
Abstract
Although many factors contribute to cellular differentiation, the role of mitochondria Ca2+ dynamics during development remains unexplored. Because mammalian embryonic epiblasts reside in a hypoxic environment, we intended to understand whether mCa2+ and its transport machineries are regulated during hypoxia. Tissues from multiple organs of developing mouse embryo evidenced a suppression of MICU1 expression with nominal changes on other MCU complex components. As surrogate models, we here utilized human embryonic stem cells (hESCs)/induced pluripotent stem cells (hiPSCs) and primary neonatal myocytes to delineate the mechanisms that control mCa2+ and bioenergetics during development. Analysis of MICU1 expression in hESCs/hiPSCs showed low abundance of MICU1 due to its direct repression by Foxd1. Experimentally, restoration of MICU1 established the periodic cCa2+ oscillations and promoted cellular differentiation and maturation. These findings establish a role of mCa2+ dynamics in regulation of cellular differentiation and reveal a molecular mechanism underlying this contribution through differential regulation of MICU1.
Similar content being viewed by others


Introduction
The molecular mechanisms and consequences of cellular hypoxia sensing factors are especially well characterized in many organisms during development and normal physiology. The initiation of these processes is rapid and involve transcriptional and posttranslational mechanisms. In higher order eukaryotes, survival is solely dependent upon the consumption of O2 as a substrate by respiratory chain complex IV, cytochrome c oxidase (COX). Upon hypoxia, the induction of COX4-2 subunit expression and a rapid degradation of COX4-1 subunit has been established in a reciprocal manner to adapt hypoxia. The HIF-1-dependent mitochondrial protease, LON degrades COX4-1 implying the well conserved O2-dependent homeostatic circuit at the mitochondrial level1,2. This phenomenon is analogues to hemoglobin F (HbF; α2γ2) vs hemoglobin A (HbA; α2β2) where HbF (~19 mmHg) has higher affinity for O2 over HbA (~27 mmHg) during development. Besides the well characterized prolyl hydroxylase family of enzymes and OXPHOS complex, ion channels have been proposed as physiological sensors of cellular hypoxia3. In the context of development, these events appear to be mediated through O2 and possibly requiring mitochondrial function. Because mitochondrial OXPHOS and energy transduction are mitochondrial Ca2+ (mCa2+) dynamics dependent processes, relatively little is known about how the cyclic switch between glycolytic and oxidative metabolism is established during development.
A characteristic feature of the MCU channel activity is driven by two major forces like a steep mitochondrial membrane potential (ΔΨm) and cytosolic Ca2+ (cCa2+) transients. A steep conductance of mCa2+ rates suggests that the channel opens only if cCa2+ attains the set-point (~2–3 μM)4,5. MCU functions as a pore subunit in that it can be regulated by a number of proteins, divalent cation and oxidants4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22. In recent years, MICU1, MICU2, and MICU3 proteins that have EF-hand Ca2+ binding motifs were shown to regulate the MCU activity4,5,7,9,10,12,13,14,23,24. Besides these, the MCU pore open modulator EMRE15,25 and an assembly factor MCUR121,26 have been identified. Although recent advances have led to a better understanding of the proteins associated with MCU complex and its role in mitochondrial bioenergetics, a large and growing body of evidence attests to the complexity of regulation of MCU activity. Until now, the regulatory role of MICU1 has been reported to control MCU activity under resting state4,5. Our knowledge about the precise function of MICU1 is still very limited, especially regarding to mechanism of Ca2+ sensing and the regulation of MCU channel activity.
A loss-of-function mutation of MICU1 has been linked to human disease characterized by proximal myopathy, learning difficulties, and a progressive extrapyramidal movement disorder27. Recently, genetic deletion of MICU1 in murine model was reported to induce higher rates of perinatal lethality4,28, with embryos antepartum at expected Mendelian ratio19. Enigmatically, MICU1−/− mice that did survive past 1 week show chorea-like movement. These data indicate the absence of MICU1 is particularly critical immediately after birth, suggesting MICU1 and thus mCa2+ signaling to be critical mediators in embryonic to postnatal developmental transition when the demand for oxidative phosphorylation surges. Indeed, it is unknown whether MICU1 regulation of MCU-mediated Ca2+ uptake is essential for the transition from the relatively hypoxic environment of the placenta to the oxygenated environment after birth.
Here we show a differential regulation of MICU1, an effect that is caused by a suboptimal growth environment and present MICU1/MCU function analogues to STIM1/Orai-dependent SOCE phenomenon where STIM1 not only senses the ER Ca2+ threshold but also senses ROS and temperature29,30,31. Importantly, tissues from brain, heart, liver, and lung of developing mouse embryo (E13.5, and E16.5) evidenced a suppression of MCU gatekeeper, MICU1 expression with minimal regulation on other MCU complex components under hypoxic milieu. We utilized human induced pluripotent stem cells (hiPSCs) and neonatal myocytes to delineate the mechanisms that control mCa2+ dynamics and mitochondrial bioenergetics during development. Analysis of MICU1 protein expression in parental fibroblast (GM942) and the corresponding re-programmed hiPSCs showed mere absence of MICU1 in hiPSCs. We also observed a differential regulation of MICU1 in NRVMs in response to hypoxia. Our data reveals that the constitutive accumulation of mCa2+, mitochondria ROS, and global proteomic changes in hiPSCs is due to the loss of MCU gatekeeping effect. In glycolytic and hypoxic hiPSCs and neonatal myocytes, MICU1 expression is under the repression of Foxd1 transcription factor. Reconstitution of MICU1 either by Foxd1 knock down or ectopic expression restores the periodic cytosolic Ca2+ transients that is a prerequisite for cellular differentiation. These findings reveal a distinct molecular mechanism underlying the contribution of mitochondrial function in low oxygen tension and the functional role for MICU1 in possibly sensing hypoxia.
Results
MICU1 expression is differentially regulated by hypoxia
Having demonstrated the importance of MICU1 in human and mouse physiology4,28, we investigated the role of MICU1 in development. We assessed the expression profile of MCU complex components in multiple metabolically active organs including brain, heart, lungs, and liver from different mouse developmental stages (E13.5, E16.5, P0, P2, P4, and P7). The western blot analysis of the fetal and postnatal tissues with antibodies specific for MCU, MICU1, and MCUR1 showed a differential expression of MICU1 with no observable changes in other MCU complex components (Fig. 1a, f). Similarly, the immunohistochemical analysis of multiple tissues showed differential expression of MICU1 (Fig. 1g, h). As depicted P0 and P7 neonate tissues showed higher expression of MICU1 while E16.5 embryos showed lower levels of MICU1 expression (Fig. 1g, h). Because we observed a differential expression of MICU1 in developmental stages, we hypothesize the increased trans-placental oxygen levels and thus modulation of tissue oxygen content to modulate MICU1 expression. As a surrogate model to test whether MICU1 is regulated by the hypoxic microenvironment, we subjected the freshly isolated neonatal rat ventricular myocytes (NRVMs) to normoxia at various time points (48–168 h) or hypoxia for 24 h followed by reoxygenation (72 and 96 h). Western blot analysis revealed a progressive increase of MICU1 protein expression under normoxic culture condition (Supplementary Fig.1a and 1b). Remarkably, the repression of MICU1 expression was observed under hypoxia and re-oxygenation restores the MICU1 protein abundance (Supplementary Fig.1a and 1b).

