
Abstract
Gallstones are responsible for one of the most common diseases in the Western world and are commonly treated with cholecystectomy. We perform a meta-analysis of two genome-wide association studies of gallstone disease in Iceland and the UK, totaling 27,174 cases and 736,838 controls, uncovering 21 novel gallstone-associated variants at 20 loci. Two distinct low frequency missense variants in SLC10A2, encoding the apical sodium-dependent bile acid transporter (ASBT), associate with an increased risk of gallstone disease (Pro290Ser: OR = 1.36 [1.25–1.49], P = 2.1 × 10–12, MAF = 1%; Val98Ile: OR = 1.15 [1.10–1.20], P = 1.8 × 10–10, MAF = 4%). We demonstrate that lower bile acid transport by ASBT is accompanied by greater risk of gallstone disease and highlight the role of the intestinal compartment of the enterohepatic circulation of bile acids in gallstone disease susceptibility. Additionally, two low frequency missense variants in SERPINA1 and HNF4A and 17 common variants represent novel associations with gallstone disease.
Similar content being viewed by others

Introduction
Gallstone disease (also known as cholelithiasis) is a common condition affecting 10–15% of the population in developed countries1, and is often complicated by cholecystitis, pancreatitis and secondary infections2. Gallstones are masses of solid material formed in the gallbladder, most commonly consisting of cholesterol2. Excess of cholesterol or a lack of bile acids increase the likelihood of gallstone formation3. Treatment by surgical removal of the gallbladder (cholecystectomy) is one of the most common surgeries in the Western world, with over 700,000 cholecystectomies performed annually in the United States1.
A twin study estimated that genetic factors account for 25% [95% CI: 9–40%] of the phenotypic variation of gallstone disease4. Sequence variants at eight loci have been reported through genome-wide association studies (GWAS) to affect the risk of gallstone disease, including missense variants in ABCG5/ABCG85, encoding ATP-binding cassette (ABC) transporters involved in the secretion of cholesterol into bile6. We have reported two rare coding variants in ABCB4 that greatly increase the risk of gallstone disease7. ABCB4 encodes the ABC transporter responsible for the secretion of lecithin into bile6. Other studies have reported sequence variants at GCKR8, TM4SF48, SULT2A18, CYP7A18, UGT1A69, and TTC39B10 to associate with risk of gallstone disease.
To search for sequence variants affecting the risk of gallstone disease, we perform a genome-wide association study (GWAS) of 27,174 cases and 736,838 controls from Iceland and the UK biobank. Subsequently, we assess the association of gallstone disease variants with acute pancreatitis, cholecystitis, fibrosis/cirrhosis of liver, cholestasis of pregnancy, gallbladder cancer, six liver biomarkers, and three cholesterol traits. We find four novel low-frequency sequence variants in SLC10A2 (two distinct variants), SERPINA1 and HNF4A that associate with gallstone disease. SLC10A2 encodes a protein that reabsorbs bile salts from the terminal ileum as part of their enterohepatic circulation. Additionally, we describe 17 novel common variants that associate with gallstone disease. Among those, two are in fucosyltransferase genes, four associate with maturity onset diabetes of the young, and six associate with lipid metabolism. We systematically assess the effects of lipid associated variants with gallstone disease risk and find inconsistent direction of effect. With the discovery of gallstone-associated variants in SLC10A2, we highlight a role of the intestinal compartment of the enterohepatic circulation of bile acids in gallstone disease susceptibility.
Results
Overview
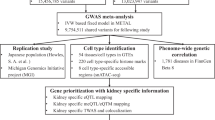
We performed a meta-analysis of gallstone disease, combining GWAS results from Iceland (8757 cases and 346,688 controls) and the UK Biobank (18,417 cases and 390,150 controls), for a total of 27,174 cases and 736,838 controls (Fig. 1, Supplementary Table 1). The GWAS from Iceland was performed using 32 million markers identified through whole-genome sequencing of 15,220 Icelanders that were subsequently imputed into 151,677 chip-typed individuals, as well as 282,894 of their first and second degree relatives7. The GWAS from the UK was performed with 40 million markers (imputation info > 0.8) from the Haplotype Reference Consortium (HRC) reference panel, imputed into chip-typed individuals of European ancestry in the UK Biobank11 (Fig. 1). Associations were considered significant if the p-value in the combined dataset was below a weighted genome-wide significance threshold based on variant annotation12. The significance thresholds used were 2.0 × 10–7 for high-impact variants (including stop-gained, frameshift, splice acceptor or donor, N = 11,465), 3.9 × 10–8 for moderate-impact variants (including missense, splice-region variants and in-frame indels, N = 197,583), 3.6 × 10–9 for low-impact variants (including upstream and downstream variants, N = 2,971,445) and 5.9 × 10–10 for lowest-impact variants (including intron and intergenic variants, N = 39,726,619). In total, 43 million markers were tested in the meta-analysis.

A flowchart describing the study design of the gallstone meta-analysis of Icelandic and UK Biobank data and results
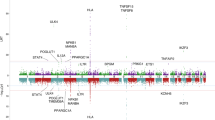
We found 32 gallstone disease association signals at 28 loci (Table 1, Supplementary Table 2, Fig. 2, and Supplementary Data 1) that reached genome-wide significance under the additive model. Of these, 21 are novel and 11 correspond to previously known variants5,8,9,13. One of the novel common variants, rs708686 upstream of FUT6, associates more significantly with gallstone disease through a recessive mode of inheritance. There was no significant heterogeneity of effects between Iceland and the UK (Table 1, Supplementary Data 1). Thirteen of the novel signals represent associations with coding variants, four of which are low frequency coding variants, with minor allele frequency (MAF) ranging from 0.6 to 4.6% (imputation info > 0.995 in both Iceland and UK). Three novel gallstone-associated non-coding variants at the LITAF and MAL2 loci and a stop gained variant at the FUT2 locus are correlated with (r2 > 0.8) or represent the strongest cis-eQTLs in their respective regions (Table 1, Supplementary Table 3).

