 CA074800), Ontario Familial Colorectal Cancer Registry (NCI/NIH grant number U01/U24 CA074783), Seattle Colorectal Cancer Family Registry (NCI/NIH grant number U01/U24 CA074794), and University of Hawaii Colorectal Cancer Family Registry (NCI/NIH grant number U01/U24 CA074806), Additional support for case ascertainment was provided from the Surveillance, Epidemiology and End Results (SEER) Program of the National Cancer Institute to Fred Hutchinson Cancer Research Center (Control Nos. N01-CN-67009 and N01-PC-35142, and Contract No. HHSN2612013000121), the Hawai’i Department of Health (Control Nos. N01-PC-67001 and N01-PC-35137, and Contract No. HHSN26120100037C, and the California Department of Public Health (contracts HHSN261201000035C awarded to the University of Southern California, and the following state cancer registries: A.Z., C.O., M.N., N.C., N.H., and by the Victoria Cancer Registry and Ontario Cancer Registry. ESTHER/VERDI was supported by grants from the Baden–Württemberg Ministry of Science, Research and Arts and the German Cancer Aid. MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. GALEON: FIS Intrasalud (PI13/01136). The MCCS was further supported by Australian NHMRC grants 509348, 209057, 251553, and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. MSKCC: the work at Sloan Kettering in New York was supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics and the Romeo Milio Foundation. Moffitt: This work was supported by funding from the National Institutes of Health (grant numbers R01 CA189184, P30 CA076292), Florida Department of Health Bankhead-Coley Grant 09BN-13, and the University of South Florida Oehler Foundation. Moffitt contributions were supported in part by the Total Cancer Care Initiative, Collaborative Data Services Core, and Tissue Core at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute-designated Comprehensive Cancer Center (grant number P30 CA076292). SEARCH: Cancer Research UK (C490/A16561). The Spanish study was supported by Instituto de Salud Carlos III, co-funded by FEDER funds –a way to build Europe– (grants PI14-613 and PI09-1286), Catalan Government DURSI (grant 2014SGR647), and Junta de Castilla y León (grant LE22A10-2). The Swedish Low-risk Colorectal Cancer Study: the study was supported by grants from the Swedish research council; K2015-55 × -22674-01-4, K2008-55 × -20157-03-3, K2006-72 × -20157-01-2 and the Stockholm County Council (ALF project). CIDR genotyping for the Oncoarray was conducted under contract 268201200008I (to K.D.), through grant 101HG007491-01 (to C.I.A.). The Norris Cotton Cancer Center - P30CA023108, The Quantitative Biology Research Institute - P20GM103534, and the Coordinating Center for Screen Detected Lesions - U01CA196386 also supported efforts of C.I.A. This work was also supported by the National Cancer Institute (grant numbers U01 CA1817700, R01 CA144040). ASTERISK: a Hospital Clinical Research Program (PHRC) and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC). COLO2&3: National Institutes of Health (grant number R01 CA060987). DACHS: This work was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1, and BR 1704/17-1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A, and 01ER1505B). DALS: National Institutes of Health (grant number R01 CA048998 to M.L.S). HPFS is supported by National Institutes of Health (grant numbers P01 CA055075, UM1 CA167552, R01 137178, and P50 CA127003), NHS by the National Institutes of Health (grant numbers UM1 CA186107, R01 CA137178, P01 CA087969, and P50 CA127003), NHSII by the National Institutes of Health (grant numbers R01 050385CA and UM1 CA176726), and PHS by the National Institutes of Health (grant number R01 CA042182). MEC: National Institutes of Health (grant numbers R37 CA054281, P01 CA033619, and R01 CA063464). OFCCR: National Institutes of Health, through funding allocated to the Ontario Registry for Studies of Familial Colorectal Cancer (grant number U01 CA074783); see Colon CFR section above. As subset of