
Abstract
The desymmetrization of meso-compounds is a useful synthetic method, as illustrated by numerous applications of this strategy in natural product synthesis. Cu-catalyzed allylic desymmetrizations enable the enantioselective formation of carbon-carbon bonds, but these transformations are limited in substrate scope and by the use of highly reactive premade organometallic reagents at cryogenic temperatures. Here we show that diverse meso-bisphosphates in combination with alkylzirconium nucleophiles undergo highly regio-, diastereo- and enantio-selective Cu-catalyzed desymmetrization reactions. In addition, C2-symmetric chiral bisphosphates undergo stereospecific reactions and a racemic substrate undergoes a Cu-catalyzed kinetic resolution. The reaction tolerates functional groups incompatible with many common organometallic reagents and provides access to a broad range of functionalized carbo- and hetero-cyclic structures. The products bear up to three contiguous stereogenic centers, including quaternary centers and spirocyclic ring systems. We anticipate that the method will be a useful complement to existing catalytic enantioselective reactions.
Similar content being viewed by others
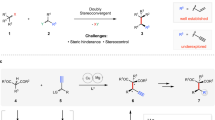

Introduction
The catalytic desymmetrization of meso-compounds is a powerful method of preparing chiral molecules. This strategy allows stereogenic features already present in symmetrical molecules to be unmasked, giving chiral molecules with multiple stereogenic centers in a single step1,2,3. These desymmetrizations have been used in the synthesis of enantiomerically enriched cyclic molecules, with metal-catalyzed asymmetric allylic addition (AAA) a proven strategy in C–C and C–X bond forming desymmetrizations4,5,6,7. Several natural product syntheses rely on a key desymmetrizing Pd-catalyzed AAA stage8,9,10,11,12. However, these processes are normally limited by the necessity to use stabilized nucleophiles (with pKa’s of less than 15), such as 1,3-dicarbonyl compounds, restricting the range of molecules that can be made using this strategy.
Cu-catalyzed AAAs enable use of nonstabilized carbon nucleophiles13,14,15,16,17,18,19, but Cu-catalyzed C–C bond forming allylic desymmetrizations are rare and have only been reported using simple unfunctionalized nucleophiles (Fig. 1a). The Cu-catalyzed desymmetrization of meso-cyclic bisphosphates using dialkylzinc reagents by Gennari and co-workers20,21 proceeds with high enantioselectivity, but requires cryogenic temperatures and is limited to the addition of just Me, Et, and iPr nucleophiles. Desymmetrizing AAA of cyclic bisphosphates using an allyl-nucleophile has been reported for two examples in 66 and 77% ee22. Recently, Feringa and co-workers reported highly enantioselective addition of alkyl lithium reagents to meso-dibromoalkenes, but this is again limited to additions of simple hydrocarbons23.

Copper-catalyzed desymmetrizations. a Previous Cu-catalyzed desymmetrizations of meso-cyclic substrates using unfunctionalized nucleophiles. b This work, using Cu-AAA and hydrozirconated alkenes to desymmetrize a wide array of meso-cyclic bisphosphates
Thus far Cu-catalyzed C–C bond forming allylic desymmetrizations have only been applied to the simplest of meso-substrates to give carbocyclic products with two tertiary stereocenters. We envisaged that the scope of these transformations could be tremendously expanded to access diverse structures by exploiting the meso-symmetry of carbo- and heterocyclic diol derivatives and introduce additional stereocenters during desymmetrization. In order to achieve this, milder AAA conditions would be required, moving away from highly reactive organometallic reagents seen in previous methods. By using zirconocene nucleophiles we envisaged that much milder reaction conditions and greater compatibility with useful functional groups could be achieved24,25,26. In principle this would represent a simple, powerful and broadly useful strategy for the synthesis of highly complex molecules bearing functional groups and multiple stereogenic centers, including stereogenic features which are difficult to make, such as quaternary centers27,28,29,30,31,32,33.
Here, we demonstrate highly enantioselective Cu-catalyzed desymmetrization of diverse meso-cyclic bisphosphates using a wide range of in situ generated hydrozirconated alkene nucleophiles. We show the reaction can be applied to desymmetrize meso-cyclic bisphosphates bearing highly desirable quaternary centers and spirocycles to give products with up to three contiguous stereocenters. Pharmaceutically relevant heterocycles can also be accessed in an enantioselective fashion (Fig. 1b).