Manhattan plot for gallstone disease meta-analysis association results (Ncases = 27,174 and Ncontrols = 736,838). Variants are plotted by chromosomal position (x-axis) and −log10 P-values (y-axis). A chi-square test was used when testing for association. Green = Novel loci represented by low-frequency variants, red = novel loci represented by common variants, blue = reported gallstone disease loci
We tested the 32 gallstone disease variants for association with five other related diseases of the biliary system (acute pancreatitis [N = 3401], cholecystitis [N = 3565], fibrosis/cirrhosis of liver [N = 1044], cholestasis of pregnancy [N = 2615], and gallbladder cancer [N = 382]), six liver biomarkers (alanine aminotransferase [ALT; N = 172,086], aspartate aminotransferase [AST; N = 164,467], gamma glutamyltransferase [GGT; N = 156,692], alkaline phosphatase [ALP; N = 154,097], albumin [N = 92,163], and bilirubin [N = 109,748]), and four lipid traits (high-density lipoprotein [HDL cholesterol; N = 136,736], low-density lipoprotein [LDL cholesterol; N = 126,220], total cholesterol [N = 150,211]) and triglycerides [N = 119,624], resulting in a total of 32 × (5 + 6 + 4) = 480 tests that had to be accounted for in the assessment of significance (Supplementary Data 2 and Supplementary Data 3). We observed five significant associations: the Pro290Ser missense variant in SLC10A2 associates with an increased risk of acute pancreatitis (see below), the previously reported8 Asp19His missense variant in ABCG8 also associates with increased risk of acute pancreatitis and cholecystitis, and the two rare coding variants we reported in ABCB4 (Leu445GlyfsTer22/rs756082276[CCT] andGly622Glu/rs756935975[T]) both associate with high risk of cholestasis of pregnancy, as previously reported7 (Supplementary Data 2). Ten gallstone disease variants associate with ALT, seven with AST, fourteen with GGT, nine with ALP, two with albumin, and two with bilirubin (Supplementary Data 3). Four variants associate with HDL cholesterol, six with LDL cholesterol, nine with total cholesterol and six with triglycerides (Supplementary Data 3).
The sibling recurrence risk of gallstone disease in the Icelandic population is 1.81 [1.71, 1.91], in line with reported estimates14 (Supplementary Data 4). Combined, the 32 gallstone disease variants account for a sibling recurrence risk of 1.13 (Supplementary Data 4).
Low-frequency coding variants in SLC10A2
We found two distinct missense variants in SLC10A2 (Pro290Ser and Val98Ile; r2 < 0.001) associating with increased risk of gallstone disease (Table 1, Supplementary Table 4). The SLC10A2 gene encodes the apical sodium-dependent bile acid transporter (ASBT), also known as the ileal sodium-dependent bile acid transporter (ISBT) that mediates the intestinal reabsorption of bile acids.
The variant in SLC10A2 with the strongest effect on gallstone disease risk is Pro290Ser (rs56398830[A]; OR = 1.36 [1.25–1.49]; P = 2.1 × 10–12, MAFIceland = 0.6%, MAFUK = 1.1%) (Table 1, Supplementary Fig. 1 and Supplementary Data 1). Others have experimentally shown that Pro290Ser nearly abolishes the bile acid transport activity of ASBT15,16 ( < 3% of the wild-type level16). Pro290Ser is in transmembrane domain seven which is highly conserved within the SLC10 family of proteins, and results in a substitution of a non-polar proline with a polar serine17 (PolyPhen-2: probably damaging, SIFT: deleterious) (Fig. 3a). We observe 62 homozygous carriers of Pro290Ser (expected number of homozygous carriers under Hardy-Weinberg (HW) equilibrium = 55); of whom eight carry a diagnosis of gallstone disease. The gallstone disease odds ratio for homozygous carriers of Pro290Ser is 3.26 [1.55–6.86], consistent with the additive model (Supplementary Fig 2a). Homozygotes are expected to have very little ASBT/SLC10A2 transport activity.

SLC10A2 transport activity and gallstone risk. a Schematic illustration of the topology of the SLC10A2 protein showing the localization of the functionally assessed missense variants Pro290Ser, Val98Ile, Val159Ile, and Ser171Ala (from PMID: 19823678). b A scatter plot showing four SLC10A2 missense variants Pro290Ser, Val98Ile, Val159Ile, and Ser171Ala. The x-axis shows the bile acid transport activity (from PMID:21649730) while the y-axis shows the odds ratio for gallstone disease in the Iceland + UK meta-analysis (Ncases = 27,174, Ncontrols = 736,838)
In a meta-analysis of acute pancreatitis in Iceland (1366 cases and 361,081 controls) and UK (1477 cases and 407,090 controls), we detected an association of Pro290Ser with increased risk of this disease (OR = 1.76 [1.38–2.24], P = 5.6 × 10–6, Supplementary Fig. 3 and Supplementary Data 2). Among the 32 gallstone-associated variants, effects on gallstone disease and acute pancreatitis were positively correlated (r = 0.81 [0.69–0.92]; P = 1.7 × 10–8, Supplementary Fig. 3). Since ASBT is a bile acid transporter that mainly transports taurocholate, we tested the effect of Pro290Ser in serum levels of taurocholate (N = 273 including four carriers of Pro290Ser). As expected, Pro290Ser associates with reduced levels of serum taurocholate (Effect: −1.6 SD [−2.56, −0.64]; P = 0.0010; Supplementary Table 5). We also tested for an effect of Pro290Ser on fibroblast growth factor 19 (FGF19, N = 293), a hormone that regulates bile acid synthesis18, and observed no effect (Effect: −0.08 SD [−1.02, 0.87]; P = 0.87; Supplementary Table 5). Despite a large sample size, we observed no association with other liver biomarkers in the Icelandic data (Supplementary Data 3).
A more common SLC10A2 missense variant, Val98Ile (rs55971546[T]), also associates with increased risk of gallstone disease (OR = 1.15 [1.10–1.20], P = 1.8 × 10–10, MAF = 4.1% in both Iceland and the UK) (Table 1, Supplementary Table 4 and Supplementary Data 1). Val98Ile is located in transmembrane domain two and has been shown experimentally by others to reduce the transport activity by 42%16.
The abovementioned study16 reported transport activity for Pro290Ser and Val98Ile, as well as two other ASBT/SLC10A2 variants, Val159Ile (rs60380208[T]) and Ser171Ala (rs188096[A]). For each of these missense variants, we compared the previously reported transport activity measurements to their effect on gallstone disease risk from the current study (Fig. 3b). Lower transport activity is correlated with greater risk of gallstone disease (r = −0.99 [−1.00, −0.66], p = 8.2 × 10–3 for OR vs. activity; r = −0.99 [−1.00, −0.77], p = 5.2 × 10–3 for log(OR) vs. activity). Additionally, we show transport activity compared to the genotypic risk of gallstone disease for different genotypes of Pro290Ser, Val98Ile and wild-type (Supplementary Fig. 4). Together these data indicate that the variants in SLC10A2 affect gallstone risk through impairment of the bile acid reabsorption function of ASBT.
We found 17 other missense variants in SLC10A2 (Supplementary Table 6). Of these, one additional variant, Pro65Leu (MAFIceland = 0.26%; MAFUK = 0.14%), associates with gallstone disease given the number of tested markers (significance threshold: P < 0.05/17 = 0.0029) (OR = 1.40 [1.15–1.72], P = 0.00096).
Finally, it is worth noting that a truncating variant in SLC10A2 (not found in our data) has been claimed to cause hypertriglyceridemia19. In our data the SLC10A2 variants Pro290Ser and Val98Ile (which were shown experimentally16 to have abolished and reduced function, respectively) do not associate (P > 0.4 for both variants) with hypertriglyceridemia, defined as having at least two serum triglyceride measurements higher than 5 mmol/L (N = 1,599; Supplementary Data 5). Thus, our data do not support the claim that loss-of-function variants in SLC10A2 cause hypertriglyceridemia.
