
Abstract
Uterine leiomyomas are common benign tumors of the myometrium. We performed a meta-analysis of two genome-wide association studies of leiomyoma in European women (16,595 cases and 523,330 controls), uncovering 21 variants at 16 loci that associate with the disease. Five variants were previously reported to confer risk of various malignant or benign tumors (rs78378222 in TP53, rs10069690 in TERT, rs1800057 and rs1801516 in ATM, and rs7907606 at OBFC1) and four signals are located at established risk loci for hormone-related traits (endometriosis and breast cancer) at 1q36.12 (CDC42/WNT4), 2p25.1 (GREB1), 20p12.3 (MCM8), and 6q26.2 (SYNE1/ESR1). Polygenic score for leiomyoma, computed using UKB data, is significantly correlated with risk of cancer in the Icelandic population. Functional annotation suggests that the non-coding risk variants affect multiple genes, including ESR1. Our results provide insights into the genetic background of leiomyoma that are shared by other benign and malignant tumors and highlight the role of hormones in leiomyoma growth.
Similar content being viewed by others

Introduction
Uterine leiomyomas, sometimes referred to as fibroids, are benign tumors that arise in the smooth muscle cells of the uterine wall1. Leiomyomas represent the most common pelvic tumors in women with prevalence by the age 50, above 80% for African–American women, and nearly 70% for Caucasian women2. Although leiomyomas are non-malignant and in majority of cases not symptomatic, about 20–25% of cases cause problems that warrant treatment. These symptoms include dysmenorrhea, pelvic pain, abnormal bleeding, infertility, and complications during pregnancy3. Leiomyomas are estrogen responsive and many of the non-surgical therapeutic options involve some form of hormonal manipulation; however, in many cases, the tumors recur when therapy is discontinued1. Leiomyomas are the most common indication for hysterectomy and the second most common cause of surgical intervention for women after cesarean section, with over 600,000 hysterectomies performed per year in the USA4.
The biological mechanisms leading to the development of uterine leiomyomas are not well understood. Risk factors include age, obesity, increased levels of estrogens, hypertension, parity, and race, with prevalence in African–American women being considerably higher than that in European–American women2. Genetic factors are also implicated in leiomyoma formation: First-degree relatives of affected women have a 2.5-fold greater risk of developing the condition than the population average5 and the concordance among monozygotic twins is almost twice that of dizygotic twins6. A genome-wide association study (GWAS) in Japanese women found variants at three loci that associate with uterine leiomyoma, at 22q13.1 (TNRC6B), 11p15.5 (BET1L), and 10q24.33 (OBFC1)7. The associations between the disease and the first two loci have been confirmed in European women8. Finally, a GWAS of leiomyoma risk among African–American women yielded a locus on 22q13.1 (CYTH4)9.
To gain insights into the genetic causes of uterine leiomyomas, we performed a meta-analysis of leiomyoma in Europeans, using GWAS data on 16,595 cases and 523,330 controls from Iceland and the UK, identifying 21 variants at 16 loci that associate with the disease.
Results
Association analysis
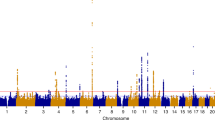
We combined the results of two GWAS‘ of uterine leiomyoma, one from Iceland and the other from the UK Biobank (UKB), using a total of 16,595 cases and 523,330 controls of confirmed Europen descent. The Icelandic GWAS consists of 6728 hospital-based, histologically confirmed leiomyoma cases and 124,542 controls, whereas the UKB leiomyoma dataset is based on 9867 hospital-based cases and 398,788 controls. A Manhattan plot of the meta-analysis results is shown in Fig. 1. As the probability of a variant impacting a phenotype differs between the functional annotation classes10, we applied genome-wide significance thresholds using a weighted Bonferroni procedure that takes this prior probability into account and corrects for all 43,421,991 variants tested (Supplementary Data 1, Methods). Using this scheme, a total of 412 variants at 16 loci reach the threshold of genome-wide significance (Supplementary Data 2). We applied conditional analysis to search for distinct signals at each locus and find additional variants at five loci that associate with leiomyoma independent of the main signal (with Pcond < 10−6); three of the secondary signals reach genome-wide significance. The 19 genome-wide significant markers and two significant secondary signals are listed in Table 1 and Supplementary Data 3, locus plots for all the markers are presented in Supplementary Fig. 1, and all association signals with P value < 5.0 × 10−8 are listed in Supplementary Data 4. We also tested 21 markers for association with 20 different tumor types using information on 42,331 cancer cases from the Icelandic Cancer Registry and several phenotypes that are strongly affected by estrogen exposure, i.e., endometriosis (1,857 cases), bone mineral density (BMD, 28,900 individuals), and age at menopause (10,216 individuals) (Supplementary Data 5 and 6).