MICU1 is differentially expressed during embryonic development. a–c Representative western blot for lysates from brain (a), heart (b), and liver (c) harvested from embryo (E13.5), and neonates (P0, P2, and P4). The western blots were probed with antibody specific for MCU, MICU1, MCUR1, and Tom20. d–f Quantification of relative MCU, MICU1, and MCUR1 protein abundance from a–c. g Representative zoomed microscopic images of the tissue microarray probed with MICU1 antibody. h Quantification of MICU1 expression levels represented as percent MICU1 expression as a function of tissue staining. i Representative western blot for lysates from control fibroblasts (GM942), hiPSCs (SeV3, SeV5, and SV20), and hESCs probed with antibody specific for MCU, MICU1, Tom20, and β-actin. j Quantification of relative MICU1 protein abundance from i. k Quantification of MICU1 mRNA levels in different tissues (brain, liver, and heart) harvested from embryo and neonates (E13.5, P0, P2, and P4). l Quantification of MICU1 mRNA levels in two independent clonal hiPSCs lines (SeV3 and SV20) and one clonal line of SeV3 (SeV5). Data represents Mean ± SEM; **P < 0.01; ***P < 0.001; n = 3–5 (one-way ANOVA)
Because stem and adult cells reside in their specific niches or anatomic locations that modulate cellular differentiation32,33,34,35,36, we next asked whether a differential expression of MICU1 was observed in human embryonic stem cells (hESCs). Subsequently, hiPSCs have replaced the well-known hESCs as they are generated in an individual-matched manner. To assess the expression of MICU1 in hiPSCs, two independent clonal hiPSC lines (SeV3 and SeV5) derived from the same skin fibroblasts (GM942), one hiPSC line: SV2037 generated from blood and the hESC line H9 were used (Supplementary Fig.1c-1e). Western blot analysis indicated high levels of MICU1 protein expression in control fibroblasts (GM942) while hiPSCs and hESCs showed low abundance of MICU1 protein (Fig. 1i, j, Supplementary Fig. 1f and 1g). The decrease in MICU1 protein expression was consistent with decreased mRNA abundance in the embryonic tissues (Fig. 1k) and hiPSCs (Fig. 1l).
Foxd1 mediated transcriptional regulation of MICU1
Next, we asked whether differential regulation of MICU1 expression is transcriptionally controlled under hypoxic micro-environment. hiPSCs or NRVMs were subjected to both hypoxia (5% O2) or normoxia (20% O2) and MICU1 expression was evaluated. As expected MICU1 mRNA and protein abundance were lower under hypoxic culture condition in hiPSCs (Fig. 2a–c, Supplementary Fig. 1h). Strikingly, MICU1 mRNA levels by qPCR analysis show an increased expression of MICU1 transcript in hiPSC clonal lines when cultured at normoxic condition (Fig. 2a). In line with increased MICU1 transcript, hiPSCs exhibited elevated MICU1 protein levels under normoxia with no observable changes in MCU protein (Fig. 2b, d, Supplementary Fig. 1h). Similar to hiPSCs, MICU1 mRNA abundance was observed in NRVMs exposed to normoxia (Supplementary Fig. 1i). Analysis of the promoter sequence of 411 genes upregulated in reprogrammed iPSCs revealed two transcription factors Foxd1 and Foxo1 to be overrepresented38. Though Foxo1 is a well-known regulator of longevity, stress response39,40, and essential for maintaining pluripotency of ESCs41, Foxd1 has been shown to function in the development of kidney42,43,44. In line with these findings, we investigated the expression levels of Foxd1 in control fibroblasts and hiPSCs. hiPSCs have suggestively increased levels of Foxd1 that was reciprocal to the MICU1 expression (Supplementary Fig. 1j and 1k). Because we observed MICU1 to be transcriptionally regulated under normoxic and hypoxic conditions (Fig. 2a, c), we next investigated whether Foxd1 is the major transcriptional regulator for MICU1 under conditions of hypoxia. Foxd1 mRNA abundance was determined in hiPSCs under conditions of normoxia/hypoxia. Foxd1 transcript levels reflected the changes in Foxd1 protein abundance (Fig. 2e, g, Supplementary Fig. 1l). Of interest, hiPSCs maintained at 20% oxygen showed striking decrease in Foxd1 mRNA (Fig. 2e) and protein levels (Fig. 2f, g, Supplementary Fig. 1l). In addition, we observed a reciprocal relationship between the Foxd1 /MICU1 mRNA (Fig. 2h) and protein levels (Fig. 2i, j, Supplementary Fig. 1m) in embryonic/neonatal tissues that were correlated with hypoxia and normoxia conditions. Taken together, these results indicate Foxd1 to transcriptionally repress MICU1 expression under condition of hypoxia.

MICU1 is transcriptionally regulated by Foxd1 under hypoxia. a Quantification of MICU1 mRNA abundance in hiPSCs exposed to normoxia (20% O2) and hypoxia (5% O2). b Representative western blot for lysates from control fibroblasts and hiPSCs grown under hypoxic/normoxic conditions and probed with antibody specific for MICU1, MCU, ATP5A, and Tom20. c, d Quantification of relative MICU1 (c) and MCU (d) protein abundance in hiPSCs grown under hypoxic/normoxic conditions. e Quantification of Foxd1 mRNA abundance in hiPSCs under normoxia/hypoxia. f Representative western blot for lysates from control fibroblasts and hiPSCs grown under normoxic/ hypoxic conditions and probed with antibody specific for Foxd1 and β-actin. g Quantification of relative protein abundance of Foxd1 and MICU1 quantified from f and b. h Quantification of Foxd1 mRNA abundance in various tissues (brain, heart, liver, and lung) harvested from different stages of embryonic development. i Representative western blot for lysates from brain, heart, liver, and lung harvested from embryos/neonates and probed with antibodies specific for MICU1, Foxd1, and β-actin. j Quantification of relative protein abundance of Foxd1 and MICU1 quantified from i. k ChIP-assay was performed in hiPSCs exposed to normoxia and hypoxia. Antibody specific for Foxd1 was used to immunoprecipitate the chromatin and the fold enrichment of micu1 promoter relative to the matched input control was quantified by qRT-PCR. Bar represents Mean ± SEM, **P < 0.01, *P < 0.05, ***P < 0.001; n = 3–5 (One-Way ANOVA)
Next, we asked whether micu1 promoter have a binding site for Foxd1. Bioinformatic analysis of micu1 promoter sequence predicted a conserved putative binding site for Foxd1 at −302–297 base pairs (bp) (TCAACA). To investigate the functional interactions between the micu1 promoter and Foxd1 during hypoxia, we performed a ChIP assay in hiPSCs under conditions of hypoxia/normoxia. Nuclear extracts from hiPSC clones maintained in hypoxic conditions showed increased Foxd1 binding to the micu1 promoter (Fig. 2k), which parallels decreased MICU1 expression (Fig. 2a, b). Additionally, to ensure the consistency of increased Foxd1 binding to micu1 promoter, we performed ChiP assay in human pulmonary microvascular endothelial cells (HPMVECs) under conditions of hypoxia/normoxia. Similar to hiPSCs, HPMVECs exposed to hypoxia display increased binding of Foxd1 to the micu1 promoter and reoxygenation restored Foxd1 binding (Supplementary Fig. 1n). After confirming the ability to replicate the phenomenon of Foxd1 binding to micu1 promoter in different cell types, we generated luciferase reporter constructs of micu1 promoter that harbors the Foxd1 binding site (Supplementary Fig. 1o). The luciferase constructs were expressed in HPMVECs and 48 h post-transfection, luciferase activity was measured after exposure of HPMVECs to hypoxia and reoxygenation. HPMVECs expressing micu1 promoter construct (-302-297 bp) exhibited differential expression of luciferase in response to hypoxia and reoxygenation (Supplementary Fig. 1p). Conversely, in cells expressing micu1Δ-302–297 the luciferase expression was not regulated by hypoxia, indicating Foxd1- mediated transcriptional repression of MICU1 during hypoxia. Further, to systematically show Foxd1 to be the transcriptional repressor for MICU1, we adopted RNAi based knockdown of Foxd1 in hiPSCs clonal lines. In agreement with the promoter and ChIP analysis, hiPSCs transfected with Foxd1 siRNA restored MICU1 expression levels, validating Foxd1-mediated transcriptional repression of MICU1 expression (Supplementary Fig. 1q and 1r).