Low-frequency coding variant in SERPINA1
A low-frequency missense variant Glu366Lys (rs28929474[T]) in SERPINA1, encoding the protease inhibitor alpha-1-antitrypsin (AAT), associates with increased risk of gallstone disease (OR = 1.33 [1.25–1.42], P = 1.8 × 10–17, MAFIceland = 0.8%, MAFUK = 1.9%) (Table 1, Supplementary Fig. 6 and Supplementary Data 1). Gly366Lys is also known as the Protease Inhibitor (PI) Z allele and causes severe alpha-1 antitrypsin deficiency (AATD) in homozygotes20. The most common clinical manifestations of AATD are emphysema and chronic obstructive pulmonary disease (COPD), and a subset of AATD cases develop liver cirrhosis and fibrosis21. Consistent with this, in a meta-analysis of Icelandic and UK Biobank emphysema and COPD data, the PI Z allele greatly increases the risk of emphysema (OR = 27.7 [17.5–43.7], P = 6.8 × 10–46) and COPD (OR = 4.84 [2.97–7.90], P = 2.8 × 10–10) in homozygotes. As previously reported22,23, heterozygous carriers of PI Z also are at increased risk of emphysema (OR = 1.44 [1.21–1.70], P = 3.0 × 10–5, Table 2).
We tested the association of PI Z with liver biomarkers in the Icelandic data (Supplementary Table 7 and Supplementary Data 3). PI Z associates with decreased alpha-1-antitrypsin and alpha-fetoprotein levels; and increased levels of alkaline phosphatase, alanine transaminase, gamma glutamyl transpeptidase and increased platelet count, consistent with previous reports20,24.
We observe four homozygous carriers (PI ZZ) in Iceland and 156 in the UK, compared to 10 and 145 expected under HW equilibrium, respectively. None of the Icelanders and nine of the UK individuals have a gallstone disease diagnosis, and all but one have undergone cholecystectomy indicating symptomatic disease (Supplementary Table 8). To investigate to what extent the observed associations are driven by PI Z allele homozygotes, we calculated the genotypic odds ratios for gallstone disease, emphysema and COPD, based on genotypes of the classical M, S, and Z alleles (Table 2, Supplementary Fig. 2b, 7 and 8). In line with the literature, the genotypic effect was consistent with a recessive trend and a compound heterozygote effect of MZ for emphysema and COPD (OMIM: 613490). However, the genotypic effect on gallstone disease was consistent with an additive model.
In addition to PI Z, the PI S allele in SERPINA1 (Glu288Val), which is one of the most common AATD deficiency alleles, associates with an increased risk of gallstone disease under the additive model (OR = 1.08 [1.03–1.13], P = 4.8 × 10–4).
Low-frequency coding variant in HNF4A
A low-frequency missense variant, Thr139Ile (rs1800961[T]), in HNF4A associates with increased risk of gallstone disease (OR = 1.29 [1.23–1.35], P = 5.7 × 10–26). Thr139Ile has a MAF of 4.6% in Iceland and 3.0% in the UK (Table 1, Supplementary Data 1 and Supplementary Fig. 9). HNF4A encodes the hepatocyte nuclear factor 4α, a master regulator of liver-specific gene expression, especially of genes involved in lipid transport and glucose and bile metabolism25. Thr139Ile has been associated with a reduction in high density lipoprotein (HDL) cholesterol and total cholesterol levels26. In addition, we observe an association of Thr139Ile with increased hemoglobin, bilirubin and gamma glutamyl transpeptidase levels (Supplementary Table 9). In-vitro reporter assays indicate that Thr139Ile decreases the ability of HNF4A to activate transcription27,28.
Common variant associations
Seventeen novel common sequence variants (MAF > 5%) associate with gallstone disease (Supplementary Data 1). One of these, rs708686[T] upstream of FUT6, fits better a recessive rather than an additive model (MAFIceland = 23%, homozygous frequency = 5.3%), and associates with increased risk of gallstone disease (ORrecessive = 1.26 [1.20–1.32], Precessive = 7.4 × 10–20; ORadditive = 1.08 [1.06–1.10], Padditive = 2.3 × 10–12). There is no difference between the risk to heterozygous carriers of rs708686[T] and non-carriers (Supplementary Fig. 10). We have previously reported rs708686[T] to associate with increased levels of vitamin B1229 (Supplementary Data 6). Similarly with what we see for gallstone disease, we observed a trend towards recessive association of rs708686 with vitamin B12 levels (Supplementary Fig. 11).
Ten of the 17 common signals represent coding variants: one stop-gained (FUT2) and nine missense (Table 1 and Supplementary Data 1). We observe an association of the variant rs601338[G] in FUT2, corresponding to the primary Lewis and ABO(H) histo-blood group antigen secretor allele, with reduced risk of gallstone disease (OR = 0.91 [0.90–0.93], P = 9.4 × 10–22). The wild-type secretor allele rs601338[G] has been implicated in susceptibility to gastrointestinal infections30,31, decreased vitamin B12 levels32 and with effects on several diseases and other traits33 (Supplementary Data 6). Consistent with previous observations, rs601338[G] associates with increased risk of gastrointestinal infections (ICD-10 code A08*) in our data (OR = 1.56 [1.34–1.82], P = 7.2 × 10–9) (Supplementary Table 10). In Iceland we also observe an effect of rs601338[G] on decreased gamma glutamyl transferase and increased folate (Supplementary Table 10). FUT2 is a paralog of the previously mentioned FUT6 and they both belong to the gene family of fucosyltransferases (63% identical on the protein level).
Of note, Ala311Val (rs17240268[A]) in ANPEP (aka CD13) associates with decreased risk of gallstone disease (OR = 0.90 [0.87–0.93], P = 6.0 × 10–11). This gene encodes Aminopeptidase N, which has been reported to promote cholesterol crystallization in in-vitro systems34 and through that may affect cholesterol gallstone formation35. The minor allele rs17240268[A] would be expected to have a reducing effect on protein function for the observed protective effect on gallstone disease to be consistent with the reported promoting effect of ANPEP on cholesterol crystallization.
In addition to the hepatocyte nuclear factor HNF4A (see above), we observe signals in two other hepatocyte nuclear factor genes. The missense variant Ile27Leu (rs1169288[C]) in HNF1A associates with decreased risk of gallstone disease (OR = 0.92 [0.91–0.94], P = 6.0 × 10–11). HNF1A encodes the hepatocyte nuclear factor 1α and is involved in the regulation of the expression of several liver-specific genes36 and bile acid transporters, including ASBT, in the enterocytes of the terminal ileum37. Ile27Leu has been associated with higher LDL cholesterol levels38,39 and increased risk of coronary artery disease40 (Supplementary Data 6). Diabetes, hypercholesterolemia and increased bile acid and cholesterol synthesis were observed in mice deficient for HNF1A37. An intron variant rs17138478[A] in HNF1B associates with increased risk of gallstone disease (OR = 1.09 [1.06–1.12], P = 5.1 × 10–10). The variant falls within a liver specific enhancer element controlling the expression of itself and has HNF4A among its DNA binding proteins (Supplementary Data 7).