Results
Desymmetrization development
Initial experiments applied conditions from AAA’s with racemic allylic chlorides34,35,36 to the meso-cyclic bisdiethylphosphate 1a; hydrozirconated 4-phenyl-1-butene in the presence of CuI and phosphoramidite ligand (S,S,S)-A37 (Table 1, entry 1). Good conversion to desymmetrized product 1b was observed (71%), but as a racemate. The relative stereochemistry of product 1b suggests the nucleophile adds via an SN2’ pathway, giving almost exclusively the trans-isomer (no SN2 product observed). The reaction proved sensitive to the copper source and in situ generated Lewis-acidic Cu(I) salts such as CuNTf2 gave up to 76% conversion and 69% ee using ligand A (entry 4). Attention then moved towards the effect of ligands.
Phosphoramidite ligands bearing a BINOL scaffold and indane moiety on nitrogen performed well in the reaction. Qualitatively, as the size of the ligand’s amido alkyl group increased so did the ee (entries 5–9). The best performing ligand was F38,39, giving 70% isolated yield, >20:1 trans:cis ratio and 86% ee (entry 9). A solvent screen revealed that this variable had little effect on the reaction, hence CH2Cl2 was retained. The dialkylphosphate group was also shown to have minimal impact on results (see Supplementary Figure 1). Lowering the temperature of the reaction increased the ee; up to 90% at 0 °C (entry 10). Lowering the temperature further (entry 11) resulted in diminished conversion, hence 0 °C was used to balance yield, ee and ease of reaction setup.
In order to show the reaction was general across different ring sizes, 6- and 7-membered meso-bisphosphates (2a and 3a, respectively) were desymmetrized with 4-phenyl-1-butene. Both systems gave >90% ee (Fig. 2). A lower yield was observed for 6-membered 2b (55% yield), hence the reaction was done at room temperature (70% yield). A range of alkene pro-nucleophiles were investigated in the desymmetrizations of 1a–3a. Simple linear and branched alkenes performed well, consistently providing good yields and high enantioselectivity, with larger substituents such as tBu reducing the yield. (c–h). Functionalized nucleophiles were investigated; a silyl protected alcohol and alkyl chloride was well tolerated (i, j). Styrenes also performed well in the reaction (k, l), with aryl bromides 1l and 2l allowing further manipulation if desired. A reaction using 1a and ethylene to give 633 mg of 1c on 4 mmol scale (1.48 g 1a, 67%, 90% ee) illustrates the reaction is scalable.

Initial reaction scope. Examination of alkene pro-nucleophiles and unsubstituted bisphosphates of different ring sizes
We next explored the effect of further substitution on the bisphosphate electrophile, as this may provide products with additional stereogenic centers. Substrate 4a with a methyl group anti to the phosphates and a benzyl group syn was prepared as a single diastereomer and subjected to the desymmetrization conditions with ligand F (Fig. 3a). The SN2’ addition product was obtained as the trans diastereomer (4b) in 56% yield and 93% ee. Here, the use of ligand E maintained the ee and improved the yield to 70%. Additionally, room temperature reactions gave high enantioselectivity in forming the quaternary center containing product. 4a was then desymmetrized with nucleophiles bearing functional groups very likely incompatible with asymmetric addition procedures of alkyl zinc and lithium reagents (Fig. 3b). A benzyl protected alcohol (4c) and an alkyl bromide (4d) were well tolerated. In addition, an (arbitrarily) disubstituted styrene bearing aryl fluoride and bromide moieties (4e) was tolerated, as was the use of a basic nitrogen atom (4f), a group normally problematic in Cu-catalysis (63% yield, 86% ee).

Further exploration of desymmetrization reaction. a Ligand optimization for electrophiles bearing a pro-quaternary center. b Substrate scope for electrophiles bearing a pro-quaternary center. c Desymmetrization of heterocyclic bisphosphates
A range of substituted bisphosphates bearing pro-quaternary centers (5a–10a), were prepared and examined (Fig. 3b). Useful functional groups such as allyl, and an aryl chloride were all well tolerated on the bisphosphate. These products are obtained as a single diastereoisomers in good yields with >90% ee (5b–7b). A substrate featuring a benzyl protected alcohol is similarly effective (61%, 90% ee, 8b).