ARCTIC, OFCCR is supported by a GL2 grant from the Ontario Research Fund, the Canadian Institutes of Health Research, and the Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society Research Institute. T.J.H. and B.W.Z. are recipients of Senior Investigator Awards from the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation. PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Additionally, a subset of control samples was genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) Prostate Cancer GWAS, Colon CGEMS pancreatic cancer scan (PanScan), and the Lung Cancer and Smoking study. The prostate and PanScan study datasets were accessed with appropriate approval through the dbGaP online resource (http://cgems.cancer.gov/data/) accession numbers phs000207.v1.p1 and phs000206.v3.p2, respectively, and the lung datasets were accessed from the dbGaP website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093.v2.p2. Funding for the Lung Cancer and Smoking study was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438. For the lung study, the GENEVA Coordinating Center provided assistance with genotype cleaning and general study coordination, 23 and the Johns Hopkins University Center for Inherited Disease Research conducted genotyping. PMH: National Institutes of Health (grant number R01 CA076366). VITAL: National Institutes of Health (grant number K05-CA154337). WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. The head and neck cancer genome-wide association analyses: The study was supported by NIH/NCI: P50 CA097190, and P30 CA047904, Canadian Cancer Society Research Institute (no. 020214) and Cancer Care Ontario Research Chair to R.H. The Princess Margaret Hospital Head and Neck Cancer Translational Research Program is funded by the Wharton family, Joe’s Team, Gordon Tozer, Bruce Galloway and the Elia family. Geoffrey Liu was supported by the Posluns Family Fund and the Lusi Wong Family Fund at the Princess Margaret Foundation, and the Alan B. Brown Chair in Molecular Genomics. This publication presents data from Head and Neck 5000 (H&N5000). H&N5000 was a component of independent research funded by the UK National Institute for Health Research (NIHR) under its Programme Grants for Applied Research scheme (RP-PG-0707-10034). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Human papillomavirus (HPV) in H&N5000 serology was supported by a Cancer Research UK Programme Grant, the Integrative Cancer Epidemiology Programme (grant number: C18281/A19169). National Cancer Institute (R01-CA90731); National Institute of Environmental Health Sciences (P30ES10126). The authors thank all the members of the GENCAPO team/The Head and Neck Genome Project (GENCAPO) was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Grant numbers 04/12054-9 and 10/51168-0). CPS-II recruitment and maintenance is supported with intramural research funding from the American Cancer Society. Genotyping performed at the Center for Inherited Disease Research (CIDR) was funded through the U.S. National Institute of Dental and Craniofacial Research (NIDCR) grant 1 × 01HG007780-0. The University of Pittsburgh head and neck cancer case-control study is supported by National Institutes of Health grants P50 CA097190 and P30 CA047904. The Carolina Head and Neck Cancer Study (CHANCE) was supported by the National Cancer Institute (R01-CA90731). The Head and Neck Genome Project (GENCAPO) was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Grant numbers 04/12054-9 and 10/51168-0). The authors thank all the members of the GENCAPO team. The HN5000 study was funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research scheme (RP-PG-0707-10034), the views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The Toronto study was funded by the Canadian Cancer Society Research Institute (020214) and the National Cancer Institute (U19-CA148127) and the Cancer Care Ontario Research Chair. The alcohol-related cancers and genetic susceptibility study in Europe (ARCAGE) was funded by the