Lipid variants and gallstone disease
The biological functions of the 32 gallstone associated variants were assessed by gene enrichment analysis using ToppGene41. Variants were assigned to genes if they were located inside the gene or located within 5 kbs upstream and downstream from a gene, on the basis of mappings from previous GWAS studies or based on evidence from eQTL data (GTEx). In total 31 variants could be assigned to 27 genes that were included in the subsequent analysis (see Supplementary Data 8 for inclusion criteria). The analysis revealed enrichment of genes at loci reported to associate with lipid levels (PMID:24097068, P = 2.5 × 10–18, 11 out of 212 genes), in addition to genes linked to bile secretion (KEGG:193146, P = 7.6 × 10–8, 5 out of 71 genes) and maturity onset diabetes of the young (KEGG:83096, P = 9.0 × 10–6, 3 out of 26 genes). Based on these results and because of the high cholesterol content of most gallstones, we tested 203 variants representing signals reported to associate with either HDL cholesterol, LDL cholesterol, total cholesterol or triglyceride serum levels for risk of gallstone disease (Supplementary Data 9). There is an excess of gallstone associated variants among reported HDL cholesterol, LDL cholesterol and total cholesterol associated variants: eight out of 83 HDL cholesterol variants (P = 5.3 × 10–19 based on a binomial test, see Methods), nine out of 65 LDL cholesterol variants (P = 1.1 × 10–22), fourteen out of 77 total cholesterol variants (P = 2.5 × 10–36) and five out of 49 triglyceride variants (P = 1.2 × 10–12) also associate with gallstone disease when all 203 tests are accounted for in the assessment of significance. In total, 23 of the 203 unique variants associating with one or more of HDL cholesterol, LDL cholesterol, total cholesterol and triglycerides also associate with gallstone disease. However, there is no consistency in the directions of the effects on serum cholesterol and gallstone disease risk (Fig. 4). This indicates that lipid serum levels are not by themselves causative factors in gallstone formation, even though cholesterol metabolism appears to have an impact on gallstone risk.

203 variants reported to be associated with either HDL cholesterol, LDL cholesterol, total cholesterol or triglyceride levels: Effect of the allele associated with an increased level of serum lipids vs. odds ratio (OR) for gallstone disease, for a) HDL cholesterol, b) LDL cholesterol, c) total cholesterol, and d) triglycerides. Crosses indicate 95% confidence intervals. Variants with a significant OR for gallstone disease (P < 0.05/203 = 2.6 × 10‒4) are shown in red
Of note is that a missense variant Gln535Arg in DAGLB (rs2303361[C]), encoding diacylglycerol lipase beta, associates with an increased risk of gallstone disease (OR = 1.06 [1.04–1.08], P = 4.7 × 10–8). The variant is in strong LD (r2 = 0.95) with the intron variant rs1880118[C] that was recently reported to associate with increased HDL cholesterol levels and increased DAGLB expression in subcutaneous adipose tissue42. We replicate the association of rs1880118[C] with increased HDL cholesterol in our data (effect = 0.15 SD [0.10–0.20], P = 2.4 × 10–9).
Diabetes and gallstone disease
The relationship between gallstone disease and diabetes is not clear43. To explore whether there is a difference in the effect of gallstone associated sequence variants between diabetics and non-diabetics, we tested whether the 32 gallstone disease associated variants confer different risk on diabetics and non-diabetics and found no significant differences (Supplementary Fig. 12). To further explore the relationship of gallstone disease and genes linked to maturity onset diabetes of the young (MODY), we tested 12,809 variants in 12 genes linked to MODY (including 36 known pathogenic MODY variants) for association with gallstone disease; no additional signals associating with gallstone disease were observed (Supplementary Data 10).
Self-report vs. hospital diagnosis
Since 4067 of the 18,417 UK cases are based only on self-reporting, we have looked for a difference between association results for self-reported and ICD10-diagnosed cases. There is no difference between effect sizes in the two groups (ICD10 and self-reported) for 30 of the 32 variants found. The two variants with a significant difference between ICD10-based and self-reported-based effects are the novel gallstone disease associated variant SERPINA1 Glu366Lys (PI Z; rs28929474[T]) (P-value for test for heterogeneity: Phet = 2.2 × 10–4) and the previously reported ABCG8 Asp19His (rs11887534[C], Phet = 1.6 × 10–9) missense variants, which both showed a larger effect when using self-reported cases than ICD10 code cases. However, both these variants associate with both ICD10-based and self-reported gallstone disease (Supplementary Fig. 13, Supplementary Table 11).
Discussion
Through a GWAS of gallstone disease in Iceland and UK, we found 21 novel sequence variants that associate with the disease, including four low frequency coding variants in SLC10A2 (two distinct variants), HNF4A and SERPINA1 with relatively large effects.
Most notably, we demonstrate that two distinct low-frequency missense variants in the bile acid transporter SLC10A2 — Pro290Ser and Val98Ile — associate with higher risk of gallstone disease. The variants have experimentally been shown to reduce the transport activity of the protein encoded by the gene, commonly known as ASBT16. We observe a tendency to a higher risk of gallstone to associate with a lower transport activity of different variants of SLC10A2. The main function of ASBT is to reabsorb bile salts (up to 95%) from the terminal ileum into ileocytes, after which the bile salts are transported back to the liver through the enterohepatic circulation44. We postulate that impairment of ASBT leads to disturbances of the enterohepatic circulation, causing imbalances of the relative amounts of bile acids, cholesterol and phospholipids, thus increasing the risk of gallstone formation. Because of its role in the enterohepatic circulation, a connection between SLC10A2 and gallstone disease risk has been postulated. In a candidate gene study45 with 240 gallstone cases and 255 controls, the rs9514089[C] SLC10A2 intron variant was reported to associate with an increased risk of gallstone disease (OR = 2.04 [1.19–3.55]; P = 7.7 × 10–3). The reported p-value does not survive multiple testing correction accounting for the 30 markers tested in their manuscript. A subsequent study46 found no association of rs9514089 with gallstone disease (OR = 0.83 [0.63–1.09]; P = 0.19), and this variant has not been reported to associate with gallstone disease by others7,8,9,10. In our Icelandic and UK data (with 27,174 cases) we find no association between rs9514089[C] and gallstone disease risk (OR = 0.991 [0.973–1.010]; P = 0.36; Supplementary Table 12). Thus, we conclude that the claimed association of rs9514089 with gallstone disease is a false positive. Other coding variants in SLC10A2 have been implicated in primary bile acid malabsorption, based on a single pedigree with one identified compound heterozygous carrier of a missense and a splice-donor variant47. SLC10A2 inhibitors have previously been considered as a therapeutic candidate for type 2 diabetes48, but are now pursued as a treatment of chronic constipation due to the initially unwanted diarrhea and steatorrhea side-effects49. Our results indicate that inhibition of SLC10A2 associates with higher risk of gallstone disease as homozygotes for Pro290Ser (expected to have less than 3% transport activity16) have an odds ratio of 3.26 [1.55–6.86]. The genes ABCB4 and SULT2A1, previously reported to be associated with gallstone disease, are also involved in bile homeostasis8,13.