Manhattan plot of the association results of meta-analysis of leiomyoma of the uterus. The Manhattan plot shows variants with P-value < 1.0 × 10−3 in a meta-analysis of GWAS data from 20,621 leiomyoma patients and 280,541 controls of European ancestry. Shown are negative log10-transformed P values (y-axis) over 22 autosomes (x-axis). Red font color indicates leiomyoma loci previously reported in the Japanese population
Variants at 16 loci associate with leiomyoma
Examination of the meta-analysis results suggests that at least two genetic pathways play a role in the development of leiomyomas; one linked to tumorigenesis and the other linked to hormone-related traits (Table 1, Supplementary Data 3). The most significant association with leiomyoma is with a low-frequency 3’UTR variant in TP53, rs7837822_G (P = 4.03 × 10−37, meta-analysis of logistic regression, OR = 1.74). The same allele of this variant was previously reported to associate with increased risk of several malignant and benign tumor types, including basal cell skin cancer, glioma, prostate cancer, neuroblastoma, and colorectal adenoma11,12,13. Four additional leiomyoma variants in our meta-analysis, one at the TERT locus, two in ATM, and one at the OBFC1 locus, have previously been associated with cancer risk. At the TERT locus, rs10069690_T was previously reported to increase the risk of thyroid cancer14, estrogen and progesterone receptor-negative breast cancer15,16, CLL17, and glioma18 and decrease the risk of testicular, prostate, bladder, and pancreatic cancers18. Two uncorrelated (r2 = 0.007) missense variants in the ATM gene, rs1801516 (Asp1853Asn) and rs1800057 (Pro1054Arg), independently associate with leiomyoma. rs1801516_A was previously reported to associate with decreased melanoma risk19 and response to chemotherapy and radiotherapy20,21. This variant shows significant association with increased risk of squamous cell skin cancer in Iceland (P = 4.35 × 10−5, logistic regression, OR = 1.21) after correction for the number of phenotypes tested (P < 0.05/29 = 1.72 × 10−3), the direction of effect being opposite to the reported melanoma effect (Supplementary Data 6). The other leiomyoma missense variant in ATM, rs1800057_G, has been linked to increased risk of chronic lymphocytic leukemia22, prostate cancer23, and renal cell carcinoma24. Finally, at the OBFC1 locus, the strongest leiomyoma risk variant, rs7907606_G, has previously been associated with the risk of basal cell carcinoma25 and is strongly correlated with variants that have been reported for risk of melanoma26, adenocarcinoma of the lung27, thyroid cancer14, renal cell carcinoma24, serous ovarian cancer28, glioma29, and CLL30, as well as to telomere length31.
Several of the leiomyoma variants and loci emerging from our meta-analysis have been linked to hormone-related traits. Variants at the CDC4/WNT4 locus have been associated with endometriosis, BMD, bone size, and ovarian cancer32,33,34,35. The leiomyoma variant rs10917151 at this locus is correlated with the strongest reported endometriosis variant (r2 = 1), as well as the ovarian cancer variant (r2 = 0.97), and one of the BMD variants (r2 = 0.81) (Table 1, Supplementary Data 3). The missense variant in MCM8, rs16991615_A, that associates with increased risk of leiomyoma in our study, also associates with delayed onset of menopause36, breast cancer37, and with increased BMD in Iceland (Supplementary Data 6). At the SYNE1/ESR1 locus, the strongest leiomyoma variant, rs58415480, is correlated with a reported endometriosis variant rs71575922 (r2 = 0.94)32. Notably, the leiomyoma variant is not correlated to any of the six independent breast cancer signals reported in European populations at this locus, represented by the variants rs9397435/rs9397437, rs3757322, rs851984, rs9918437, rs78796841, and rs274765237,38,39 (all r2 < 0.03 in CEU). Two variants at the GREB1 locus, the missense variant rs10929757 (Asn77Thr) and rs148143917 (upstream variant), associate with leiomyoma. Two association signals for endometriosis have been reported at this locus32, but neither are strongly correlated with the leiomyoma variants (Table 1). The leiomyoma variant rs10929757 (Asn77Thr) associates with endometriosis, whereas only one of the endometriosis variants, rs77294520, associates with leiomyoma in the Icelandic data (Supplementary Data 6 and 7). Two additional variants have potential links to hormone metabolism. First, rs2202282 at 4q13.3 is located in a cluster of sulfotransferase genes, including SULT1E1, which catalyzes the sulfation of estrogens40. Second, the low-frequency variant rs765333492 (SCFD2, MAF = 0.3%) found only in the Icelandic dataset also shows suggestive association with endometriosis in Iceland (P = 0.018, logistic regression, OR = 1.92) (Supplementary Data 6).