Loss of MICU1 regulates mCa2+ uptake and bioenergetics
Because, MICU1 is the gate keeper for MCU that determines the set-point for MCU-mediated mCa2+ uptake, and prevent mCa2+ overload, we next asked whether the repression of MICU1 in hiPSCs result in constitutive MCU-mediated mCa2+ accumulation even at [Ca2+]i below the threshold (>3 μM)5. We performed simultaneous measurement of mCa2+ uptake and mitochondrial membrane potential (ΔΨm) in permeabilized fibroblast and hiPSC clone, SeV3. Digitonin permeabilized cells were bathed in intracellular-like media (ICM) containing thapsigargin (Tg) to prevent Ca2+ uptake through SERCA and loaded with FuraFF (bath Ca2+ indicator) and JC-1 (ΔΨm indicator) (Fig. 3a, c). A single bolus of extramitochondrial Ca2+ pulse (1 μM) within the functional inhibitory range of MICU1, was added and the rate of mCa2+ uptake was measured as a function of decrease in bath Ca2+ fluorescence. SeV3 was able to rapidly clear 1 μM Ca2+ pulse indicating loss of MICU1 inhibition on MCU activity. Conversely, fibroblasts did not clear the 1 μM Ca2+ and evidenced intact MICU1 function (Fig. 3c, d). An uncoupler CCCP was added at 750 s to depolarize the mitochondrial membrane and release accumulated matrix Ca2+. Consistent with the absence of MICU1 in SeV3, we observed constitutive activation of MCU and increased mCa2+accumulation (Fig. 3e, f). Next, we asked whether excessive accumulation of mCa2+ have an impact on ΔΨm. We observed increased accumulation of mCa2+ to result in decreased ΔΨm further substantiating MCU is relieved from MICU1 gatekeeping effect (Fig. 3a, b; Supplementary Fig. 2a and 2b). Next, we restored MICU1 expression in hiPSCs either by ectopic expression of MICU1 using adenovirus or by knocking down Foxd1. We measured mCa2+ uptake and ΔΨm in MICU1 restored SeV3 (Fig. 3). Either ectopic expression of MICU1 (Fig. 3a, f) or silencing Foxd1 (Supplementary Fig. 1q and 1r; Fig. 3g, k) re-established the gatekeeping effect on MCU-mediated Ca2+ uptake and maintained ΔΨm. Next, we asked whether constitutive activation of MCU and excessive accumulation of mCa2+ results in increased mROS generation. In line with constitutive MCU activation, hiPSCs showed elevated mROS compared to control fibroblasts (Supplementary Fig. 2c and 2d).

Loss of MICU1 in hiPSCs augment MCU-mediated Ca2+ uptake and overloads matrix Ca2+. An equal number of fibroblasts (GM942), hiPSCs (SeV3), and SeV3 expressing MICU1cells (6 × 106) were permeabilized with digitonin (40 μM), loaded with JC-1 (800 nM) and Fura-FF (1 μM) and changes in ΔΨm and mCa2+ uptake was measured using a multi-wavelength excitation dual-wavelength emission spectrofluorimeter. a Mean traces of ΔΨm in fibroblasts and hiPSCs. b Quantification of basal ΔΨm before addition of an extramitochondrial Ca2+ bolus. c Mean traces of [Ca2+]out measured in permeabilized fibroblasts and hiPSCs. d Quantification of the rate of mCa2+ uptake as a function of decrease in bath Ca2+ fluorescence after an extramitochondrial Ca2+ pulse (1 μM). e Mean traces of [Ca2+]out after addition of CCCP. f Quantification of total mCa2+ released after the addition of CCCP as a function of increase in bath Ca2+ fluorescence. Foxd1 knock down restores MICU1 gate keeping effect. g–k SeV3 cells were transiently transfected with control and Foxd1 siRNA and 48 h post transfection, ΔΨm and mCa2+ uptake was measured simultaneously in permeabilized cells. g Mean traces of ΔΨm in control (Neg siRNA) and Foxd1 KD hiPSCs. h Mean traces of [Ca2+]out measured in permeabilized control (Neg siRNA) and Foxd1 KD hiPSCs. i Quantification of basal ΔΨm before addition of an extramitochondrial Ca2+ bolus. j Quantification of the rate of mCa2+ uptake as a function of decrease in bath Ca2+ fluorescence after an extramitochondrial Ca2+ pulse (1 μM). k Quantification of total mCa2+ released after the addition of CCCP. Data represents Mean ± SEM; **P < 0.01, ***P < 0.001; n = 4–6 (One-way ANOVA)
Because we were able to mimic the regulation of MICU1 by hypoxia in NRVMs (Supplementary Fig. 1a and 1b), we then performed the simultaneous measurement of mCa2+ uptake and ΔΨm in permeabilized NRVMs exposed to hypoxia and reoxygenation (Supplementary Fig. 2e-2j). Consistent with the rapid clearance of 1 μM Ca2+ bolus by hiPSCs, NRVMs exposed to hypoxia also show a constitutive activation of MCU with a functional phenotype similar to loss of MICU15. Matrix Ca2+ is known to activate the dehydrogenases and increase the mitochondrial oxygen consumption rate (OCR). On the other hand, excessive accumulation of matrix Ca2+ results in mitochondrial permeability transition pore opening and bioenergetics crisis. Next, we assessed the cellular bioenergetics by exposing NRVMs sequentially to the inhibitors of the complex activity (Supplementary Fig. 2k). Consistent with mCa2+ overload, basal, and maximal OCR were significantly reduced in NRVMs exposed to hypoxia (Supplementary Fig. 2l-2n). Additionally, NRVMs exposed to hypoxia show increased proton leak (Supplementary Fig. 2n), further validating uncoupled Ca2+ activated OXPHOS during hypoxia.
Reconstitution of MICU1 establish iCa2+ transients in hiPSCs
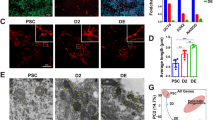
Intracellular Ca2+ (iCa2+) transients are maintained by the simultaneous interplay of counteracting processes, the on and off mechanisms45,46. iCa2+ oscillations play a pivotal role in intracellular signaling and is also a control oscillatory regime for many cellular processes. Recent studies have shown hiPSCs to have functional IP3-regulated intracellular Ca2+ stores and store-operated Ca2+ entry (SoCE) for refiling of their stores. They are sensitive to stimulation with ATP, histamine, and platelet-derived growth factor. It has long been studied and accepted that a fine regulation of iCa2+ transients are maintained by mitochondria, that has a unique ability to decode and transduce Ca2+ signals into an energy output (ATP). Since we observed low abundance of MICU1 in hiPSCs (Fig. 1i, 4a, b, Supplementary Fig. 3a), we next asked whether the loss of MICU1 impairs mCa2+ buffering and thus alters the iCa2+ oscillatory phenotype. Both control fibroblasts, hiPSCs (SeV3, SeV5, and SV20), and hESCs were loaded with Fluo-4 and spontaneous iCa2+ oscillations were measured. Fibroblasts showed an oscillatory phenotype, while hiPSCs and hESCs exhibited no iCa2+ oscillations implicating perturbed iCa2+ buffering possibly by mitochondria (Fig. 4c, e and Supplementary Fig. 3b and 3c). To substantiate the loss of MICU1 as a cause for perturbed iCa2+ transients, we reconstituted MICU1 either by ectopic expression or by knocking down Foxd1 (Fig. 4a, b, Supplementary Fig. 3a) in hiPSCs. The reconstitution of MICU1 re-established the iCa2+ oscillatory phenotype corroborating MICU1’s function in maintaining cellular Ca2+ transients (Fig. 4c, e and Supplementary Fig. 3b and 3c).