European Commission’s 5th Framework Program (QLK1-2001-00182), the Italian Association for Cancer Research, Compagnia di San Paolo/FIRMS, Region Piemonte, and Padova University (CPDA057222). The Rome Study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) IG 2011 10491 and IG2013 14220 to S.B., and Fondazione Veronesi to S.B. The IARC Latin American study was funded by the European Commission INCO-DC programme (IC18-CT97-0222), with additional funding from Fondo para la Investigacion Cientifica y Tecnologica (Argentina) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (01/01768-2). We thank Leticia Fernandez, Instituto Nacional de Oncologia y Radiobiologia, La Habana, Cuba and Sergio and Rosalina Koifman, for their efforts with the IARC Latin America study São Paulo center. The IARC Central Europe study was supported by European Commission’s INCO-COPERNICUS Program (IC15- CT98-0332), NIH/National Cancer Institute grant CA92039, and the World Cancer Research Foundation grant WCRF 99A28. The IARC Oral Cancer Multicenter study was funded by grant S06 96 202489 05F02 from Europe against Cancer; grants FIS 97/0024, FIS 97/0662, and BAE 01/5013 from Fondo de Investigaciones Sanitarias, Spain; the UICC Yamagiwa-Yoshida Memorial International Cancer Study; the National Cancer Institute of Canada; Associazione Italiana per la Ricerca sul Cancro; and the Pan-American Health Organization. Coordination of the EPIC study is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The lung cancer genome-wide association analyses: Transdisciplinary Research for Cancer in Lung (TRICL) of the International Lung Cancer Consortium (ILCCO) was supported by (U19-CA148127, CA148127S1, U19CA203654, and Cancer Prevention Research Institute of Texas RR170048). The ILCCO data harmonization is supported by Cancer Care Ontario Research Chair of Population Studies to R. H. and Lunenfeld-Tanenbaum Research Institute, Sinai Health System. The TRICL-ILCCO OncoArray was supported by in-kind genotyping by the Centre for Inherited Disease Research (26820120008i-0-26800068-1). The CAPUA study was supported by FIS-FEDER/Spain grant numbers FIS-01/310, FIS-PI03-0365, and FIS-07-BI060604, FICYT/Asturias grant numbers FICYT PB02-67 and FICYT IB09-133, and the University Institute of Oncology (IUOPA), of the University of Oviedo and the Ciber de Epidemiologia y Salud Pública. CIBERESP, SPAIN. The work performed in the CARET study was supported by the National Institute of Health/National Cancer Institute: UM1 CA167462 (PI: Goodman), National Institute of Health UO1-CA6367307 (PIs Omen, Goodman); National Institute of Health R01 CA111703 (PI Chen), National Institute of Health 5R01 CA151989-01A1(PI Doherty). The Liverpool Lung project is supported by the Roy Castle Lung Cancer Foundation. The Harvard Lung Cancer Study was supported by the NIH (National Cancer Institute) grants CA092824, CA090578, CA074386. The Multi-ethnic Cohort Study was partially supported by NIH Grants CA164973, CA033619, CA63464, and CA148127. The work performed in MSH-PMH study was supported by The Canadian Cancer Society Research Institute (020214), Ontario Institute of Cancer and Cancer Care Ontario Chair Award to R.J.H. and G.L. and the Alan Brown Chair and Lusi Wong Programs at the Princess Margaret Hospital Foundation. NJLCS was funded by the State Key Program of National Natural Science of China (81230067), the National Key Basic Research Program Grant (2011CB503805), the Major Program of the National Natural Science Foundation of China (81390543). The Norway study was supported by Norwegian Cancer Society, Norwegian Research Council. The Shanghai Cohort Study (SCS) was supported by National Institutes of Health R01 CA144034 (PI: Yuan) and UM1 CA182876 (PI: Yuan). The Singapore Chinese Health Study (SCHS) was supported by National Institutes of Health R01 CA144034 (PI: Yuan) and UM1 CA182876 (PI: Yuan). The work in TLC study has been supported in part the James & Esther King Biomedical Research Program (09KN-15), National