Variants at three loci previously reported for uterine fibroids in Japanese women7, 22q13.1 (TNRC6B), 11p15.5 (BET1L), and 10q24.33 (OBFC1), reached genome-wide significance in our meta-analysis. Our top TNCRB6 variant is strongly correlated with the variant reported in Japanese women (r2 = 0.97). At the BET1L locus, we found two distinct signals, one of which is fully correlated with the reported BET1L variant in Japanese women (Table 1, Supplementary Data 3). At the OBFC1 locus, the previously discussed cancer-associated variant rs7907606 is not correlated with the Japanese leiomyoma variant reported (rs7913069, P = 0.98, meta-analysis of logistic regression, OR = 1.0, r2 = 0.001 in Europeans and 0.014 in Japanese). To further analyze the Japanese signal in our data, we tested 278 variants that correlate with rs7913069 (with r2 > 0.1) in the Japanese population for association with leiomyoma in our dataset (Supplementary Data 8). The lowest P-value observed is 0.00015 for rs78381949, a variant that correlates with r2 = 0.23 with rs7913069 in East-Asians, but has very little correlation with rs7907606 (r2 = 0.04) in Caucasians. It is thus unlikely that rs7907606 and rs7913069 are tagging the same association signal. Finally, the variant at the CYTH4 locus identified in African–American women9, rs739187, does not associate with leiomyoma in our data (P = 0.51, meta-analysis of logistic regression, OR = 1.01). rs739187 is about 3 Mb away from the leiomyoma variant rs12484951 (TNRC6B) and the 2 variants are not correlated either in Europeans (r2 = 0.0064) or African–Americans (r2 = 0.017).
Overlap of leiomyoma, endometriosis, and endometrial cancer
Although a large fraction of hysterectomies are due to leiomyoma, hysterectomies performed for other clinical conditions such as endometriosis, uterine prolapse, and cancer may also lead to diagnosis of leiomyoma. We assessed whether the leiomyoma signals are driven by endometriosis cases within the leiomyoma group. To this end, we removed all known endometriosis cases from the Icelandic leiomyoma sample set and compared the association results with the results obtained for the full Icelandic leiomyoma group. The effects of leiomyoma associations were not diminished for any of the variants (Supplementary Data 6). We note that since we only have information on surgically diagnosed endometriosis cases, it is possible that there may be some undiagnosed cases left in the leiomyoma group. We also assessed the association of all 19 reported endometriosis variants32 with the endometriosis-excluded leiomyoma group. Only rs12037376 (CDC42/WNT4), rs71575922 (SYNE1/ESR1), and rs77294520 (GREB1) associate with leiomyoma after correction for the number of tests (P < 0.05/19 = 2.6 × 10−3), with all three variants showing the same direction of effect for endometriosis and leiomyoma (logistic regression, Supplementary Data 7).
We tested if the leiomyoma variants associate with endometrial cancer, using 2462 endometrial cancer cases (1009 from Iceland and 1453 from UKB) and 336,978 controls (218,565 from Iceland and 118,413 from UKB) (Supplementary Data 9). Only rs10917151 (CDC42/WNT4) associates with endometrial cancer (P = 4.5 × 10−4, logistic regression, OR 1.14) after correcting for the number of tests (P = 0.05/21 = 0.0024). We also tested the association between leiomyoma and seven variants reported in GWAS studies of endometrial cancer41. None of the endometrial cancer variants associate with leiomyoma (Supplementary Data 10).
Functional annotation of non-coding leiomyoma variants
To assess the functional role of the non-coding leiomyoma variants, we annotated the lead non-coding variants along with correlated variants (r2 > 0.8, N = 438), using data on regulatory regions in uterine tissue from the Encode project (www.encodeproject.org). Specifically, we focused on acetylation of lysine K27 of histone H3 (H3K27ac) and open chromatin regions (DNase Hypersensitivity Sites; DHS). This analysis identified 51 out of 438 variants that intersected with H3K27ac marked regions (Supplementary Data 11). In addition, 14 variants were found within DHS sites, of which 10 are also within H3K27ac regions. To search for gene targets of variants in regulatory regions, we analyzed chromatin interaction maps and identified 85 potential target genes that associate with one or more of the candidate regulatory variants (Supplementary Data 11, Fig. 2, Supplementary Fig. 2). Genes whose transcription start sites (TSS) are found within 1 kb of a regulatory variant are listed (Supplementary Data 11).