Reconstitution of MICU1 in hiPSCs establishes iCa2+ transients and modulates lineage-specific cell differentiation. a Representative western blot for lysates from hiPSCs with and without MICU1 expression. The western blots were probed with antibody specific for MICU1, MCU, ATP5A, Cytochrome C, Foxd1, Tom20, and β-actin. b Quantification of relative protein abundance of Foxd1 and MICU1. c Traces of spontaneous cytosolic Ca2+ oscillations in control fibroblasts and hiPSCs with and without MICU1 expression. Intact cells were loaded with fluo4 and spontaneous Ca2+ oscillations were observed at 488 nm. d Quantification of peak fluo-4 fluorescence. e Quantification of the frequency of cCa2+oscillations. f Representative western blot for lysates from hiPSCs induced for lineage specific differentiation for the specified days with and without MICU1 expression. The western blots were probed with antibody specific for MICU1, MCU, Foxd1, and β-actin. g Traces of spontaneous cCa2+ oscillations in hiPSCs upon induction of lineage specific differentiation with and without MICU1 expression. Ca2+ transients were measured at different days of differentiation: Day 3, 4, and 5 for endoderm and mesoderm; Day 5, 6, and 7 for ectoderm. h Quantification of peak fluo-4 fluorescence. i Quantification of the frequency of cCa2+oscillations. Data indicate quantified peak fluorescence/frequency of oscillation. Mean ± SEM is represented in the figures; **P < 0.01, *P < 0.05, ***P < 0.001; n = 3–4 (One-Way Anova) individual experiments. Dots represents mean data from group of single cells
iCa2+ transients modulate cell differentiation
The link between the spatiotemporal pattern of iCa2+ transients and expression of developmental genes is a key phenomenon, and that the frequency as well as the amplitude of Ca2+ transients is important for the regulation of gene expression47. The features of Ca2+-dependent expression are not only restricted to cells but also crucial in embryo development. During Xenopus embryonic development, the expression of early neural genes ZiC3 and geminin were downregulated by Ca2+ transients in a spatiotemporal pattern which was confirmed by the blockade of l-type Ca2+ channels48,49. Given the restoration of the Ca2+ oscillatory phenotype upon MICU1 expression in hiPSCs, we asked whether reconstitution of MICU1 is accompanied by a switch in hiPSCs to a more differentiated phenotype. To this end, we mapped the proteomes of fibroblasts (GM942), and hiPSCs with and without MICU1 expression (SeV3, and SeV3 + AdMICU1) using label-free proteomics (GeLC-MS/MS). 6220 proteins were identified when analyzing the fold-change expression ratios comparing fibroblasts and hiPSCs. Of these, 662 proteins were expressed exclusively in fibroblasts and control hiPSCs, 985 in fibroblasts and hiPSCs expressing MICU1, and 1202 proteins in common (Supplementary Fig. 3d). This data infers the differential expression of a relatively small number of proteins in cells that express MICU1. Gene Ontology (GO) enrichment analysis revealed that cell cycle and related processes including DNA replication, nucleotide biosynthesis, cell proliferation, organ development, and function genes were overrepresented in MICU1 reconstituted hiPSCs (Supplementary Fig. 3e). Also, the mapping of fibroblasts and hiPSCs revealed enrichment of developmental markers (embryonic, tissue, and cardiovascular) by MICU1 expression (Supplementary Fig. 3f and 3g). Indeed, protein-protein interaction network analysis highlighted major Ca2+-cellular signaling pathways including the MAPK and NF-κB signaling proteins being expressed at elevated levels in hiPSCs expressing MICU1 (Supplementary Fig. 3h). Additionally, the epithelial to mesenchymal transition (EMT) pathway proteins, N-Cadherin, Vimentin, MMPs, nuclear β-catenin, and fibronectin were enriched in hiPSCs expressing MICU1 (Supplementary Fig. 3i and 3j). Of the transcriptional factors involved in EMT, ZEB was highly elevated in hiPSCs expressing MICU1. Because, hiPSCs share similarity to ESCs and undergo same trend of EMT during differentiation, we asked whether the ectopic expression of MICU1 change the pluripotent nature of hiPSCs. Surprisingly, MICU1 expression did not alter the mRNA levels of OCT4, and NANOG (Supplementary Fig. 3k) when cells were cultured under pluripotency-maintaining conditions. EMT transcript Zeb2 was indicated to modulate cell-fate decision during the transformation of ESCs to primary germ layer differentiation50,51. Like gastrulation where most of the cells undergo EMT, we expect ectopic expression of MICU1 in hiPSCs to result in a hybrid EMT that enable cells to identify an external signal and acquire maximum cellular transition. It has been known that a fully transformed mesenchymal fate to be associated with mesoderm development, but neural crest delamination in the ectoderm or partial endoderm formation are also consequences of EMT52,53.
Having observed the pluripotency markers to remain unaltered in hiPSCs expressing MICU1 and EMT pathway proteins to be over-represented, we asked whether ectopic expression of MICU1 regulate iPSC-derived early lineage specification. SV20 hiPSCs cultured in pluripotency-maintaining media were infected with AdMICU1. 48 h post-infection, cells were replated and induced for early lineage commitment by separately applying cytokines that specify each of the three germ layers: endoderm, mesoderm, and ectoderm. While the ectopic expression of MICU1 (Fig. 4f, Supplementary Fig. 4a) significantly upregulated genes of the endoderm lineage (Supplementary Fig. 4b), but had little or no effect on mesoderm or ectoderm genes (Supplementary Fig. 4c and 4d).
We next asked whether MICU1 expression is restored in hiPSCs as they differentiate into different cell lineages. The differentiation of cells was accompanied by the up-regulation of MICU1 protein (Fig. 4f, Supplementary Fig. 4a) and restored the iCa2+ transients which is a prerequisite for cell differentiation. Spontaneous Ca2+ oscillation was observed in hiPSCs upon induction of lineage specific differentiation, with observable stimulation in endodermal and mesodermal lineages (Fig. 4g, i). Next, we asked whether ectopic expression of MICU1 cause a leftward shift (early) in the induction of Ca2+ oscillation and could explain the early lineage selection. The ectopic expression of MICU1 resulted in a significant difference in the amplitude of Ca2+ oscillation in mesoderm and endoderm, while the ectodermal Ca2+ oscillation remain unaffected (Fig. 4g, i).
Expression of MICU1 promotes cardiomyocyte maturation
EMT has been identified as one of the first steps of cardiac differentiation52,53,54 with Ca2+ as an essential signal integrator for differentiation of ESCs to functional cardiomyocytes. Because, we observed EMT pathway proteins to be over-represented during ectopic expression of MICU1, to further evaluate the role of MICU1 in cardiomyocyte differentiation and maturation, we generated hiPSCs-derived cardiomyocytes (hiPSC-CMs), using established protocol which results in immature myocytes with mixed subtypes55,56 and ectopically expressed MICU1 in these cells for 5 days. Not surprisingly, hiPSCs-CMs had low levels of MICU1 (Fig. 5a, b, Supplementary Fig. 5a) which correlated with the MICU1 levels of rat neonatal myocytes (Supplementary Fig. 5b-5d). In effect the differentiation-day 24 hiPSCs-CMs exhibited immature spontaneous cCa2+ transients (Fig. 5c, e) resembling neonatal (P0) cardiomyocytes (Supplementary Fig. 5f and 5g). Whereas the recorded transients were consistent in amplitude and frequency in the hiPSCs-CMs expressing MICU1 (Fig. 5c-e). Analysis of spontaneous iCa2+ transients at day 46 show no observable changes between day 24 and 46 in the frequency and the amplitude. Conversely, ectopic expression of MICU1 show an increase in the frequency and amplitude of iCa2+ transients mimicking NRVMs isolated from neonates (Fig. 5c-e and Supplementary Fig. 5f and 5g). Surprisingly, quantitative qPCR analysis showed a more differentiated phenotype in MICU1 expressing d24 hiPSC-CMs, where expression of genes encoding cardiac transcription factor, Mef2c (Fig. 5f) and cardiac structural and contractile proteins: MYH6 and MYH7 (Fig. 5g) were significantly regulated when compared to hiPSCs-CMs. This cardiac transcript profile of the hiPSCs-CMs expressing MICU1 is indicative of maturing myocytes57,58,59.