Institutes of Health Specialized Programs of Research Excellence (SPORE) Grant (P50 CA119997), and by a Cancer Center Support Grant (CCSG) at the H. Lee Moffitt Cancer Center and Research Institute, an NCI designated Comprehensive Cancer Center (grant number P30-CA76292). The Vanderbilt Lung Cancer Study—BioVU dataset used for the analyses described was obtained from Vanderbilt University Medical Center’s BioVU, which is supported by institutional funding, the 1S10RR025141-01 instrumentation award, and by the Vanderbilt CTSA grant UL1TR000445 from NCATS/NIH. Dr. Aldrich was supported by NIH/National Cancer Institute K07CA172294 (PI: Aldrich) and Dr. Bush was supported by NHGRI/NIH U01HG004798 (PI: Crawford). The Copenhagen General Population Study (CGPS) was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council and Herlev Hospital. The NELCS study: Grant Number P20RR018787 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). The Kentucky Lung Cancer Research Initiative was supported by the Department of Defense [Congressionally Directed Medical Research Program, U.S. Army Medical Research and Materiel Command Program] under award number: 10153006 (W81XWH-11-1-0781). Views and opinions of, and endorsements by the author(s) do not reflect those of the US Army or the Department of Defense. This research was also supported by unrestricted infrastructure funds from the UK Center for Clinical and Translational Science, NIH grant UL1TR000117 and Markey Cancer Center NCI Cancer Center Support Grant (P30 CA177558) Shared Resource Facilities: Cancer Research Informatics, Biospecimen and Tissue Procurement, and Biostatistics and Bioinformatics. The M.D. Anderson Cancer Center study was supported in part by grants from the NIH (P50 CA070907, R01 CA176568) (to X.W.), Cancer Prevention & Research Institute of Texas (RP130502) (to X.W.), and The University of Texas MD Anderson Cancer Center institutional support for the Center for Translational and Public Health Genomics. The deCODE study of smoking and nicotine dependence was funded in part by a grant from NIDA (R01- DA017932). The study in Lodz center was partially funded by Nofer Institute of Occupational Medicine, under task NIOM 10.13: Predictors of mortality from non-small cell lung cancer—field study. Genetic sharing analysis was funded by NIH grant CA194393. The research undertaken by M.D.T., L.V.W., and M.S.A. was partly funded by the National Institute for Health Research (NIHR). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. M.D.T. holds a Medical Research Council Senior Clinical Fellowship (G0902313). The work to assemble the FTND GWAS meta-analysis was supported by the National Institutes of Health (NIH), National Institute on Drug Abuse (NIDA) grant number R01 DA035825 (Principal Investigator [PI]: DBH). The study populations included COGEND (dbGaP phs000092.v1.p1 and phs000404.v1.p1), COPDGene (dbGaP phs000179.v3.p2), deCODE Genetics, EAGLE (dbGaP phs000093.vs.p2), and SAGE. dbGaP phs000092.v1.p1). See Hancock et al. Transl Psychiatry 2015 (PMCID: PMC4930126) for the full listing of funding sources and other acknowledgments. The Resource for the Study of Lung Cancer Epidemiology in North Trent (ReSoLuCENT)study was funded by the Sheffield Hospitals Charity, Sheffield Experimental Cancer Medicine Centre and Weston Park Hospital Cancer Charity. The ovarian cancer genome-wide association analysis: The Ovarian Cancer Association Consortium (OCAC) is supported by a grant from the Ovarian Cancer Research Fund thanks to donations by the family and friends of Kathryn Sladek Smith (PPD/RPCI.07). The scientific development and funding for this project were in part supported by the US National Cancer Institute GAME-ON Post-GWAS Initiative (U19-CA148112). This study made use of data generated by the Wellcome Trust Case Control consortium that was funded by the Wellcome Trust under award 076113. The results published here are in part based upon data generated by The Cancer Genome Atlas Pilot Project established