Leiomyoma risk variants intersecting with regulatory regions and their candidate target genes. Two loci on chromosomes 6 (upper panel) and 13 (lower panel) are shown as examples. Acetylation of lysine residue K27 of histone H3 (H3K27ac) and open chromatin (DHS, DNase Hypersensitivity Sites) in the uterus samples are shown as tick marks at the top. At chromosome 6, the lead variant rs58415480 is shown along with the only other variant in the same LD class (r2 > 0.8), rs71575922. At chromosome 13, rs117245733 is in an LD class by itself, i.e., does not have strongly correlated markers. Candidate target genes were identified by the analysis of available chromatin interaction maps, shown on the figure as orange arcs. The shaded regions represent regions found in contact with the regulatory variants
The variant rs58415480 at 6q25.2 (SYN1/ESR1) is in strong LD with only one other variant, rs71575922 (r2 = 0.96) and both reside in a regulatory region in the uterine tissue. According to chromosomal interaction data, rs58415480 and rs71575922 are candidate regulators of seven target genes including ESR1, which is important in the growth of leiomyomas (Fig. 2, upper panel). The variant on chromosome 13, rs117245733, does not have a strongly correlated marker. This variant resides in a region densely marked by H3K27ac in the uterine tissue, indicative of regulatory activity (Fig. 2, lower panel). Out of the eleven candidate target genes regulated by rs117245733, the FOXO1 gene is notable, as its expression in primary human endometrial stromal cells is regulated by ESR142 and it is a direct protein–protein interaction partner of ESR1 in breast cancer cells43. The secondary signal at 13q14.11, rs7986407, also targets FOXO1 (Supplementary Fig. 2). At 11p13, the well-known tumor suppressor gene WT1 interacts with several variants, with rs10835884, rs11031716, and rs11828433 representing the top-ranking causal variants (Supplementary Data 11, Supplementary Fig. 2). According to the Human Protein Atlas (www.proteinatlas.org), WT1 expression is limited to smooth muscle cells of the uterus, in addition to Fallopian tube, testis, and kidney tissue.
Heritability and polygenic score (PGS)
We used LD score regression44 to estimate the SNP heritability of leiomyoma. Using the Icelandic population, we estimate the SNP heritability to be 13% (95% CI 4–22%). We estimate the sibling recurrence risk in Iceland to be 1.74 (95% CI 1.63, 1.85, P < 0.0001) and find that the proportion of sibling recurrence risk for leiomyoma explained by the 21 variants is 10.8%.
The sharing of susceptibility loci of leiomyoma with different tumor types on one hand and hormone-related traits on the other suggests that leiomyomas are genetically correlated with these phenotype groups. We used a polygenic score (PGS) to map the genetic correlation between leiomyoma and 28 selected tumor/hormone-related phenotypes (Table 2). To avoid confounding, risk alleles, P-values, and effect estimates were extracted from the UKB study on leiomyoma and used to calculate a standardized PGS for the genotyped cases in the Icelandic dataset. As expected, the PGS associates with leiomyoma in the Icelandic dataset (OR = 1.25, P = 3.2 × 10−55), which may be interpreted such that one standard deviation increase in the PGS leads to a 25% increased chance of leiomyoma. After correction for the number of phenotypes tested (P < 0.05/29 = 1.72 × 10−3), the PGS was also significantly correlated with the risk of being diagnosed with cancer (all cancer types together), thyroid cancer and prostate cancer (likelihood ratio test, Table 2).
Discussion
Results from this meta-analysis highlight two distinct genetic pathways that play a role in the development of leiomyomas; one of genes linked to tumorigenesis (TP53, TERT, ATM, and OBFC1) and the other of variants and loci linked to hormone metabolism (CDC42/WNT4, GREB1, MCM8, and SYN1/ESR1). It is tempting to speculate that the tumor-associated variants provide genetic background for leiomyoma development, whereas the hormone-associated variants enhance the growth of the tumors, causing them to become symptomatic. A common genetic etiology between cancer and leiomyoma is further suggested by the leiomyoma PGS that associates with several cancers, including a phenotype consisting of individuals affected by any type of cancer. We did not have the power to test the association of the variants with leiomyosarcoma—the malignant tumor originating in the myometrium— because of the rarity of this tumor type (44 cases in this study).
The association between leiomyoma and the 3′UTR variant in TP53 is of particular interest. Contrary to most pathogenic mutations in TP53, the variant does not change the amino acid sequence of the protein, but instead disrupts normal polyadenylation and reduces the abundance of mRNA11. This results in a cancer risk spectrum quite distinct from the Li–Fraumeni tumor syndrome. Notably, the variant predisposes to some non-lethal tumor types such as basal cell skin cancer and colorectal adenomas, but not colorectal cancer11. The association between missense variants in ATM and leiomyoma is also unexpected. We have previously shown that loss-of-function mutations in ATM confer high risk of gastric cancer in the Icelandic population45. These same loss-of-function variants do not associate with leiomyoma risk, suggesting that the missense variants may have a mild effect on some specific functions of ATM in uterine tissue. Here, we report for the first time an association between the missense variant Asp1853Asn in ATM (rs1801516) and increased risk of squamous cell skin cancer.