MICU1 expression promotes iPSC-Cardiomyocyte maturation. a Representative western blot for lysates from SeV3 and SeV3-derived cardiomyocytes (SeV3-CMs) with and without MICU1 expression. The western blots were probed with antibody specific for MICU1, MCU, Foxd1, Tom20, and β-actin. b Quantification of the MICU1 mRNA abundance in SeV3 and day 24 SeV3-CMs. c Traces of spontaneous cytosolic Ca2+ oscillations in SeV3-CMs with and without MICU1 expression at day 24 and day 46 post-differentiation. d Quantification of peak fluo-4 fluorescence. e Quantification of the frequency of cCa2+oscillations. Data indicate Mean ± SEM; **P < 0.001, ***P < 0.001; n = 3–4 independent experiments. Dots represents data from group of single cells. f Quantification of mRNA abundance of the early cardiac transcription factor Mef2c in SeV3-CMs with or without MICU1 expression at day 24 of differentiation. g Quantification of mRNA abundance of the cardiac contractility proteins, MYH6, and MYH7 in SeV3-CMs with or without MICU1 expression at day 24 of differentiation. Data indicate Mean ± SEM; *P < 0.05, ***P < 0.001; n = 3–4. h, j Measurement of oxygen consumption rate (OCR) in SeV3-CMs with or without MICU1 expression using glucose (glucose oxidation) (h) or carnitine (fatty acid oxidation) (j) as substrate (glucose oxidation). After basal OCR measurement, oligomycin (A), FCCP (B), and rotenone + Antimycin A (C) were added as indicated. h, j Representative traces of OCR in SeV3-CMs. i, k Quantification of basal and maximal OCR, and spare capacity in SeV3-CMs. Data indicate Mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.0001; n = 3
Despite the very high energy demand, the adult heart has essentially no energy reserves and continuously produce ATP to meet the work load. Mitochondrial oxidative phosphorylation provides over 90% of the energy demand with fatty acid being the major source of oxidative metabolism. In contrast to the mature cardiomyocyte, energy metabolism during myocyte development differ quite dramatically. In the early embryonic stage, the cardiomyocyte precursor seems to be dependent on glycolysis as a source of energy. However successful transition of the ESCs to cardiomyocytes requires a switch from glycolytic metabolism to mitochondrial oxidative phosphorylation. Maintaining high glycolytic rates facilitate the proliferative state of the developing cardiomyocyte, whereas increase in mitochondrial oxidative capacity marks a more terminally differentiated myocyte. To determine if modulation of MICU1 levels play a role in metabolic phenotype, OCR was measured in hiPSCs-CMs with or without ectopic expression of MICU1 using glucose (Fig. 5h) or carnitine (Fig. 5j) as substrate. The ectopic expression of MICU1 markedly increased the contribution of fatty acid oxidation to overall cardiac energetics of hiPSCs-CMs (Fig. 5j, k) approaching levels observed in myocytes isolated from P5 (Supplementary Fig. 5h-5l). The increase in fatty acid oxidation was paralleled by the decrease in glucose oxidation (Fig. 5h, i). Our data provide an experimental confirmation that mitochondrial calcium signaling plays an important role in cardiac development and maturation.
Discussion
Since mitochondria establish a huge thermodynamic force, the ions and metabolites that are fluxed into mitochondria are used for metabolic cascades. In parallel, over 90% of the cellular O2 is consumed for ATP generation. Having established mCa2+ dynamics, oxygen consumption and energy production are well coupled sequential cellular processes evolutionarily, there must be several check points that could be sensed and controlled during the developmental processes. In the developing heart, the cardiac energetic demand increases with increasing cardiac performance required for embryonic growth60,61,62. The morphological changes occurring between an early embryonic stage (E9.5) and late embryonic stage (E13.5) in the heart coincide with significant changes in mechanical load, contractile force generation, and blood flow. These epigenetic factors including changes in intracardiac hemodynamics and mechanical stress could impact mitochondrial maturation and myocyte differentiation. A study establishes mechanical stretch to promote ES cell differentiation into myocytes63. Evidently, between E8.5 and E10 the pO2 in the mouse heart is lower than 10 mm Hg64. Because the embryos at this stage have lower O2 levels due to lack of placental-embryonic circulation64, we expect absence of MICU1 in early embryonic myocytes due its transcriptional regulation. We also expect a restoration of MICU1 in myocytes as the heart establishes the placental-embryonic circulation after E10. Such a controlled regulation of MICU1 expression should influence cellular differentiation and maturation. While MICU1 reconstitution promoted myocytes maturation, we did not observe a significant effect on early lineage differentiation. One possibility is that the inductive cues that drive the initial lineage commitment are very dominant and are overriding the impact of ectopic MICU1. Further analysis of the effect of MICU1 on lineage specification will need to be carried out to address this possibility. Nonetheless, our study is the first to establish the role of mitochondrial calcium signaling during development and cellular differentiation.
Our study also have implications on maturation of hiPSC-derived cardiomyocytes and other cell types. It is well appreciated that a common limitation of using hiPSC-derived cell lineages for disease modeling is the immaturity of the cells compared to their fully developed in vivo counterparts65,66. Functional maturation is a difficult obstacle to overcome in vitro despite long term culture and is the subject of intense investigation. Our finding that modulation of mCa2+ dynamics in immature cardiomyocytes promotes a more mature phenotype suggests that ectopic expression of MICU1 in newly formed iPSC-CMs may be utilized in combination with other favorable engineered microenvironments to generate more mature tissues in vitro. This approach may also potentially be applied to other metabolically active hiPSC-derived lineages such as neurons and hepatocytes.
An intriguing question that arises from our study is how embryonic myocytes compensate for the phenotypes of ablated MICU1 expression resulting in constitutive MCU activation, accumulation of matrix Ca2+ and elevated mROS generation. It is possible that there could be a compensatory mechanism that may result in the opening of the mitochondrial permeability transition pore (mPTP) in early embryonic heart67. Because MCU-mediated Ca2+ uptake (anterograde) and mROS generation (reterograde) are often interdependent phenomenon, we believe a complex role for both mCa2+ and redox signaling during embryonic development. In the early embryonic stage mCa2+-mediated mROS is essential for initial commitment of progenitor cells towards differentiated cell lineage through regulated mCa2+ and mROS. Having described how the hypoxic signals could be decoded as ion regulatory signals in maintaining developmental plasticity, thereby facilitating adaptation to stress conditions and to allow a well-integrated tissue repair response, future studies are warranted to better understand the role of MCU-mediated Ca2+ signaling during development.
Methods
Animals and cell culture
Wild type C57BL/6 mice were maintained in our animal facility in accordance and with approval from Institutional animal care and use committee. Tissues including brain, heart, and liver were harvested from deeply anesthetized mice with a mixture of xylazine (40 mg/kg) and ketamine (80 mg/kg). The harvested tissues were perfused with PBS and stored until further use.