Spirocyclic systems, which can be challenging to prepare with either diastereo- or enantio-control40,41,42,43, performed well in the reaction effectively giving a single isomer product when both indane (9b) and chromane (10b) based ring systems were examined.
As well as carbocyclic structures, we envisaged that medicinally relevant 7-membered heterocyclic44,45 meso-bisphosphates could be accessed, allowing synthesis of substituted oxepenes and azepenes. Direct catalytic asymmetric manipulation of these ring systems to form new C–C bonds appears to be unexplored. In this case, it was not obvious that these reactions would be successful as tolerance to heteroatoms is often problematic in asymmetric Cu-catalyzed reactions46. However, we found that oxepene 11a and Boc protected azepene 12a performed very well under our desymmetrization conditions, and hydrometallation-addition of 4-phenyl-1-butene gave 11b and 12b in over 70% yield with >92% ee (Fig. 3c).
Further stereocontrolled additions to bisphosphates were explored by examining C2-symmetric trans-cyclopentene epimers; enantiopure (S,S)-13 and rac-trans-13. Cu-catalyzed kinetic resolutions of racemic starting material are also rare, and to the best of our knowledge there have been no reports of Cu-catalyzed asymmetric addition to cyclic C2-symmetric trans-diol derivatives. Both trans-bisphosphates were synthesized from the trans-diol, which can be prepared as a single enantiomer by enzymatic desymmetrization47. To our delight both substrates underwent stereospecific allylic alkylation (SAA). (Fig. 4). It was hypothesized that these SAA products would have cis-stereochemistry, rather than the trans exclusively obtained in the above meso-desymmetrization if the Cu-catalyst was mediating anti-SN2’ type processes.

Cu-SAA/KR using trans-cyclic phosphates. Reaction conditions: Cu-SAA and Cu-KR see Table 1, entry 9; a LiAlH4 (2.2 eq), Et2O, rt; b HCO2H (2.05 eq), DIAD (2.0 eq), PPh3 (2.0 eq), Toluene 0 °C; c NEt3 (0.05 eq), MeOH, rt; d (EtO)2P(O)Cl (2.0 eq), NEt (2.0 eq), DMAP (0.33 eq), CH2Cl2, rt. Selectivity factor S calculated using S = ln[1 − c(1 + ee)]/ln[1 − c(1 − ee)]
Enantiopure (S,S)-13 stereospecifically reacts with hydrozirconated 4-phenyl-1-butene under desymmetrization conditions to give 14 in 65% yield, as a single isomer. Both the (S,S) and (R,R) forms of ligand F give the same product 14 in >20:1 cis:trans ratio with >99% ee. Inversion of the phosphate stereocenter of 14 in a multistep process gave (1R, 2R) 1b, confirming the absolute configuration of 1b produced in the enantioselective meso-desymmetrization. All other absolute configurations were assigned by analogy to 1b.
Use of rac-13 led to a rare example of a Cu-catalyzed asymmetric kinetic resolution of racemic starting materials. Under desymmetrization conditions, if conversion is stopped at 43%, then 14 is obtained with 40% yield and 80% ee, and enantioenriched 13 is recovered in 53% yield with 71% ee.
The downstream reactivity of the reaction products were briefly investigated to demonstrate that they could be further elaborated. (Fig. 5). Reduction of phosphate 1c to give alcohol 15 proceeded in excellent yield. Aryl bromide 2l was also able to undergo a Heck reaction to give cis-fused tricycle 16 in 80% yield. In addition, dihydroxylation of 4b furnished 17, a diol with five contiguous stereocenters, in 75% yield with 4:1 diastereoselectivity.

Further functionalization of products. a Reduction of the phosphate to give an alcohol. b Heck reaction to form tricyclic scaffold. c Alkene dihydroxylation to give a fully substituted cyclopentane
Mechanistically, we tentatively postulate that a Cu-phosphoramidite complex forms a hybrid species I with the hydrozirconated alkene, which can co-ordinate to the bisphosphate and form an alkene donor complex (II). We suggest species I as in situ NMR spectroscopic studies of reactant and catalyst mixtures revealed no explicit transmetallation from Zr to Cu. II may react to form Cu-(III) σ-allyl species III, where the copper displaces the phosphate in a stereodetermining anti-SN2’ like process16,48. Reductive elimination of intermediate III would give the product and regenerate the Cu-phosphoramidite catalyst (Fig. 6).