In conclusion, we report the first GWAS of leiomyoma in European populations. Our results show a genetic overlap between leiomyoma and various cancers, and highlight the role of estrogen in tumor growth.
Methods
Datasets
The meta-analysis combined the results of two GWAS of uterine leiomyoma. The Icelandic dataset consisted of 6,728 histologically confirmed cases of leiomyoma of the uterus (ICD codes ICD10 D25 and ICD9 218 and 219) diagnosed at the Department of Pathology, Landspitali University Hospital, compared to 124,542 female controls. The UK Biobank (UKB) leiomyoma dataset consisted of 9,867 cases with ICD10 D25 compared to 398,788 controls of both sexes. The UKB phenotype is based on diagnoses codes recorded for a participant during all visits to a hospital. Given the high prevalence of leiomyoma, it is clear that a fraction of the controls in both datasets will have undiagnosed leiomyoma. This under-diagnosis of the controls weakens the power of the GWAS, but should not lead to false-positive results.
The Icelandic cancer case–control datasets used were described previously46. The primary source of information on cancer is the Icelandic Cancer Registry (ICR), which has registered all solid, non-cutaneous cancers in the entire population of Iceland since 195547. Registration of skin and hematological cancers started in 1980s. ICR registration is based on the ICD system and includes information on histology (systemized nomenclature of medicine, SNOMED). Over 94% of diagnoses in the ICR have histological confirmation. The endometriosis32, bone phenotype48, and age at menopause49 datasets have been described previously. For sex-specific phenotypes in Iceland, only controls for the relevant sex were included in the analysis.
The study was approved by the Icelandic National Bioethics Committee (reference number. 17–124). Written informed consent was obtained from all genotyped subjects.
Genotyping
The Icelandic part of the study is based on the genotypes of 150,656 Icelanders, who have been genotyped using Illumina SNP chips along with whole-genome sequence (WGS) data from 15,220 Icelanders. The genotypes for the 150,656 individuals were long-range phased and imputed using information from the WGS individuals50. Using genealogic information, the sequence variants were imputed into 282,894 relatives of the genotyped individuals to further increase the sample size for association analysis and to increase the power to detect the associations51. A total of 34.5 million variants were used in the Icelandic GWAS. Datasets were constructed as a part of disease association efforts at deCODE genetics. For further information regarding genotyping and imputation, we refer to Gudbjartsson et al.52
Genotyping of UKB samples was performed using a custom-made Affymetrix chip, UK BiLEVE Axiom53, and with the Affymetrix UK Biobank Axiom array54. Imputation was performed by the Wellcome Trust Centre for Human Genetics, using the Haplotype Reference Consortium (HRC) and the UK10K haplotype resources55. This yields a total of 96 million imputed variants, however only 27 million variants imputed using the HRC reference set passed the quality filters used in our study.
GWAS and meta-analysis
Logistic regression assuming an additive model was used to test for association between variants and disease, treating disease status as the response and expected genotype counts from imputation as covariates, and using the likelihood ratio test to compute P-values. The association analysis for both the Icelandic and UKB datasets was done using a software developed at deCODE genetics52. For the Icelandic study group, patients and controls are matched on gender and age at diagnosis or age at inclusion, and information on county of origin within Iceland are included as covariates to adjust for possible population stratification. For the UK datasets, cases and controls are restricted to individuals of genetically confirmed white British origin, and 40 principle components are included in the analysis to adjust for population substructure. To account for inflation in test statistics due to cryptic relatedness and stratification, we applied the method of linkage disequilibrium (LD) score regression44 to estimate the inflation in test statistics and adjusted all P values accordingly. The estimated correction factor for Leiomyoma based on LD score regression was 1.13 for the Icelandic and 1.10 for the UK datasets, respectively.
Variants in the UKB imputation dataset were mapped to NCBI Build38 positions and matched to the variants in the Icelandic dataset based on allele variation. The results from the two cohorts were combined using a fixed-effect model, in which the cohorts were allowed to have different population frequencies for alleles and genotypes, but were assumed to have a common OR and were weighted with the inverse of the variance. We selected a threshold of 0.9 imputation info for variants available in the Icelandic dataset and 0.8 imputation info for variants only available in the UKB dataset. A total of 43,421,991 variants were used in the analysis. Heterogeneity was tested by comparing the null hypothesis of the effect being the same in all populations to the alternative hypothesis of each population having a different effect using a likelihood ratio test.