Ventricular cardiomyocytes from embryos (E9.5, and E13.5) or neonates (P0, P1, and P4) heart (NRVMs) were prepared as previously described68. Embryonic and neonatal mycoytes were cultured in Ham’s F-10 supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/ml) at 37 °C in a 95% air/5% CO2 humidified atmosphere for 4 days. Human microvascular pulmonary endothelial cells (HPMVECs) was purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM-high glucose media supplemented with 20% FBS and penicillin/streptomycin (100 U/ml) at 37 °C in a 95% air/5% CO2 humidified atmosphere.
hiPSCs derivation and culture of hiPSCs/hESCs
Experiments involving the use of hESCs and hiPSCs have been approved by the University of Pennsylvania Embryonic Stem Cell Research Oversight Committee. Human primary fibroblasts (GM00942) from an apparently healthy 5 year old female Caucasian donor were obtained from Coriell Institute which operates the National Institute of General Medical Sciences (NIGMS) Human Genetic Cell Repository. The NIGMS Informed Consent was obtained from patient at time of sample submission. Coriell also obtained a Certificate of Confidentiality from the National Institutes of Health to help ensure patient’s privacy. iPSC cell lines iPS-GM942-SeV3 and iPS-GM942-SeV5 were derived from GM942 fibroblasts using Sendai Viral reprogramming vectors (CytotuneTM, Thermofisher Scientific) according to manufacturer’s instructions. The iPSC-SV20 cell line has been described previously37 and has been deposited at WiCell Institute under the name PENN123i-SV20. The hESC cell line H9 was purchased from WiCell Institute. hiPSCs/hESCs were maintained on Geltrex (Thermofisher Scientific)—coated plates in either mTeSR1 (Stem Cell Technologies) or iPS-Brew XF (Miltenyi Biotec) media at 37 °C in 5% CO2/5% O2/90% air humidified atmosphere. Cells were passaged every 4–5 days using StemMACS Passaing Solution (Miltenyi Biotec). For adenovirus transduction experiments, day 2–3 cells were infected for 3 h to overnight and harvested at various time points after infection.
hiPSC characterization
For flow cytometric analysis of hiPSCs, cells were dissociated into single cells with Accutase Enzyme Cell Detachment Medium (Innovative Cell Technologies) and stained with AlexaFluor 647 anti-human SSEA-4 (Biolegend) and PE mouse anti-human TRA-1-60 (Pharmingen) antibodies. Samples were analyzed using BD Accuri C6 (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, Ashland, OR). For karyotype analysis, iPSCs were cultured in T25 flasks on mouse feeders and live cultures were sent to Cell Line Genetics (Madison, WI) for cytogenetic analysis using G-banded Karyotyping. An average of 20 cells per cell line were analyzed for chromosome integrity.
Plasmids
Light Switch Promoter Reporter GoClone plasmid with MICU1 promoter sequence cloned (SwitchGear Genomics; S712264) and the corresponding control plasmids were used for luciferase assay. The mutant micu1Δ3027-297 were custom synthesized as gBlock gene fragments from IDT Inc. and subcloned into appropriate vectors for further use. The plasmid was confirmed by sequencing before use.
RNA interference
hiPSCs (0.5 × 106/well) grown on six-well plates were transfected with pools of 5 distinct siRNAs against Foxd1 (ON-TARGETplus SMARTpool, Dharmacon, USA) (50 nM) using RNAiMAX transfection reagent (Thermo Scientific). As controls, non-targeting siRNA duplexes (Dharmacon) were used.
Hypoxia/reoxygenation exposure
Freshly isolated NRVMs (rat neonates), hiPSCs, and HPMVEs were subjected to varying hours of hypoxia (5% O2–5% CO2) followed by reoxygenation (20% O2–5%CO2) to study the differential expression of MICU1 by hypoxia.
Western blotting
Cell extracts were prepared using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1 mM EDTA, 1% NP-40, protease inhibitor cocktail (Complete, Roche), and Halt phosphatase inhibitor cocktail (Thermo Scientific). Equal amounts of protein (25 μg/lane) were separated on 4–12% Bis-Tris polyacrylamide gel, transferred to a PVDF membrane using iBlot 2 PVDF regular stacks (Thermo Scientific), and probed with antibodies specific for MCU (1:500, Sigma Aldrich: HPA016480), MICU1 (1:500, in house), MCUR1 (1:500, In house), MICU2 (1:1000, Sigma Aldrich: HPA045511), Actin (1:5000, Santa Cruz: sc-47778), TOM20 (1: 2000, Santa Cruz: sc-17764), cytochrome c (1:5000, Santa Cruz: sc-13156), ATP5A (1:2000, Abcam: ab14748), and Foxd1 (1:500, GeneTex: GTX100271).
ChIP, quantitative PCR, and luciferase activity
hiPSCs/HPMVECs were grown to ~80 to 90% confluency in a 100-mm culture dish containing the appropriate growth media at normoxic/hypoxic environment. ChIP assay was performed using PierceTM Magnetic ChIP kit. In brief, DNA-protein complexes were crosslinked and immunoprecipitated using ChIP-validated antibodies against RNA polymerase II, Foxd1 and negative control IgG. Immune complexes were extracted and analyzed by quantitative PCR using primers (Supplementary Table 1) that flank specific regions in the MICU1 promoter. Values were normalized by input DNA. Results were depicted as the fold enrichment over basal expression.
HPMVECs (1 × 106) were transfected with luciferase reporter plasmids (4 μg) containing MICU1 promoter sequence with or without binding elements for Foxd1. After 48 h, cells were exposed to hypoxia (6 h) and reoxygenation (18 h). The cells were lysed, and luciferase activity was measured (LightSwitch Luciferase Assay Reagent) using a plate reader (Infinite M1000 PRO, Tecan).
Measurement of spontaneous cytoplasmic Ca2+ oscillation
Fibroblasts/hiPSCs/cell lineage induced hiPSCs/hiPSCs-CMs with or without MICU1 expression were grown on 0.2% gelatin coated 25 mm glass coverslips. The cells were loaded with 5 μM Fluo-4/AM (30 min) in extracellular medium at room temperature. Coverslips were mounted in an open perfusion microincubator (PDMI-2; Harvard Apparatus) and imaged. Spontaneous Ca2+ oscillations were recorded every 3 s (510 Meta; Carl Zeiss, Inc.) at 488 excitations using a ×63 oil objective. Images were analyzed and quantified by using ImageJ (NIH).
Simultaneous measurement of Ca2+ uptake and ΔΨ m
An equal number of cells (6 × 106 cells) were washed in Ca2+ free PBS, pH 7.4, resuspended and permeabilized with 40 µg/ml digitonin in 1.5 ml of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Hepes-Tris, pH 7.2 and 2 µM thapsigargin to block the SERCA pump. All the measurements were performed in the presence of 5 mM succinate. The simultaneous measurement of ΔΨm and extramitochondrial Ca2+ ([Ca2+]out) clearance as an indicator of mCa2+ uptake was achieved by loading the permeabilized cells with JC-1 (800 nM) and Fura2-FF (0.5 µM), respectively. Fluorescence was monitored in a multi-wavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI). [Ca2+]out is represented as the excitation ratio (340 nm/380 nm) of Fura2-FF/FA fluorescence and ΔΨm as the ratio of the fluorescence of J-aggregate (570 nm excitation/595 nm emission) and monomer (490 nm excitation/535 nm emission) forms of JC-1. A single Ca2+ bolus (1 μM) and mitochondrial uncoupler, CCCP (2 μM), were added at the indicated time points. All the experiments were performed at 37 °C with constant stirring5,7,11,26.