A number of the novel variants described here to associate with gallstone disease are at loci linked to Mendelian diseases: Interestingly, the hepatocyte nuclear factors HNF1A, HNF1B, and HNF4A are all linked to autosomal dominant forms of maturity-onset diabetes of the young (MODY3, OMIM:600496; MODY5, OMIM:137920; and MODY1, OMIM:125850; respectively). In addition, among the reported gallstone loci, the glucokinase regulatory protein (GCKR) is a regulator of glucokinase (GCK), that is linked to MODY2 (OMIM:125851).
Among the novel gallstone variants, two are in fucosyltransferase genes (FUT2 and FUT6). Fucosyltransferases mediate fucosylation which is an abundant posttranslational modification of glycosylated proteins and lipids. FUT2 is responsible for the majority of fucosylation in the gastrointestinal tract50,51. Interestingly, we observe opposing effects of the FUT2 variant rs601338 on the risk of gallstone disease and susceptibility to gastrointestinal infection. This constitutes an example of antagonistic pleiotropy where a variant has opposing effects on the risk of two diseases. The increased risk of gastrointestinal infection observed for the wild-type secretor allele rs601338[G] is likely due to the presence of fucosylated glycans in the gastrointestinal tract that serve as adhesion sites for pathogens. FUT2 may influence biliary disease progression through interaction with the biliary and intestinal microbiota52,53. However, the mechanism through which the rs601338 variant influences the risk of gallstone disease is unclear, as the FUT2 locus is highly pleiotropic and is the subject of balancing selection in humans54,55. Further investigation is needed to explain the role of fucosylation in the pathogenesis of gallstone disease.
We show that the PI Z allele of SERPINA1 associates with increased risk of gallstone disease. Severe AAT deficiency in PI ZZ homozygotes predisposes to emphysema and, less commonly, to liver disease20. The precise pathomechanism of liver disease associated with severe AAT deficiency is not well understood but histopathological findings of liver parenchyma in PI ZZ homozygotes indicate that liver abnormalities result from toxic intracellular accumulation of AAT in hepatocytes56. Moderate AAT deficiency in PI MZ heterozygotes has been associated with slightly increased risk of liver disease and intracellular accumulation of AAT in hepatocytes57. We speculate that MZ heterozygosity may result in general liver dysfunction as result of toxic accumulation of AAT in hepatocytes over time, thus predisposing to gallstone disease.
Biliary cholesterol secretion plays a major role in cholesterol homeostasis58. Variants in genes involved in cholesterol homeostasis, including ABCG5/ABCG8 and CYP7A1, are reported to associate with gallstone disease8. Six of the novel gallstone associated variants, or highly correlated variants (r2 > 0.8), have been reported to associate with blood cholesterol levels (in HNF4A, HNF1A, FUT2, FADS2, MARCH8, and JMJD1C) (Supplementary Data 9). Most notably, HNF4A is a nuclear receptor highly expressed in the liver, and has been shown to control the expression of the closely linked cholesterol transporters ABCG5 and ABCG8 that share a promotor59. Despite a clear role of the aforementioned genes in cholesterol homeostasis, it remains unclear whether serum cholesterol levels affect risk of gallstone disease60. When considering 203 variants reported to associate with serum HDL cholesterol, LDL cholesterol, total cholesterol or triglyceride levels we do not observe a consistent direction of effect of lipid increasing alleles on the risk of gallstone disease. This suggests that there is not a direct path between serum lipid levels and the production of gallstones, which is in line with observations from previous studies61.
While our data, as well as data from others61, do not support a direct path between serum cholesterol levels and gallstone formation, some aspects of cholesterol metabolism may contribute to gallstone disease. Biliary cholesterol super-saturation is considered a major factor in promoting cholesterol nucleation and gallstone formation62,63,64. Thus sequence variants affecting the amount of cholesterol secreted into bile, or the cholesterol/bile acid ratio, are likely to impact gallstone formation. Depending on the mechanism of action of the cholesterol regulating genes, the allele associated with gallstone risk may not consistently decrease or increase serum cholesterol levels. For example, increased activity of the sterol-transporter ABCG5/8 that has a direct role in promoting biliary cholesterol secretion and in reducing intestinal absorption of dietary cholesterol is expected to increase the risk of gallstones while lowering circulating cholesterol. In contrast, reduced function of CYP7A1, the rate-limiting enzyme in the conversion of cholesterol to bile acids in the liver, likely predisposes to gallstone formation through a decrease in bile acid synthesis. It has been postulated that the increase in serum cholesterol levels that has been observed in carriers of CYP7A1 gallstone risk variants may be mediated through down-regulation of hepatic LDL receptors65.
In summary, we have discovered 21 novel gallstone disease variants in the largest gallstone disease GWAS to date. The associations presented emphasize the role of sequence variants in genes involved in cholesterol homeostasis and specifically highlight the intestinal compartment of the enterohepatic circulation in the pathogenesis of gallstone disease. We conclude that sequence variants affecting the amount of cholesterol secreted into bile, or the cholesterol/bile acid ratio, are likely to cause gallstone formation.
Methods
Study subjects from Iceland
This study is based on whole-genome sequence data from the whole blood of 15,220 Icelanders participating in different disease projects at deCODE genetics. Additionally, 151,677 Icelanders have been genotyped using Illumina SNP chips and genotype probabilities for untyped relatives have been calculated based on Icelandic genealogy. The process used to whole-genome sequence the Icelandic population, and the subsequent imputation from which the data for this analysis were generated, has been comprehensively described in a recent publication7. We note that selection of individuals for whole-genome sequencing and chip genotyping was not performed with regards to gallstone case status.
All individuals who donated blood, or their guardians, provided written informed consent. All sample identifiers have been encrypted, in accordance with the regulations of the Icelandic Data Protection Authority. The study was approved by the National Bioethics Committee (ref:VSNb2015100030/03.03). Personal identities of the participants and biological samples were encrypted using a third-party system approved and monitored by the Icelandic Data Protection Authority. The National Bioethics Committee approved the study, including the protocol, methodology and all documents presented to the participants. All methods were performed in accordance with the relevant guidelines and regulations.
To identify gallstone cases, we searched for patients with International Classification of Diseases (ICD) codes, diagnosis code ICD-10 K80 Cholelithiasis and ICD-9 574, indicative of gallstone disease, at Landspitali—The National University Hospital of Iceland in Reykjavik (LUH), and the Icelandic Medical Center in Mjodd (Laeknasetrid). A total of 8757 gallstone cases were included in the association analysis; 892 of these were whole-genome sequenced, 5840 were genotyped using various Illumina chips and imputed using long-range phased haplotypes, and genotype probabilities for 2026 were imputed on the basis of information from genotyped close relatives. In addition, cases of five diseases of the biliary system were identified by the ICD codes K85 for acute pancreatitis, K81 for cholecystitis, K74 for fibrosis/cirrhosis of liver, O26.6 for cholestasis of pregnancy and C23 for gallbladder cancer. Individuals recruited through different genetic research projects at deCODE were used as controls, and individuals in the gallstone cohort were excluded from the control group. Among the controls, 13,204 were whole-genome sequenced, 133,026 were genotyped by chip, and 200,458 were imputed on the basis of the genotypes of close relatives. In total, the number of controls was 346,688.