We used the weighted Holm–Bonferroni method56 to account for all 43,421,991 variants being tested (P-value < (0.05×weight)/43,421,991). Using the weighing scheme given in Sveinbjornsson et al.10, this procedure controls the family-wise error rate at 0.05; P ≤ 2.1 × 10−7 for high-impact variants (including stop-gained and loss, frameshift, splice acceptor or donor and initiator codon variants, n = 13,264), P ≤ 5.0 × 10−8 for missense, splice-region variants and in-frame-indels (n = 257,401), P ≤ 4.6 × 10−9 for low-impact variants (including synonymous, 3′ and 5′ UTR, and upstream and downstream variants, n = 3,303,568), P ≤ 2.0 × 10−9 for deep intronic and intergenic variants in DNase I hypersensitivity sites (DHS) (n = 5,999,736), and P ≤ 7.6 × 10−10 for other non-DHS deep intronic and intergenic variants (n = 33,998,502).
Conditional analysis
We applied approximate conditional analyses, implemented in the GCTA software57, to the meta-analysis summary statistics to look for additional association signals at each of the genome-wide significant loci. LD between variants was estimated using a set of 8700 whole-genome sequenced Icelandic individuals. The analysis was restricted to variants present in both the Icelandic and UKB datasets and within 1 Mb from the index variants. We tested 16 loci and about 30,000 variants in the conditional analysis and reported variants with conditional P value < 10−6. The results from GCTA were verified by conditional analysis using genotype data in the Icelandic and UK datasets separately, and the results presented in Table 1 are obtained by meta-analyzing those results.
Polygenic score, heritability, and explained variance
We derived a leiomyoma polygenic risk score (PGS) for the Icelandic individuals and, to avoid bias, we only used effect estimates from the GWAS summary results for leiomyoma from the UK dataset. The PGS was calculated using genotypes for about 630,000 autosomal markers included on the Illumina SNP chips to avoid uncertainty due to imputation quality. We estimated linkage disequilibrium (LD) between markers using 14,938 phased Icelandic samples and used this LD informaton to calculate adjusted effect estimates using LDpred58,59. We created several PGSs assuming different fractions of causal markers (the P parameter in Ldpred) and selected the best one based on the prediction of leiomyoma in the Icelandic dataset (0.3% causal variants). This PGS was then used when we calculate the correlation of the PGS with other phenotypes in the Icelandic dataset.
We computed LD scores for genotyped individuals in the Icelandic cohort using only high-quality markers and estimated the heritability explained by all markers, with MAF above 1% using LD score regression44. We estimated the risk ratio among siblings (λS) of Icelandic leiomyoma cases (N = 6728) by cross-matching with a genealogy database that covers the entire Icelandic nation. The risk ratio among siblings (λS) was estimated at 1.74 [1.63, 1.85] (P < 0.0001) using an approach previously described by Edvardsson et al60. This allows us to calculate the proportion of sibling recurrence risk of leiomyoma explained by the signals identified in the present study, log(λS[i])/log(λS)61.
Functional annotation of leiomyoma variants
Variants in linkage disequilibrium (LD) with the lead variants were identified on the basis of in-house genotype data using r2 > 0.8 for pairwise comparison of the nearest 100,000 variants to define an LD class. These variants were then annotated by intersection with chromatin immunoprecipitation (ChIP) signal data for uterine tissue. The ChIP-seq data was derived from the ENCODE project (www.encodeproject.org) downloaded in pre-processed (MACS v2 algorithm) bigWig format representing analysis of acetylation of lysine K27 of histone H3 (H3K27ac) with accession number ENCFF407ITR. The signal P-values were adjusted by the Benjamini-Hochberg procedure to account for multiple hypothesis and thresholded at the 1% FDR significance level. DNase hypersensitivity data (DHS) for uterus tissue were also downloaded in pre-processed format, accession number ENCFF604WBU, and intersected with variant position in each LD class.
Chromatin interaction map data were derived from Hi-C sequencing for various cell types and primary tissue samples62. The data were downloaded from Omnibus, accession number GSE87112, in pre-processed format (Fit-Hi-C algorithm) representing false-discovery rates (FDR) for contact regions at 40 kb resolution. To define statistically significant contacts we used a threshold value of FDR < 10−6 in mesoendoderm cells62. We confined this analysis to variants residing within candidate regulatory regions based on Encode data for H3K27ac and DHS in uterus tissue. The regions found to interact with the variant-containing regions were then queried for Refseq genes and intersected with a database for cancer genes (NCG 5.0; ncg.kcl.ac.uk) and tissue-specific expression patterns as provided in the Human Protein Atlas (www.proteinatlas.org), v18 downloaded in December 2017.
Data availability
The Icelandic population WGS data has been deposited at the European Variant Archive under accession code PRJEB8636. The authors declare that the data supporting the findings of this study are available within the article, its Supplementary Data files and upon request. The UK Biobank data can be obtained upon application (ukbiobank.ac.uk).