Proteomic analysis
SeV3 cells were infected with Adcontrol andAdMICU1 virus. Cell extracts were prepared using RIPA buffer. The whole cell lysate was identified by label-free proteomics (GeLC-MS/MS)26. Mass spectra processing was performed using Proteome Discoverer 1.4.0.288 (DBVersion:79). The generated de-isotoped peak list was submitted to an in-house Mascot server 2.2.07 for searching against the Swiss-Prot database (Release 2013_01, version 56.6, 538849 sequences). Mascot search parameters were set as follows: species, Homo sapiens (Homo sapiens = 20233 sequences); enzyme, trypsin with maximal 2 missed cleavage; fixed modification, cysteine carboxymethylation; variable modification, methionine oxidation; 10 ppm Da mass tolerance for precursor peptide ions; and 0.2 Da tolerance for MS/MS fragment ions. All peptide matches were filtered using an ion score cutoff of 20. Following identifications, MS spectra were uploaded to the LC-MS ProGenesis 4.1 software to generate list of proteins that are differentially expressed in SeV3 with ectopic expression of MICU1.
Measurement of mitochondrial superoxide
Mitochondrial superoxide was measured by using the mitochondrial oxygen free radical indicator MitoSOX Red (molecular probes; Invitrogen). Briefly, cells grown on 0.2% gelatin coated glass coverslips were loaded with 5 μM MitoSOX Red for 30 min at 37 °C, and coverslips were mounted in an open perfusion microincubator (PDMI-2; Harvard Apparatus) and imaged. Confocal (510 Meta; Carl Zeiss, Inc.) images were obtained at 561 nm excitation by using a ×63 oil objective. Images were analyzed, and the mean MitoSOX Red fluorescence was quantified by using Image J software (NIH).
Mitochondrial oxygen consumption rate
Intact SeV3-CMs/NRVMs were subjected to oxygen consumption rate (OCR) measurement at 37 °C in an XF96 extracellular flux analyzer (Seahorse Bioscience). SeV3-CMs/NRVMs (3 × 105) were sequentially challenged with oligomycin, FCCP, and rotenone plus antimycin A for measurement of OCR69. For measurement of glucose or fatty acid oxidation glucose or carnitine were used as substrates respectively.
Induction of lineage-specific differentiation of hiPSCs
Exponentially growing SV20 hiPSCs were infected with MICU1 adenovirus or control virus overnight. Cells were harvested 2 days later and replated on Geltrex-coated 24-well plates at 1 × 105 cells/well (ectoderm) and 4 × 105 cells/well (endoderm and mesoderm) and cultured overnight. STEMdiffTM Trilineage Differentiation Kit (Stem Cell Technologies) was used to induce specification of cells of the three germ layers. Differentiating cells were assayed at various time points of the differentiation time course (5 days for endoderm and mesoderm; 7 days for ectoderm).
Quantitative measurement of lineage-specific markers
Differentiating hiPSCs were harvested and total RNA was isolated using a PureLink RNA mini kit (Thermofisher Scientific). cDNA was generated using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Semi-quantitative real-time PCR was performed using SYBR green reagents and primers against lineage-specific marker genes (Supplementary Table 1).
Generation of hiPSCs-derived cardiomyocytes (iPSCs-CMs)
hiPSC-cardiomyocytes (iPSCs-CMs) were generated using standard protocols55,56. In general, cells used were >95% positive for cTnT staining by FACS. Differentiated cells were re-plated at 6 × 105 cells/well in 6-well plates for calcium transients measurement and 2 × 104 cells/well for Seahorse experiments and maintained in RPMI media plus B27 Supplement (cat#17504044, Thermofisher Scientific) before they were analyzed. Differentiated iPSCs-CMs were harvested and total RNA was isolated using a PureLink RNA mini kit (Thermofisher Scientific). cDNA was generated using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Semi-quantitative real-time PCR was performed using SYBR green reagents and primers against Mef2C, MYH7, and MYH6 (Supplementary Table 1).
Quantification and statistical analysis
Data from multiple experiments were quantified and expressed as mean ± SEM., and differences between groups were analyzed by using two-tailed paired Student’s t-test. Differences in the means among multiple data sets were analyzed using 1-way ANOVA with the Kruskal–Wallis test, followed by pairwise comparison using the Dunn test. P value <0.05 was considered significant in all analysis. The data were computed with either Graphpad Prism version 7.0 or SigmaPlot 11.0 Software.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The proteomics data that support the findings of this study are available from peptideatlas, with the accession code PASS01222.
References
Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 (2007).
Ngo, J. K., Pomatto, L. C. & Davies, K. J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 1, 258–264 (2013).
Lopez-Barneo, J., del Toro, R., Levitsky, K. L., Chiara, M. D. & Ortega-Saenz, P. Regulation of oxygen sensing by ion channels. J. Appl. Physiol. 96, 1187–1195; discussion 1170–1182 (2004).
Liu, J. C. et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16, 1561–1573 (2016).
Mallilankaraman, K. et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644 (2012).
Baughman, J. M. et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011).
Hoffman, N. E. et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 5, 1576–1588 (2013).
Hoffman, N. E. et al. SLC25A23 augments mitochondrial Ca(2)( + ) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 25, 936–947 (2014).
Kamer, K. J., & Mootha, V. K. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307 (2014).
Kevin Foskett, J., & Madesh, M. Regulation of the mitochondrial Ca(2 + ) uniporter by MICU1 and MICU2. Biochem. Biophys. Res. Commun. 449, 377–383 (2014).
Mallilankaraman, K. et al. MCUR1 is an essential component of mitochondrial Ca2 + uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 (2012).
Patron, M. et al. MICU1 and MICU2 finely tune the mitochondrial Ca2 + uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737 (2014).
Perocchi, F. et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2 + ) uptake. Nature 467, 291–296 (2010).
Plovanich, M. et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 8, e55785 (2013).
Sancak, Y. et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382 (2013).
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I. & Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 (2011).
Chaudhuri, D., Sancak, Y., Mootha, V. K. & Clapham, D. E. MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704 (2013).
Dong, Z. et al. Mitochondrial Ca2 + uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell 65, 1014–1028 e1017 (2017).
Kirichok, Y., Krapivinsky, G. & Clapham, D. E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 (2004).
Pan, X. et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472 (2013).
Vais, H. et al. MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell. Metab. 22, 533–535 (2015).
Lee, S. K. et al. Structural insights into mitochondrial calcium uniporter regulation by divalent cations. Cell Chem. Biol. 23, 1157–1169 (2016).
Csordas, G. et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987 (2013).
Payne, R., Hoff, H., Roskowski, A. & Foskett, J. K. MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Rep. 21, 3141–3154 (2017).
Vais, H. et al. EMRE is a matrix Ca(2 + ) sensor that governs gatekeeping of the mitochondrial Ca(2 + ) uniporter. Cell Rep. 14, 403–410 (2016).
Tomar, D. et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15, 1673–1685 (2016).
Logan, C. V. et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 46, 188–193 (2014).
Antony, A. N. et al. MICU1 regulation of mitochondrial Ca(2 + ) uptake dictates survival and tissue regeneration. Nat. Commun. 7, 10955 (2016).
Xiao, B., Coste, B., Mathur, J. & Patapoutian, A. Temperature-dependent STIM1 activation induces Ca(2) + influx and modulates gene expression. Nat. Chem. Biol. 7, 351–358 (2011).
Hawkins, B. J. et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 190, 391–405 (2010).
Soboloff, J., Madesh, M., & Gill, D. L. Sensing cellular stress through STIM proteins. Nat. Chem. Biol. 7, 488–492 (2011).
Jones, D. L. & Wagers, A. J. No place like home: anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 9, 11–21 (2008).