by the National Cancer Institute and National Human Genome Research Institute (dbGap accession number phs000178.v8.p7). The OCAC OncoArray genotyping project was funded through grants from the U.S. National Institutes of Health (CA1X01HG007491-01 (C.I.A.), U19-CA148112 (T.A.S.), R01-CA149429 (C.M.P.), and R01-CA058598 (M.T.G.); Canadian Institutes of Health Research (MOP-86727 (L.E.K.) and the Ovarian Cancer Research Fund (A.B.). The COGS project was funded through a European Commission’s Seventh Framework Programme grant (agreement number 223175 - HEALTH-F2-2009-223175) and through a grant from the U.S. National Institutes of Health (R01-CA122443 (E.L.G)). Funding for individual studies: AAS: National Institutes of Health (RO1-CA142081); AOV: The Canadian Institutes for Health Research (MOP-86727); AUS: The Australian Ovarian Cancer Study Group was supported by the U.S. Army Medical Research and Materiel Command (DAMD17-01-1-0729), National Health & Medical Research Council of Australia (199600, 400413 and 400281), Cancer Councils of New South Wales, Victoria, Queensland, South Australia and Tasmania and Cancer Foundation of Western Australia (Multi-State Applications 191, 211, and 182). The Australian Ovarian Cancer Study gratefully acknowledges additional support from Ovarian Cancer Australia and the Peter MacCallum Foundation; BAV: ELAN Funds of the University of Erlangen-Nuremberg; BEL: National Kankerplan; BGS: Breast Cancer Now, Institute of Cancer Research; BVU: Vanderbilt CTSA grant from the National Institutes of Health (NIH)/National Center for Advancing Translational Sciences (NCATS) (ULTR000445); CAM: National Institutes of Health Research Cambridge Biomedical Research Centre and Cancer Research UK Cambridge Cancer Centre; CHA: Innovative Research Team in University (PCSIRT) in China (IRT1076); CNI: Instituto de Salud Carlos III (PI12/01319); Ministerio de Economía y Competitividad (SAF2012); COE: Department of Defense (W81XWH-11-2-0131); CON: National Institutes of Health (R01-CA063678, R01-CA074850; and R01-CA080742); DKE: Ovarian Cancer Research Fund; DOV: National Institutes of Health R01-CA112523 and R01-CA87538; EMC: Dutch Cancer Society (EMC 2014-6699); EPC: The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom); GER: German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401) and the German Cancer Research Center (DKFZ); GRC: This research has been co-financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)—Research Funding Program of the General Secretariat for Research & Technology: SYN11_10_19 NBCA. Investing in knowledge society through the European Social Fund; GRR: Roswell Park Cancer Institute Alliance Foundation, P30 CA016056; HAW: U.S. National Institutes of Health (R01-CA58598, N01-CN-55424, and N01-PC-67001); HJO: Intramural funding; Rudolf-Bartling Foundation; HMO: Intramural funding; Rudolf-Bartling Foundation; HOC: Helsinki University Research Fund; HOP: Department of Defense (DAMD17-02-1-0669) and NCI (K07-CA080668, R01-CA95023, P50-CA159981 MO1-RR000056 R01-CA126841); HUO: Intramural funding; Rudolf-Bartling Foundation; JGO: JSPS KAKENHI grant; JPN: Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare; KRA: This study (Ko-EVE) was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), and the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (HI16C1127; 0920010); LAX: American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124; LUN: ERC-2011-AdG 294576-risk factors cancer, Swedish Cancer Society, Swedish Research Council, Beta Kamprad Foundation; MAC: National Institutes of Health (R01-CA122443, P30-CA15083, P50-CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; Fraternal Order of Eagles; MAL: Funding for this study was provided by research grant R01- CA61107 from the National Cancer Institute, Bethesda, MD, research grant 94 222 52 from the Danish Cancer Society, Copenhagen, Denmark; and the Mermaid I project; MAS: Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Initiatives Foundation; MAY: National Institutes of Health (R01-CA122443, P30-CA15083, and P50-CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; MCC: Cancer Council Victoria, National Health and Medical Research Council of Australia (NHMRC) grants number 209057, 251533, 396414, and 504715; MDA: DOD Ovarian Cancer Research Program (W81XWH-07-0449); MEC: NIH (CA54281, CA164973, CA63464); MOF: Moffitt Cancer Center, Merck Pharmaceuticals, the state of Florida, Hillsborough County, and the city of Tampa; NCO: National Institutes of Health (R01-CA76016) and the Department of Defense (DAMD17-02-1-0666); NEC: National Institutes of Health R01-CA54419 and P50-CA105009 and Department of Defense W81XWH-10-1-02802; NHS: UM1 CA186107, P01 CA87969, R01 CA49449, R01-CA67262, UM1 CA176726; NJO: National Cancer Institute (NIH-K07 CA095666, R01-CA83918, NIH-K22-CA138563, and P30-CA072720) and the Cancer Institute of New Jersey; If Sara Olson and/or Irene Orlow is a co-author, please add NCI CCSG award (P30-CA008748) to the funding sources; NOR: Helse Vest, The Norwegian Cancer Society, The Research Council of Norway; NTH: Radboud University Medical Centre; OPL: National Health and Medical Research Council (NHMRC) of Australia (APP1025142) and Brisbane Women’s Club; ORE: OHSU Foundation; OVA: This work was supported by Canadian Institutes of Health Research grant (MOP-86727) and by NIH/NCI 1 R01CA160669-01A1; PLC: Intramural Research Program of the National Cancer Institute; POC: Pomeranian Medical University; POL: Intramural Research Program of the National Cancer Institute; PVD: Canadian Cancer Society and Cancer Research Society GRePEC Program; RBH: National Health and Medical Research Council of Australia; RMH: Cancer Research UK, Royal Marsden Hospital; RPC: National Institute of Health (P50-CA159981, R01-CA126841); SEA: Cancer Research UK (C490/A10119 C490/A10124); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; SIS: NIH, National Institute of Environmental Health Sciences, Z01-ES044005 and Z01-ES049033; SMC: The bbSwedish Research Council-SIMPLER infrastructure; the Swedish Cancer Foundation; SON: National Health Research and Development Program, Health Canada, grant 6613-1415-53; SRO: Cancer Research UK (C536/A13086, C536/A6689) and Imperial Experimental Cancer Research Centre (C1312/A15589); STA: NIH grants U01 CA71966 and U01 CA69417; SWE: Swedish Cancer foundation, WeCanCureCancer and VårKampMotCancer foundation; SWH: NIH (NCI) grant R37-CA070867; TBO: National Institutes of Health (R01-CA106414-A2), American Cancer Society (CRTG-00-196-01-CCE), Department of Defense (DAMD17-98-1-8659), Celma Mastery Ovarian Cancer Foundation; TOR: NIH grants R01-CA063678 and R01 CA063682; UCI: NIH R01-CA058860 and the Lon V Smith Foundation grant LVS39420; UHN: Princess Margaret Cancer Centre Foundation-Bridge for the Cure; UKO: The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre; UKR: Cancer Research UK (C490/A6187), UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; USC: P01CA17054, P30CA14089, R01CA61132, N01PC67010, R03CA113148, R03CA115195, N01CN025403, and California Cancer Research Program (00-01389V-20170, 2II0200); VAN: BC Cancer Foundation, VGH & UBC Hospital Foundation; VTL: NIH K05-CA154337; WMH: National Health and Medical Research Council of Australia, Enabling Grants ID 310670 & ID 628903. Cancer Institute NSW Grants 12/RIG/1-17 & 15/RIG/1-16; WOC: National Science Centren (N N301 5645 40). The Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academia Reserve. The prostate cancer genome-wide association analyses: we pay tribute to Brian Henderson, who was a driving force behind the OncoArray project, for his vision and leadership, and who sadly passed away before seeing its fruition. We also thank the individuals who participated in these studies enabling this work. The ELLIPSE/PRACTICAL (http//:practical.icr.ac.uk) prostate cancer consortium and his collaborating partners were supported by multiple funding mechanisms enabling this current work. ELLIPSE/PRACTICAL