Postulated reaction mechanism. Catalytic cycle illustrating anti-SN2’ addition of alkylcopper species generated from alkylzirconium intermediates
In conclusion, we have developed a broadly applicable highly regio-, diastereo-, and enantio-selective desymmetrization of meso-cyclic bisphosphates using alkylzirconocene nucleophiles. A range of substituted bisphosphates can be used in the reaction, including heterocycles, and the protocol was also applied to C2-symmetric trans-cyclicbisphosphates. The products bear up to three stereocenters, including quaternary centers and spirocyclic ring systems.
Methods
General methods
For synthetic details and analytical data for all reaction products see Supplementary Methods.
General procedure for enantioselective desymmetrization
Note: All manipulations are carried out under an Ar atmosphere and in the absence of light where possible. A flame-dried 5 mL round bottomed flask was charged with CuCl (4 mg, 0.04 mmol, 0.1 eq) and phosphoramidite ligand E (24.7 mg, 0.048 mmol, 0.12 eq). Dry CH2Cl2 (1.3 mL) was added to the mixture which was left to stir at room temperature for 1 h. AgNTf2 (23 mg, 0.06 mmol, 0.15 eq) was then added to the flask and left to stir at room temperature for 15 min. A pale brown suspension was formed. Meanwhile a separate 5 mL flame-dried round bottomed flask was charged with Cp2ZrHCl (206 mg, 0.8 mmol, 2.0 eq) and suspended in dry CH2Cl2 (0.8 mL). Alkene (1.0 mmol, 2.5 eq) was added to the mixture which was left to stir at room temperature until the mixture became a homogenous yellow solution (approx. 30 min, varies with alkene). The flask containing Cu-catalyst was transferred to the alkylzirconium reagent using a syringe equipped with a Camlab PTFE syringe filter (13 mm size, pore diameter 0.22 µm). The combined flask contents formed a black mixture which was left to stir at room temperature in the absence of light for 10 min. Meso-cyclic bisphosphate (0.4 mmol) in CH2Cl2 (0.2 mL) was added to the mixture which was left to stir at room temperature in the absence of light for 16 h. The mixture was quenched with 1 M aq. NH4Cl (1 mL) and left to stir for 10 min. The organic layer was separated, filtered through a plug of Celite and evaporated in vacuo to give an off-white mixture of solid and oil. The crude product was purified on silica to give the product.
References
Manna, M. S. & Mukherjee, S. Catalytic asymmetric desymmetrization approaches to enantioenriched cyclopentanes. Org. Biomol. Chem. 13, 18–24 (2015).
Zeng, X.-P., Cao, Z.-Y., Wang, Y.-H., Zhou, F. & Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev. 116, 7330–7396 (2016).
Merad, J., Candy, M., Pons, J.-M. & Bressy, C. Catalytic enantioselective desymmetrization of meso compounds in total synthesis of natural products: towards an economy of chiral reagents. Synthesis 49, 1938–1954 (2017).
Lu, Z. & Ma, S. Metal-catalysed enantioselective allylation in asymmetric synthesis. Angew. Chem. Int. Ed. 47, 258–297 (2008).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactins of organometallic reagents to contruct C–C bonds. Chem. Rev. 115, 9587–9652 (2015).
Mernard, F., Chapman, T. M., Dockendorff, C. & Lautens, M. Rhodium-catalysed asymmetric allylic substitution with boronic acid nucleophiles. Org. Lett. 8, 4569–4572 (2006).
Mernard, F., Perez, D., Roman, D. S., Chapman, T. M. & Lautens, M. Ligand-controlled selectivity in the desymmetrization of meso cyclopenten-1,4-diols via rhodium(I)-catalyzed addition of arylboronic acids. J. Org. Chem. 75, 4056–4068 (2010).
Trost, B. M. Desymmetrization of prochiral diesters via transition metal catalyzed reactions. Isr. J. Chem. 37, 109–118 (1997).