Study subjects from the UK
The UK Biobank project is a large prospective cohort study of over 500,000 individuals from across the United Kingdom, aged between 40–69 at recruitment66. Genotyping was performed using a custom-made Affimetrix chip, UK BiLEVE Axiom67 in the first 50,000 participants, and with Affimetrix UK Biobank Axiom array in the remaining participants68, 95% of the signals are on both chips. Imputation was performed by Wellcome Trust Centre for Human Genetics using the Haplotype Reference Consortium (HRC) and the UK10K haplotype resources11. This yields a total of 96 million imputed variants, however only 40 million variants imputed using the HRC reference set were used in this study due to quality issues with the remaining variants.
The population from UK Biobank consisted of 18,417 cases and 390,150 controls, all individuals of European ancestry. Gallstone disease was ascertained based on self-reported (self-reported non-cancer illness, Data-Field 20002) and ICD diagnoses obtained from primary or secondary diagnoses codes a participant has had recorded across all their episodes in hospital. Diagnoses are coded according to ICD-9 code 574 or ICD-10 code K80. The UKB Resource was accessed under Application Number ‘24711’.
Association testing and meta-analysis
We used logistic regression to test for association between variants and disease, assuming a multiplicative model, treating disease status as the response and expected genotype counts as covariates. For the Icelandic cohort, the association testing was done using software developed at deCODE genetics7. The threshold for genome-wide significance was corrected for multiple testing using a class-specific Bonferroni procedure based on predicted functional impact of classes of variants12. This yielded significance thresholds of 2.0 × 10–7 for high-impact variants (including stop-gained, frameshift, splice acceptor or donor, N = 11,465), 3.9 × 10–8 for moderate-impact variants (including missense, splice-region variants and in-frame indels, N = 197,583), 3.6 × 10–9 for low-impact variants (including upstream and downstream variants, N = 2,971,445) and 5.9 × 10–10 for lowest-impact variants (including intron and intergenic variants, N = 39,726,619). We have previously demonstrated that the variant annotation based multiple testing method provides greater power to detect associations than Bonferroni correction where all variants are treated equally, while maintaining an overall family-wise error rate of 0.05. All variants except loss-of-function variants (including stop-gained, frameshift, splice acceptor or donor) have p-value thresholds stricter than the common threshold of 5.0 × 10–8, while loss-of-function variants were given a threshold of 2.0 × 10–7 due to their higher prior probability of representing true positive signals. In total, 42,907,111 variants were tested in the meta-analysis. Variants in the UK Biobank imputation dataset have been mapped to NCBI Build38 positions and subsequently matched to the variants in the Icelandic dataset based on allele variation. Results from UK and Iceland were combined using a Mantel-Haenszel model69 in which the groups were allowed to have different population frequencies for alleles and genotypes but were assumed to have a common OR. We tested for heterogeneity by comparing the null hypothesis of the effect being the same in all populations to the alternative hypothesis of each population having a different effect using a likelihood ratio test. I2 describes the proportion of total variation in study estimates due to heterogeneity.
Sibling recurrence risk
Sibling recurrence risk ratios (λS[i]) for the risk alleles of the identified signals associated with gallstone disease in the present meta-analysis. We estimated the risk ratio among siblings (λS) of Icelandic patients with gallstone disease (N = 2628) by cross-matching with a genealogy database that covers the entire Icelandic nation. Risk ratio among siblings (λS) was estimated at 1.81 [1.71, 1.91] (P < 0.0001), using the approach suggested by Edvardsson et al.70. Also, we calculated the proportion of sibling recurrence risk of gallstone disease explained by the signals identified in the present study, which is given by log(λS[i])/log(λS)71.
Population attributable fraction
Population attributable fraction is defined as the fraction of cases that would be eliminated from the population if the risks of all individuals carrying the risk variant could be contained, e.g., through a treatment, to be the same as non-carriers of the at-risk variant(s). It can be calculated for a variant using the following formula:
PAF = 1 – (1/W) where W = (1 − p)2 + 2p(1 − p)RR + p2RR2
Here ‘p’ denotes the at-risk allele frequency and RR is the relative risk of for a disease. Relative risks are estimated by odds ratios under the assumption of the multiplicative model. Accordingly, the RR for carrying two risk variants is RR2.
Gene set enrichment analysis
Gene set enrichment analysis was conducted using the online ToppGene tool41.
Binomial test for excess of cholesterol variants
Excess of gallstone associated variants among reported HDL cholesterol, LDL cholesterol and total cholesterol variants was tested using a binomial model, as follows: Since 170 variants were tested, a Bonferroni adjusted significance level of 0.05/170 = 2.9 × 10–4 was used. Thus, under the null hypothesis of no association with gallstone disease, the probability of getting a significant association is 2.9 × 10–4 for each variant, since the P-values for association with gallstone disease have a Uniform[0,1] distribution under the null. Therefore, the number of significant associations with gallstone disease in each group has a Binomial (N, P) distribution, where N is the number of tested variants in the group and P = 2.9 × 10–4, and this can be used as the basis for a binomial test. For example, for the HDL cholesterol variants, eight out of 83 variants associate with gallstone disease, and the P-value for the binomial test of excess of HDL variants is therefore Prob(X ≥ 8) = 2.2 × 10–18, where X has a Binomial(83, 2.9 × 10–4) distribution.
URLs
For Online Mendelian Inheritance in Man (OMIM), see https://www.omim.org/. For GWAS catalog, see https://www.ebi.ac.uk/gwas/. For GTEx Portal, see https://www.gtexportal.org/.
Code availability
We used publicly available software (URLs listed below) in conjunction with the above described algorithms in the sequencing processing pipeline (Whole-genome sequencing, Association testing, RNA-seq mapping and analysis): for BWA 0.7.10 mem, https://github.com/lh3/bwa; for
GenomeAnalysisTKLite 2.3.9, https://github.com/broadgsa/gatk/; for
Picard tools 1.117, https://broadinstitute.github.io/picard/; for
SAMtools 1.3, http://samtools.github.io/; for Bedtools v2.25.0-76-g5e7c696z, https://github.com/arq5x/bedtools2/; for Variant Effect Predictor, https://github.com/Ensembl/ensembl-vep; for BOLT-LMM, https://data.broadinstitute.org/alkesgroup/BOLT-LMM/downloads/.
Data availability
Sequence variants passing GATK filters have been deposited in the European Variation Archive, accession number PRJEB15197.
References
Shaffer, E. A. Epidemiology of gallbladder stone disease. Best. Pract. Res. Clin. Gastroenterol. 20, 981–996 (2006).
Portincasa, P., Moschetta, A. & Palasciano, G. Cholesterol gallstone disease. Lancet 368, 230–239 (2006).