Li, L. & Xie, T. Stem cell niche: structure and function. Annu. Rev. Cell Dev. Biol. 21, 605–631 (2005).
Scadden, D. T. The stem-cell niche as an entity of action. Nature 441, 1075–1079 (2006).
Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4, 7–25 (1978).
Yin, T. & Li, L. The stem cell niches in bone. J. Clin. Invest. 116, 1195–1201 (2006).
Yang, W. et al. Generation of iPSCs as a pooled culture using magnetic activated cell sorting of newly reprogrammed cells. PLoS ONE 10, e0134995 (2015).
Koga, M. et al. Foxd1 is a mediator and indicator of the cell reprogramming process. Nat. Commun. 5, 3197 (2014).
Carter, M. E. & Brunet, A. FOXO transcription factors. Curr. Biol. 17, R113–R114 (2007).
Kloet, D. E. & Burgering, B. M. The PKB/FOXO switch in aging and cancer. Biochim. Biophys. Acta 1813, 1926–1937 (2011).
Zhang, Y., Gan, B., Liu, D. & Paik, J. H. FoxO family members in cancer. Cancer Biol. Ther. 12, 253–259 (2011).
Humphreys, B. D. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85–97 (2010).
Levinson, R. S. et al. Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development 132, 529–539 (2005).
Hatini, V., Huh, S. O., Herzlinger, D., Soares, V. C. & Lai, E. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 10, 1467–1478 (1996).
Nicholls, D. G. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem. J. 176, 463–474 (1978).
Williams, G. S., Boyman, L., Chikando, A. C., Khairallah, R. J. & Lederer, W. J. Mitochondrial calcium uptake. Proc. Natl Acad. Sci. USA 110, 10479–10486 (2013).
Berridge, M. J., Lipp, P. & Bootman, M. D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21 (2000).
Leclerc, C., Lee, M., Webb, S. E., Moreau, M. & Miller, A. L. Calcium transients triggered by planar signals induce the expression of ZIC3 gene during neural induction in Xenopus. Dev. Biol. 261, 381–390 (2003).
Leclerc, C., Webb, S. E., Daguzan, C., Moreau, M. & Miller, A. L. Imaging patterns of calcium transients during neural induction in Xenopus laevis embryos. J. Cell Sci. 113(Pt 19), 3519–3529 (2000).
Chng, Z., Teo, A., Pedersen, R. A. & Vallier, L. SIP1 mediates cell-fate decisions between neuroectoderm and mesendoderm in human pluripotent stem cells. Cell Stem Cell 6, 59–70 (2010).
Goossens, S. et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood 117, 5620–5630 (2011).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Thiery, J. P. & Sleeman, J. P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 (2006).
Kovacic, J. C., Mercader, N., Torres, M., Boehm, M. & Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation 125, 1795–1808 (2012).
Palpant, N. J. et al. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat. Protoc. 12, 15–31 (2017).
Laflamme, M. A. et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 25, 1015–1024 (2007).
Ronaldson-Bouchard, K. et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018).
Uosaki, H. et al. Transcriptional landscape of cardiomyocyte maturation. Cell Rep. 13, 1705–1716 (2015).
Yang, X., Pabon, L. & Murry, C. E. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 114, 511–523 (2014).
Conway, E. M. & Carmeliet, P. Cardiovascular biology: signalling silenced. Nature 425, 139–141 (2003).
Conway, S. J., Kruzynska-Frejtag, A., Kneer, P. L., Machnicki, M. & Koushik, S. V. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis 35, 1–21 (2003).
Porter, G. A. Jr. et al. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 31, 75–81 (2011).
Schmelter, M., Ateghang, B., Helmig, S., Wartenberg, M. & Sauer, H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J. 20, 1182–1184 (2006).
Krishnan, J. et al. Essential role of developmentally activated hypoxia-inducible factor 1alpha for cardiac morphogenesis and function. Circ. Res. 103, 1139–1146 (2008).
Roy-Chowdhury, N., Wang, X., Guha, C. & Roy-Chowdhury, J. Hepatocyte-like cells derived from induced pluripotent stem cells. Hepatol. Int. 11, 54–69 (2017).
Weick, J. P. Functional properties of human stem cell-derived neurons in health and disease. Stem Cells Int. 2016, 4190438 (2016).
Hom, J. R. et al. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 21, 469–478 (2011).
Pinson, A. in Cell Culture Techniques in Heart and Vessel Research (ed. Piper, H.M.) 20–35 (Springer-Verlag, Berlin-Heidelberg-New York, 1990).
Doonan, P. J. et al. LETM1-dependent mitochondrial Ca2 + flux modulates cellular bioenergetics and proliferation. FASEB J. 28, 4936–4949 (2014).
Acknowledgements
We thank Dr. Walter J. Koch for the reagents provided for the isolation of NRVMs. Daniel Martinez for tissue array sample processing, construction of the array, immunohistochemical analysis, and image acquisition. We also thank Juan Arbelaez of the iPSC Core Facility at the University of Pennsylvania for generating GM942 iPSC lines. Access to CHOP pathology core was supported by the 2017 MITO RAG CHOP core grant. S.S. and M.M., thank Dr. Marni Falk for the core grant. This research was funded by the National Institutes of Health (R01GM109882, R01HL086699, R01HL119306, R01HL142673 and 1S10RR027327 to M.M and by the Institute for Regenerative Medicine of the University of Pennsylvania to W.Y.; S.S. is supported by the NIH K99/R00 grant (1 K99 HL138268-01). E.C. and N.N., are supported by the AHA fellowships (18POST33990217, and 17PRE33660720). Z.D. is supported by China Scholarship Council (No.201403170252).
Author information
Authors and Affiliations
Contributions
S.S., Ka.C., K.N.S., Z.D., N.N., E.C., C.L. and M.M. performed and analyzed experiments involving biochemical, mitochondrial bioenergetics, molecular, and cellular experiments. K.J.S. and W.Y. characterized the GM942 iPSC lines, maintained and cultured hiPSCs, performed the tri-lineage differentiation, derived iPSCs-cardiomyocytes, and performed molecular biology experiments involving these cell types. J.I. and Ku.C. isolated and cultured myocytes from embryo and neonates. D.T. performed the western blot analysis for MICU1 expression in NRVMs (Supplementary Figs. 1a and 5a). C.B., V.N.S.G. and S.M. performed proteomic analysis of the hiPSCs. Y.T., embryo collection and tissue isolation. S.S. and S.R. cloned and generated luciferase plasmid constructs.; R.K. contributed to qPCR reagents and primers. S.S., W.Y. and M.M. conceived, designed, analyzed, and interpreted experimental data. S.S. and M.M. wrote the manuscript. Y.T. and W.Y. commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shanmughapriya, S., Tomar, D., Dong, Z. et al. FOXD1-dependent MICU1 expression regulates mitochondrial activity and cell differentiation. Nat Commun 9, 3449 (2018). https://doi.org/10.1038/s41467-018-05856-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-05856-4
This article is cited by
-
Mitochondrial calcium uniporter channel gatekeeping in cardiovascular disease
Nature Cardiovascular Research (2024)
-
Genetic lineage tracing identifies adaptive mechanisms of pancreatic islet β cells in various mouse models of diabetes with distinct age of initiation
Science China Life Sciences (2024)
-
The TRIM21-FOXD1-BCL-2 axis underlies hyperglycaemic cell death and diabetic tissue damage
Cell Death & Disease (2023)
-
Integrating spatial transcriptomics with single-cell transcriptomics reveals a spatiotemporal gene landscape of the human developing kidney
Cell & Bioscience (2022)
-
Cell type determination for cardiac differentiation occurs soon after seeding of human-induced pluripotent stem cells
Genome Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