Trost, B. M. Pd asymmetric allylic alkylation (AAA). A powerful synthetic tool. Chem. Pharm. Bull. 50, 1–14 (2002).
Trost, B. M. & Crawley, M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 103, 2921–2943 (2003).
Graening, T. & Schmalz, H.-G. Pd-catalyzed enantioselective allylic substitution: new strategic options for the total synthesis of natural products. Angew. Chem. Int. Ed. 42, 2580–2584 (2003).
Trost, B. M. Asymmetric allylic alkylation, an enabling methodology. J. Org. Chem. 69, 5813–5837 (2004).
Woodward, S. Decoding the ‘black box’ reactivity that is organocuprate conjugate addition chemistry. Chem. Soc. Rev. 29, 393–401 (2000).
Alexakis, A., Bäckvall, J. E., Krause, N., Pàmies, O. & Diéguez, M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 108, 2796–2823 (2008).
Harutyunyan, S. R., den Hartog, T., Geurts, K., Minnaard, A. J. & Feringa, B. L. Catalytic asymmetric conjugate addition and allylic alkylation with grignard reagents. Chem. Rev. 108, 2824–2852 (2008).
Falciola, C. A. & Alexakis, A. Copper-catalyzed asymmetric allylic alkylation. Eur. J. Org. Chem. 2008, 3765–3780 (2008).
Yoshikai, N. & Nakamura, E. Mechanisms of nucleophillic organocopper(I) reactions. Chem. Rev. 112, 2339–2372 (2012).
Alexakis, A., Krause, N. & Woodward, S. Copper-Catalyzed Asymmetric Synthesis (Wiley-VCH, Weinheim, Germany, 2014).
Harutyunyan, S. Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers (Springer, Switzerland, 2016).
Piarulli, U., Daubos, P., Claverie, C., Roux, M. & Gennari, C. A catalytic and enantioselective desymmetrization of meso cyclic allylic bisdiethylphosphates with organozinc reagents. Angew. Chem. Int. Ed. 42, 234–236 (2003).
Piarulli, U. et al. Copper phosphoramidite-catalyzed enantioselective desymmetrization of meso-cyclic allylic bisdiethyl phosphates. Org. Lett. 5, 4493–4496 (2003).
Yasuda, Y., Ohmiya, H. & Sawamura, M. Copper-catalyzed enantioselective allyl–allyl coupling between allylic boronates and phosphates with a phenol/N-heterocyclic carbene chiral ligand. Angew. Chem. Int. Ed. 55, 10816–10820 (2016).
Goh, S. S. et al. Desymmetrization of meso-dibromocycloalkenes through copper(I)-catalyzed asymmetric allylic substitution with organolithium reagents. J. Am. Chem. Soc. 140, 7052–7055 (2018).
Maksymowicz, R. M., Roth, P. M. C. & Fletcher, S. P. Catalytic asymmetric carbon-carbon bond formation using alkenes as alkylmetal equivalents. Nat. Chem. 4, 649–654 (2012).
Sidera, M., Roth, P. M. C., Maksymowicz, R. M. & Fletcher, S. P. Formation of quaternary centers by copper-catalyzed asymmetric conjugate addition of alkylzirconium reagents. Angew. Chem. Int. Ed. 52, 7995–7999 (2013).
Roth, P. M. C., Sidera, M., Maksymowicz, R. M. & Fletcher, S. P. Copper-catalyzed asymmetric conjugate addition of alkylzirconium reagents to cyclic enones to form quaternary centers. Nat. Protoc. 9, 104–111 (2014).
Quasdorf, K. W. & Overman, L. E. Catalytic enantioselective synthesis of quaternary carbon stereocenters. Nature 516, 181–191 (2014).
Hawner, C. & Alexakis, A. Metal-catalyzed asymmetric conjugate addition reaction: formation of quaternary stereocenters. Chem. Commun. 46, 7295–7306 (2010).
Das, J. P. & Marek, I. Enantioselective synthesis of all-carbon quaternary stereogenic centers in acyclic systems. Chem. Commun. 47, 4593–4623 (2011).
Marek, I. et al. All-carbon quaternary stereogenic centers in acyclic systems through the creation of several C-C bonds per chemical step. J. Am. Chem. Soc. 136, 2682–2694 (2014).