Genotyping of the OncoArray was funded by the US National Institutes of Health (NIH) (U19 CA148537 for ELucidating Loci Involved in Prostate Cancer SuscEptibility (ELLIPSE) project and X01HG007492 to the Center for Inherited Disease Research (CIDR) under contract number HHSN268201200008I). Additional analytical support was provided by NIH NCI U01 CA188392 (F.R.S.). Funding for the iCOGS infrastructure came from the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, and C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065, and 1U19 CA148112; the GAME-ON initiative), the Department of Defense (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. This work was supported by the Canadian Institutes of Health Research, European Commission’s Seventh Framework Programme grant agreement n° 223175 (HEALTH-F2-2009-223175), Cancer Research UK Grants C5047/A7357, C1287/A10118, C1287/A16563, C5047/A3354, C5047/A10692, C16913/A6135, C5047/A21332 and The National Institute of Health (NIH) Cancer Post-Cancer GWAS initiative grant: No. 1 U19 CA148537-01 (the GAME-ON initiative). We also thank the following for funding support: The Institute of Cancer Research and The Everyman Campaign, The Prostate Cancer Research Foundation, Prostate Research Campaign UK (now Prostate Action), The Orchid Cancer Appeal, The National Cancer Research Network UK, and The National Cancer Research Institute (NCRI) UK. We are grateful for support of NIHR funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. The Prostate Cancer Program of Cancer Council Victoria also acknowledge grant support from The National Health and Medical Research Council, Australia (126402, 209057, 251533, 396414, 450104, 504700, 504702, 504715, 623204, 940394, and 614296), VicHealth, Cancer Council Victoria, The Prostate Cancer Foundation of Australia, The Whitten Foundation, PricewaterhouseCoopers, and Tattersall’s. E.A.O., D.M.K., and E.M.K. acknowledge the Intramural Program of the National Human Genome Research Institute for their support. The BPC3 was supported by the U.S. National Institutes of Health, National Cancer Institute (cooperative agreements U01-CA98233 to D.J.H., U01-CA98710 to S.M.G., U01-CA98216 to E.R., and U01-CA98758 to B.E.H., and Intramural Research Program of NIH/National Cancer Institute, Division of Cancer Epidemiology and Genetics). CAPS GWAS study was supported by the Swedish Cancer Foundation (grant no 09-0677, 11-484, 12-823), the Cancer Risk Prediction Center (CRisP; www.crispcenter.org), a Linneus Centre (Contract ID 70867902) financed by the Swedish Research Council, Swedish Research Council (grant no K2010-70 × -20430-04-3, 2014-2269). The Hannover Prostate Cancer Study was supported by the Lower Saxonian Cancer Society. PEGASUS was supported by the Intramural Research Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health. RAPPER was supported by the NIHR Manchester Biomedical Research Center, Cancer Research UK (C147/A25254, C1094/A18504) and the EU’s 7th Framework Programme Grant/Agreement no 60186. Overall: this research has been conducted using the UK Biobank Resource (application number 16549). NHS is supported by UM1 CA186107 (NHS cohort infrastructure grant), P01 CA87969, and R01 CA49449. NHSII is supported by UM1 CA176726 (NHSII cohort infrastructure grant), and R01-CA67262. A.L.K. is supported by R01 MH107649. We would like to thank the participants and staff of the NHS and NHSII for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
Author information
Authors and Affiliations
Contributions