References
Dvorská, D., Braný, D., Danková, Z., Halašová, E. & Višňovský, J. Molecular and clinical attributes of uterine leiomyomas. Tumour Biol. 39, 1010428317710226 (2017).
Baird, D. D., Dunson, D. B., Hill, M. C., Cousins, D. & Schectman, J. M. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am. J. Obstet. Gynecol. 188, 100–107 (2003).
Gupta, S., Jose, J. & Manyonda, I. Clinical presentation of fibroids. Best. Pract. Res. Clin. Obstet. Gynaecol. 22, 615–626 (2008).
Doll, K. M., Dusetzina, S. B. & Robinson, W. Trends in inpatient and outpatient hysterectomy and oophorectomy rates among commercially insured women in the United States, 2000–2014. JAMA Surg. 151, 876–877 (2016).
Vikhlyaeva, E. M., Khodzhaeva, Z. S. & Fantschenko, N. D. Familial predisposition to uterine leiomyomas. Int. J. Gynaecol. Obstet. 51, 127–131 (1995).
Luoto, R. et al. Heritability and risk factors of uterine fibroids−the Finnish Twin Cohort study. Maturitas 37, 15–26 (2000).
Cha, P.-C. et al. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 43, 447–450 (2011).
Edwards, T. L., Michels, K. A., Hartmann, K. E. & Velez Edwards, D. R. BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum. Genet. 132, 943–953 (2013).
Hellwege, J. N. et al. A multi-stage genome-wide association study of uterine fibroids in African Americans. Hum. Genet. https://doi.org/10.1007/s00439-017-1836-1 (2017).
Sveinbjornsson, G. et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet. 48, 314–317 (2016).
Stacey, S. N. et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 43, 1098–1103 (2011).
Enciso-Mora, V. et al. Low penetrance susceptibility to glioma is caused by the TP53 variant rs78378222. Br. J. Cancer 108, 2178–2185 (2013).
Diskin, S. J. et al. Rare variants in TP53 and susceptibility to neuroblastoma. J. Natl. Cancer Inst. 106, dju047 (2014).
Gudmundsson, J. et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 8, 14517 (2017).
Haiman, C. A. et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat. Genet. 43, 1210–1214 (2011).
Palmer, J. R. et al. Genetic susceptibility loci for subtypes of breast cancer in an African American population. Cancer Epidemiol. Biomark. Prev. 22, 127–134 (2013).
Speedy, H. E. et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 46, 56–60 (2014).
Wang, Z. et al. Imputation and subset-based association analysis across different cancer types identifies multiple independent risk loci in the TERT-CLPTM1L region on chromosome 5p15.33. Hum. Mol. Genet. 23, 6616–6633 (2014).
Barrett, J. H. et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 43, 1108–1113 (2011).
Kweekel, D. M. et al. Explorative study to identify novel candidate genes related to oxaliplatin efficacy and toxicity using a DNA repair array. Br. J. Cancer 101, 357–362 (2009).
Andreassen, C. N. et al. Individual patient data meta-analysis shows a significant association between the ATM rs1801516 SNP and toxicity after radiotherapy in 5456 breast and prostate cancer patients. Radiother. Oncol. 121, 431–439 (2016).
Rudd, M. F., Sellick, G. S., Webb, E. L., Catovsky, D. & Houlston, R. S. Variants in the ATM-BRCA2-CHEK2 axis predispose to chronic lymphocytic leukemia. Blood 108, 638–644 (2006).
Angèle, S. et al. ATM polymorphisms as risk factors for prostate cancer development. Br. J. Cancer 91, 783–787 (2004).
Scelo, G. et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat. Commun. 8, 15724 (2017).
Chahal, H. S. et al. Genome-wide association study identifies 14 novel risk alleles associated with basal cell carcinoma. Nat. Commun. 7, 12510 (2016).
Law, M. H. et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 47, 987–995 (2015).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. https://doi.org/10.1038/ng.3892 (2017).
Phelan, C. M. et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 49, 680–691 (2017).
Walsh, K. M. et al. Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 6, 42468–42477 (2015).
Ojha, J. et al. Genetic variation associated with longer telomere length increases risk of chronic lymphocytic leukemia. Cancer Epidemiol. Biomark. Prev. 25, 1043–1049 (2016).
Levy, D. et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl Acad. Sci. USA 107, 9293–9298 (2010).
Sapkota, Y. et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 8, 15539 (2017).
Estrada, K. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44, 491–501 (2012).
Rivadeneira, F. et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 41, 1199–1206 (2009).
Kuchenbaecker, K. B. et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 47, 164–171 (2015).
He, C. et al. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat. Genet. 41, 724–728 (2009).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Stacey, S. N. et al. Ancestry-shift refinement mapping of the C6orf97-ESR1 breast cancer susceptibility locus. PLoS Genet. 6, e1001029 (2010).