Büschleb, M. et al. Synthetic strategies toward natural products containing contiguous stereogenic quaternary carbon atoms. Angew. Chem. Int. Ed. 55, 4156–4186 (2016).
Maciá, B. Formation of quaternary stereocenters by copper-catalysed enantioselective conjugate addition reaction. Top. Organomet. Chem. 58, 41–98 (2016).
Prusov, E. V. Construction of quaternary stereogenic centers in the total synthesis of natural products. Angew. Chem. Int. Ed. 56, 14356–14358 (2017).
You, H., Rideau, E., Sidera, M. & Fletcher, S. P. Non-stabilized nucleophiles in Cu-catalysed dynamic kinetic asymmetric allylic alkylation. Nature 517, 351–355 (2015).
Rideau, E., You, H., Sidera, M., Claridge, T. D. W. & Fletcher, S. P. Mechanistic studies on a Cu-catalyzed asymmetric allylic alkylation with cyclic racemic starting materials. J. Am. Chem. Soc. 139, 5614–5624 (2017).
Schafer, P., Sidera, M., Palacin, T. & Fletcher, S. P. Asymmetric cross-coupling of alkyl, alkenyl and (hetereo)aryl nucleophiles with racemic allyl halides. Chem. Commun. 53, 12499–12511 (2017).
Teichert, J. F. & Feringa, B. L. Phosphoramidites: privileged ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 49, 2486–2528 (2010).
Gao, Z. & Fletcher, S. P. Acyclic quaternary centers from asymmetric conjugate addition of alkylzirconium reagents to linear trisubstituted enones. Chem. Sci. 8, 641–646 (2017).
Gao, Z. & Fletcher, S. P. Construction of β to carbonyl stereogenic centres by asymmetric 1,4-addition of alkylzirconocenes to dienones and ynenones. Chem. Commun. 54, 3601–3604 (2018).
Sannigrahi, M. Stereocontrolled synthesis of spirocyclics. Tetrahedron 55, 9007–9071 (1999).
Kotha, S., Deb, A. C., Lahiri, K. & Manivannan, E. Selected synthetic strategies to spirocyclics. Synthesis 41, 165–193 (2009).
Franz, A. K., Hanhan, N. V. & Ball-Jones, N. R. Asymmetric catalysis for the synthesis of spriocyclic compounds. ACS Catal. 3, 540–553 (2013).
Ling, T. & Rivas, F. All-carbon quaternary stereocenters in natural products and medicinal chemistry: recent advances. Tetrahedron 72, 6729–6777 (2016).
Reekie, T. A., Kavanagh, M. E., Longworth, M. & Kassiou, M. Synthesis of biologically active seven-membered-ring heterocycles. Synthesis 45, 3211–3227 (2013).
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Holec, C., Sandkuhl, D., Rother, D., Kroutil, W. & Pietruszka, J. Chemoenzymatic synthesis towards the active agent travoprost. ChemCatChem 7, 3125–3130 (2015).
Persson, E. S. M., van Klaveren, M., Grove, D. M., Bäckvall, J. E. & van Koten, G. ortho-Chelating arenethiolatocopper(I) complexes as versatile catalysts in the regioselective cross-coupling of allylic derivatives with nBuMgI—an example of reversed reactivity of leaving groups. Chem. Eur. J. 1, 351–359 (1995).
Acknowledgments
We are grateful to the EPSRC for funding (EP/N022246/1) and to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship to R.J., generously supported by AstraZeneca, Diamond Light Source, Defense Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. We thank Dr. H. You for preliminary experimental work.
Author information
Authors and Affiliations
Contributions
R.J. performed experiments. R.J., R.D.C.P., and S.P.F. designed experiments and analyzed experimental results. R.J. and S.P.F. wrote and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jacques, R., Pullin, R.D.C. & Fletcher, S.P. Desymmetrization of meso-bisphosphates using copper catalysis and alkylzirconocene nucleophiles. Nat Commun 10, 21 (2019). https://doi.org/10.1038/s41467-018-07871-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07871-x
This article is cited by
-
Exosomes derived from hucMSC attenuate renal fibrosis through CK1δ/β-TRCP-mediated YAP degradation
Cell Death & Disease (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