All authors reviewed and commented on the manuscript, as well as approved the submission. Writing group: X.J., H.K.F., F.R.S., S.L.S., J.P.T., Y. Han., K. Michailidou, C.L., K.B.K., J.D., D.V.C., G. Casey, M.M.G., J. Huyghe, D. Thomas, R.J. Hung, B.D., J.M., U.P., L.H., M. Garcia-Closas, R.A.E., G. Chenevix-Trench, P.J.B., C.A.H., J. Schleutker, D.F.E., S.B.G., P.D.P., A.L.P., B.P., C.I.A., P.K., S. Lindström. Interpret results: A.A., I.L.A., A.C.A., N.N.A., S.A., B.K.A., R.B.B., J. Batra, A.B., S.I.B., S.A.B., C.B., S.E.B., M.K.B., J. Benitez, R.B., G. Cadoni, T.C., P.T.C., G. Cancel-Tassin, D.C., A.T.C., J. Chang-Claude, D.C.C., J.M. Collee, F.J.C., A.C., J.M. Cunningham, C. Chen, M.B.D., P.D., O.D., J.L.D., T.D., E.D., C.K.E., D.V.E., D.G.E., P.A.F., R.T.F., F.F., S.F., E.F., J. Garber, S.A.G., G.G.G., D.E.G., M.T.G., G.G., K.G., P.G., U.H., J. Huyghe, F.H., R. Herrero, P. Hall, M.H., D.G.H., G.I., E.N.I., C.I., P.J., M.A. Jakubowska, A. Joshi, L.E.K., T.K., E.K., A.S.K., L.A.K., J. Kim, M.K., V.N.K., P.L., N.D.L., F. Loupakis, H.L., G. Liu, D. Lambrechts, D.A.L., C.I.L., A.L., N.M.L., G. Leslie, J. Lester, L. Maehle, C.M., L.L.M., S.M., L. McGuffog, A. Mannermaa, P.M., F.M., M.M., V.M., K.B.M., L. Mucci, K. Muir, F.C.N., B.G.N., R.L.N., K.O., O.I.O., H.O., A.O., H.P., J.Y.P., M.T.P., T.P., C.M.P., A.I.P., D. Plaseska-Karanfilska, M.P., K. Stefansson, S.J.R., L.R., H.S.R., M.J. Riggan, M.A.R., K.D.R., E.S., E.J.S., M.B.S., M.K.S., V.W.S., E.M.S., M.L.S., K.D.S., M.C.S., A.B.S., V.L.S., S.S., J. Stone, K. Sundfeldt, A.T., J.A.T., M.R.T., M.B.T., K.L.T., L.B., A.E.T., P.A.T., R.C.T., N.T., C.V., A.V., Q.W., S.W., J.N.W., E.W., A.S.W., F.W., R.W., X.W., D.Y., W.Z., A.Z., J.M.L. Oversee consortium dataset: F.R.S., M.K.B., J.M.L., R. Saxena, R.J. Hung, U.P., R.A.E., G. Chenevix-Trench, D.F.E., S.B.G., C.I.A., P.K. Develop and review the analysis plan and statistical analysis: X.J., H.K.F., A.L.P., B.P., C.I.A., P.K., S. Lindström. Design and manage individual study: D.A., M.C.A., I.L.A., H.A.-C., N.N.A., K.J.A., E.V.B., D.D.B., J. Brenton, M.W.B., S. Benlloch, H. Bickeboller, S. Boccia, N.V.B., H. Brauch, H. Brenner, J. Brunet, M.B., H. Brunnstrom, D.R.B., B.B., M.A.C., I.C., L.C., N.C., A.L.C., J. Clements, S.J.C., C. Cybulski, K.B.C., F.C., J. Chang-Claude, M.C.C., K.C., A.d., M. Gago-Dominguez, J.L.D., A.M.D., M.D., D.M.E., C.E., R.L.F., T.L., J.C.F., O.F., S.J.G., P.A.G., J.A.G., G.G.G., A.K.G., M.S.G., E.L.G., M.T.G., K.G., M.H.G., H.G., J. Gronwald, N.H., P. Hillemanns, F.C.H., R.J. Hamilton, J. Hampe, A.H., E.H., Y. Hong, J.L.H., R. Houlston, P.J.H., D.J.H., E.N.I., S.A.I., A.J., P.J., Mattias. Johansson, Mikael. Johansson, E.M.J., R.K., B.Y.K., K.K., S.K.K., J.A.K., Z.K., S.K., J. Kupryjanczyk, M.L., S. Lam, D. Lessel, M.T.L., E.L., J. Lubinski, L.L., F. Lejbkowicz, J. Lubinski, A. Meindl, T.M., P.M., A. Miller, R.L.M., R.J.M., M.M., A.M.M., K.L.N., D.E.N., A.R.N., S.L.N., H.N., P.A.N., L.F.N., L.N., K.O., E.O., A.A.A.O., O.I.O., A.F.O., H.P., J.Y.P., N.P., K.L.P., W.H.P., C.M.P., D. Prokofieva, P.R., G.R., E.J.v.R., H.A.R., A.R., M.J. Roobol, B.R., K.D.R., D.P.S., J. Simard, V.W.S., H.S., E.M.S., W.S., C.F.S., J.L.S., V.L.S., R. Sutphen, A.J.S., E.H.T., C.M.T., M.D.T., S.N.T., M. Thomassen, M. Tischkowitz, D. Torres, S.S.T., C.M.U., N.U., E.V.N., M.E.A., P.M.W., C.R.W., S.W., M.C.W., C.W., H.W., F.W., A.W., P.W., M.W., A.H.W., X.W., S.Z., K.K.Z., R. Saxena.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, X., Finucane, H.K., Schumacher, F.R. et al. Shared heritability and functional enrichment across six solid cancers. Nat Commun 10, 431 (2019). https://doi.org/10.1038/s41467-018-08054-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-08054-4
This article is cited by
-
A distinct class of pan-cancer susceptibility genes revealed by an alternative polyadenylation transcriptome-wide association study
Nature Communications (2024)
-
Antihypertensive drug targets and breast cancer risk: a two-sample Mendelian randomization study
European Journal of Epidemiology (2024)
-
Context-aware single-cell multiomics approach identifies cell-type-specific lung cancer susceptibility genes
Nature Communications (2024)
-
Genome-wide association analyses of breast cancer in women of African ancestry identify new susceptibility loci and improve risk prediction
Nature Genetics (2024)
-
Investigating the tissue specificity and prognostic impact of cis-regulatory cancer risk variants
Human Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