Admirand, W. H. & Small, D. M. The physicochemical basis of cholesterol gallstone formation in man. J. Clin. Invest. 47, 1043 (1968).
Katsika, D. et al. Genetic and environmental influences on symptomatic gallstone disease: A Swedish study of 43,141 twin pairs. Hepatology 41, 1138–1143 (2005).
Buch, S. et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat. Genet. 39, 995–999 (2007).
Dawson, P. A., Lan, T. & Rao, A. Bile acid transporters. J. Lipid Res. 50, 2340–2357 (2009).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Joshi, A. D. et al. Four Susceptibility Loci for Gallstone Disease Identified in a Meta-analysis of Genome-Wide Association Studies. Gastroenterology 151, 351–363.e28 (2016).
Oussalah, A. et al. Exome-wide association study identifies new low-frequency and rare UGT1A1 coding variants and UGT1A6 coding variants influencing serum bilirubin in elderly subjects: a strobe compliant article. Medicine 94, e925 (2015).
Rodriguez, S. et al. Lipids, obesity and gallbladder disease in women: insights from genetic studies using the cardiovascular gene-centric 50K SNP array. Eur. J. Hum. Genet. 24, 106–112 (2016).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Sveinbjornsson, G. et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet. 48, 314–317 (2016).
Gudbjartsson, D. F. et al. Sequence variants from whole genome sequencing a large group of Icelanders. Sci. Data 2, 150011 (2015).
Polubriaginof, F. C. et al. Disease Heritability Inferred from Familial Relationships Reported in Medical Records. Cell 173, 1692–1704 (2018).
Wong, M. H., Oelkers, P. & Dawson, P. A. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J. Biol. Chem. 270, 27228–27234 (1995).
Ho, R. H. et al. Functional characterization of genetic variants in the apical sodium-dependent bile acid transporter {(ASBT; SLC10A2)}. J. Gastroenterol. Hepatol. 26, 1740–1748 (2011).
Geyer, J., Wilke, T. & Petzinger, E. The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmiede. Arch. Pharmacol. 372, 413–431 (2006).
Choi, M. et al. Identification of a hormonal basis for gallbladder filling. Nat. Med. 12, 1253–1255 (2006).
Love, M. W. et al. Analysis of the ileal bile acid transporter gene, SLC10A2, in subjects with familial hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 21, 2039–2045 (2001).
Stoller, J. K. & Aboussouan, L. S. A review of α 1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 185, 246–259 (2012).
Cox, D. W. & Smyth, S. Risk for liver disease in adults with alpha 1-antitrypsin deficiency. Am. J. Med. 74, 221–227 (1983).
Hersh, C. P. et al. Chronic obstructive pulmonary disease in α1-antitrypsin PI MZ heterozygotes: A meta-analysis. Thorax 59, 843–849 (2004).
Sørheim, I. C. et al. α1-antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohorts. Chest 138, 1125–1132 (2010).
Bernspång, E., Carlson, J. & Piitulainen, E. The liver in 30-year-old individuals with alpha1-antitrypsin deficiency. Scand. J. Gastroenterol. 44, 1349–1355 (2009).
Hwang-Verslues, W. W. & Sladek, F. M. HNF4α-role in drug metabolism and potential drug target? Curr. Opin. Pharmacol. 10, 698–705 (2010).
Liu, D. J. et al. Exome-wide association study of plasma lipids in > 300,000 individuals. Nat. Genet. 49, 1758–1766 (2017).
Ek, J. et al. The Functional Thr130Ile and Val255Met Polymorphisms of the Hepatocyte Nuclear Factor 4alpha (HNF 4alpha) Gene Associate with Type 2 Diabetes and Altered beta cell Function among Danes. J. Clin. Endocrinol. Metab. 90, 3054–3059 (2005).
Zhu, Q. et al. T130I mutation in HNF-4alpha gene is a loss-of-function mutation in hepatocytes and is associated with late-onset Type 2 diabetes mellitus in Japanese subjects. Diabetologia 46, 567–573 (2003).
Grarup, N. et al. Genetic Architecture of Vitamin B12 and Folate Levels Uncovered Applying Deeply Sequenced Large Datasets. PLoS. Genet. 9, e1003530 (2013).
Thorven, M. et al. A homozygous nonsense mutation (428G– > A) in the human secretor (FUT2) gene provides resistance to symptomatic norovirus (GGII) infections. J. Virol. 79, 15351–15355 (2005).
Ikehara, Y. et al. Polymorphisms of two fucosyltransferase genes (Lewis and Secretor genes) involving type I Lewis antigens are associated with the presence of anti-Helicobacter pylori IgG antibody. Cancer Epidemiol. Biomark. Prev. 10, 971–977 (2001).
Hazra, A. et al. Common variants of FUT2 are associated with plasma vitamin B12 levels. Nat. Genet. 40, 1160–1162 (2008).
Deboever, C. et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat. Commun. 9, 1612 (2018).
Núñez, L. et al. Cholesterol crystallization-promoting activity of aminopeptidase-N isolated from the vesicular carrier of biliary lipids. FEBS Lett. 329, 84–88 (1993).
Offner, G. D., Gong, D. & Afdhal, N. H. Identification of a 130-kilodalton human biliary concanavalin a binding protein as aminopeptidase N. Gastroenterology 106, 755–762 (1994).
Ekholm, E., Nilsson, R., Groop, L. & Pramfalk, C. Alterations in bile acid synthesis in carriers of hepatocyte nuclear factor 1α mutations. J. Intern. Med. 274, 263–272 (2013).
Shih, D. Q. et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 27, 375–382 (2001).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1285 (2013).
Spracklen, C. N. et al. Association analyses of East Asian individuals and trans-ancestry analyses with European individuals reveal new loci associated with cholesterol and triglyceride levels. Hum. Mol. Genet. 26, 1770–1784 (2017).
van der Harst, P. & Verweij, N. The Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. CIRCRESAHA.117.312086. https://doi.org/10.1161/CIRCRESAHA.117.312086(2017).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, 305–311 (2009).
Zhcou, D. et al. A novel variant associated with HDL-C levels by modifying DAGLB expression levels: An annotation-based genome-wide association study. Eur. J. Hum. Genet. https://doi.org/10.1038/s41431-018-0108-4 (2018).
Weikert, C. et al. Presence of gallstones or kidney stones and risk of type 2 diabetes. Am. J. Epidemiol. 171, 447–454 (2010).
Barbara Döring, T. L., Geyer, J. & Petzinger, E. The SLC10 Carrier Family: Transport Functions and Molecular Structure. in Current Topics in Membranes (ed. Bevensee, M. O.) 70, (Academic Press, 2012).
Renner, O. et al. A variant of the SLC10A2 gene encoding the apical sodium-dependent bile acid transporter is a risk factor for gallstone disease. PLoS One 4, e7321 (2009).
Tönjes, A. et al. Effects of SLC10A2 variant rs9514089 on gallstone risk and serum cholesterol levels-meta-analysis of three independent cohorts. BMC Med. Genet. 12, 149 (2011).