Dunning, A. M. et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 48, 374–386 (2016).
Rižner, T. L. The important roles of steroid sulfatase and sulfotransferases in gynecological diseases. Front. Pharmacol. 7, 1–16 (2016).
Cheng, T. H. et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat. Genet. 48, 667–674 (2016).
Kaya Okur, H. S., Das, A., Taylor, R. N., Bagchi, I. C. & Bagchi, M. K. Roles of estrogen receptor-α and the coactivator MED1 during human endometrial decidualization. Mol. Endocrinol. Baltim. Md 30, 302–313 (2016).
Lombardi, M. et al. Hormone-dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells. J. Cell Biol. 182, 327–340 (2008).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Helgason, H. et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nat. Genet. 47, 906–910 (2015).
Rafnar, T. et al. Association of BRCA2 K3326* with small cell lung cancer and squamous cell cancer of the skin. J. Natl Cancer Inst. https://doi.org/10.1093/jnci/djy002 (2018).
Sigurdardottir, L. G. et al. Data quality at the Icelandic Cancer Registry: comparability, validity, timeliness and completeness. Acta Oncol. Stockh. Swed. 51, 880–889 (2012).
Styrkarsdottir, U. et al. Sequence variants in the PTCH1 gene associate with spine bone mineral density and osteoporotic fractures. Nat. Commun. 7, 10129 (2016).
Sulem, P. et al. Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche. Nat. Genet. 41, 734–738 (2009).
Kong, A. et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 40, 1068–1075 (2008).
Styrkarsdottir, U. et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 497, 517–520 (2013).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Wain, L. V. et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med. 3, 769–781 (2015).
Welsh, S., Peakman, T., Sheard, S. & Almond, R. Comparison of DNA quantification methodology used in the DNA extraction protocol for the UK Biobank cohort. BMC Genom. 18, 26 (2017).
Bycroft, C. et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv https://doi.org/10.1101/166298 (2017).
Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6, 65–70 (1979).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012). S1-3.
Kong, A. et al. Selection against variants in the genome associated with educational attainment. Proc. Natl Acad. Sci. USA 114, E727–E732 (2017).
Vilhjálmsson, B. J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015).
Edvardsson, V. O., Palsson, R., Indridason, O. S., Thorvaldsson, S. & Stefansson, K. Familiality of kidney stone disease in Iceland. Scand. J. Urol. Nephrol. 43, 420–424 (2009).
Witte, J. S., Visscher, P. M. & Wray, N. R. The contribution of genetic variants to disease depends on the ruler. Nat. Rev. Genet. 15, 765–776 (2014).
Schmitt, A. D. et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059 (2016).
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number ‘24711’. We thank the individuals who participated in the study and whose contribution made this work possible. We acknowledge the Icelandic Cancer Registry for assistance in the ascertainment of the cancer patients.
Author information
Authors and Affiliations
Contributions
T.R., B.G., P.S., B.V.H., G.T, and K.S. designed the study and interpreted the results. A.S, A.T., E.A.H., K.K., O.I., V.G., R.T.G., R.A., J.G.J., and K.O. carried out the subject ascertainment, recruitment, and collection of clinical data. U.S., V.S., S.N.S., J.G., G.A.A., A.O., F.Z., G.H., R.P.K., O.B.D., G.M., V.T., F.W.A., and U.T. collected, processed, and analyzed the genotype and phenotype data. O.A.S. performed the functional annotations. A.I., M.L.F., L.S., J.K.S., G.S., D.F.G., B.G., and B.V.H., performed the statistical and bioinformatics analyses. T.R., B.G., O.A.S, G.T., and K.S., drafted the manuscript. All authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors that are affiliated with deCODE are employees of deCODE genetics/Amgen. The remaining authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rafnar, T., Gunnarsson, B., Stefansson, O.A. et al. Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat Commun 9, 3636 (2018). https://doi.org/10.1038/s41467-018-05428-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-05428-6
This article is cited by
-
A common allele increases endometrial Wnt4 expression, with antagonistic implications for pregnancy, reproductive cancers, and endometriosis
Nature Communications (2024)
-
Integrating leiomyoma genetics, epigenomics, and single-cell transcriptomics reveals causal genetic variants, genes, and cell types
Nature Communications (2024)
-
Evidence of a causal effect of genetic tendency to gain muscle mass on uterine leiomyomata
Nature Communications (2023)
-
Uterine fibroid polygenic risk score (PRS) associates and predicts risk for uterine fibroid
Human Genetics (2022)
-
Emerging Roles of Long Non-coding RNAs in Uterine Leiomyoma Pathogenesis: a Review
Reproductive Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