Oelkers, P., Kirby, L. C., Heubi, J. E. & Dawson, P. A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Invest. 99, 1880–1887 (1997).
Chen, L. et al. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am. J. Physiol. Endocrinol. Metab. 302, E68–E76 (2012).
Mosińska, P., Fichna, J. & Storr, M. Inhibition of ileal bile acid transporter: an emerging therapeutic strategy for chronic idiopathic constipation. World J. Gastroenterol. 21, 7436–7442 (2015).
Hurd, E. A., Holmén, J. M., Hansson, G. C. & Domino, S. E. Gastrointestinal mucins of Fut2-null mice lack terminal fucosylation without affecting colonization by Candida albicans. Glycobiology 15, 1002–1007 (2005).
Sakamoto, J. et al. Expression of Lewis a, Lewis b, Lewis x, Lewis y, Sialyl-Lewis a, and Sialyl-Lewis x Blood Group Antigens in Human Gastric Carcinoma and in Normal Gastric Tissue. Cancer Res. 49, 745–752 (1989).
Verdier, J., Luedde, T. & Sellge, G. Biliary Mucosal Barrier and Microbiome. Visz. Gastrointest. Med. Surg. 31, 156–161 (2015).
Swidsinski, A. & Lee, S. P. The role of bacteria in gallstone pathogenesis. Front. Biosci. 6, E93–E103 (2001).
Koda, Y. et al. Contrasting patterns of polymorphisms at the ABO-Secretor gene (FUT2) and plasma α(1,3)fucosyltransferase gene (FUT6) in human populations. Genetics 158, 747–756 (2001).
Ferrer-Admetlla, A. et al. A natural history of FUT2 polymorphism in humans. Mol. Biol. Evol. 26, 1993–2003 (2009).
Tomashefski, J. F., Crystal, R. G., Wiedemann, H. P., Mascha, E. & Stoller, J. K. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: Findings of the Death Review Committee of the national registry for individuals with severe deficiency of alpha-1 antitrypsin. Hum. Pathol. 35, 1452–1461 (2004).
de Serres, F. & Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 276, 311–335 (2014).
Dikkers, A. & Tietge, U. J. F. Biliary cholesterol secretion: More than a simple ABC. World J. Gastroenterol. 16, 5936–5945 (2010).
Sumi, K. et al. Cooperative Interaction between Hepatocyte Nuclear Factor 4 and GATA Transcription Factors Regulates ATP-Binding Cassette Sterol Transporters ABCG5 and ABCG8. Mol. Cell. Biol. 27, 4248–4260 (2007).
Banim, P. J. R. et al. The aetiology of symptomatic gallstones quantification of the effects of obesity, alcohol and serum lipids on risk. Epidemiological and biomarker data from a UK prospective cohort study (EPIC-Norfolk). Eur. J. Gastroenterol. Hepatol. 23, 733–740 (2011).
Stender, S., Frikke-Schmidt, R., Benn, M., Nordestgaard, B. G. & Tybjærg-Hansen, A. Low-density lipoprotein cholesterol and risk of gallstone disease: A Mendelian randomization study and meta-analyses. J. Hepatol. 58, 126–133 (2013).
Marschall, H.-U. & Einarsson, C. Gallstone disease. J. Intern. Med. 261, 529–542 (2007).
von Kampen, O. et al. Genetic and functional identification of the likely causative variant for cholesterol gallstone disease at the ABCG5/8 lithogenic locus. Hepatology 57, 2407–2417 (2013).
Fuchs, M. et al. Biliary cholesterol hypersecretion in gallstone-susceptible mice is associated with hepatic up-regulation of the high-density lipoprotein receptor SRBI. Hepatology 33, 1451–1459 (2001).
Qayyum, F. et al. Genetic variants in CYP7A1 and risk of myocardial infarction and symptomatic gallstone disease. Eur. Heart J. 39, 2106–2116 (2018).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS. Med. 12, e1001779 (2015).
Wain, L. V. et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med. 3, 769–781 (2015).
Welsh, S., Peakman, T., Sheard, S. & Almond, R. Comparison of DNA quantification methodology used in the DNA extraction protocol for the UK Biobank cohort. BMC Genom. 18, 26 (2017).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl Cancer Inst. 22, 719–748 (1959).
Edvardsson, V. O., Palsson, R., Indridason, O. S., Thorvaldsson, S. & Stefansson, K. Familiality of kidney stone disease in Iceland. Scand. J. Urol. Nephrol. 43, 420–424 (2009).
Witte, J. S., Visscher, P. M. & Wray, N. R. The contribution of genetic variants to disease depends on the ruler. Nat. Rev. Genet. 15, 765–776 (2014).
Acknowledgements
We thank the individuals who participated in this study and whose contribution made this work possible. We also thank our valued colleagues who contributed to the data collection and phenotypic characterization of clinical samples, as well as to the genotyping and analysis of the data.
Author information
Authors and Affiliations
Contributions
E.F., A.O., A.M.D., A.H., H.H., D.F.G., P.S., and K.S. designed the study and interpreted the results. S.G., I.J., S.O., T.S., and E.S.B. carried out subject ascertainment and recruitment. I.O., G.I.E., and O.S. contributed to data acquisition. E.F., A.O., S.B., G.T., A.M.D., S.J., O.A.S., F.Z., B.G., G.H.H., G.S., D.A.S., G.M., D.F.G., and P.S. performed statistical and bioinformatics analyses. E.F., A.O., S.B., G.T, A.M.D., O.A.S., G.L.N., G.A.A., B.O.J., R.P.K, T.R., U.T., D.F.G., P.S., and K.S. drafted the manuscript. All authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors E.F., A.O., S.G., S.B., G.T., A.M.D., S.J., O.A.S., G.L.N., F.Z., G.A.A., B.G., G.H.H., A.H., B.O.J., R.P.K., G.S, D.A.S., G.M., H.H., I.J., T.R., U.T., D.F.G., P.S., and K.S., who are affiliated with deCODE genetics/AMGEN, declare competing financial interests as employees. The remaining authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferkingstad, E., Oddsson, A., Gretarsdottir, S. et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun 9, 5101 (2018). https://doi.org/10.1038/s41467-018-07460-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07460-y
This article is cited by
-
Membrane transporters in drug development and as determinants of precision medicine
Nature Reviews Drug Discovery (2024)
-
The impact of diagnostic delay on survival in alpha-1-antitrypsin deficiency: results from the Austrian Alpha-1 Lung Registry
Respiratory Research (2023)
-
Genome-Wide Association Study of Gallstone Disease Identifies Novel Candidate Genomic Variants in a Latino Community of Southwest USA
Journal of Racial and Ethnic Health Disparities (2023)
-
Genetic studies of paired metabolomes reveal enzymatic and transport processes at the interface of plasma and urine
Nature Genetics (2023)
-
Multi-ancestry genome-wide association analyses improve resolution of genes and pathways influencing lung function and chronic obstructive pulmonary disease risk
Nature Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
