
Abstract
Recently, loading ligand-protected gold (Au) clusters as visible light photosensitizers onto various supports for photoredox catalysis has attracted considerable attention. However, the efficient control of long-term photostability of Au clusters on the metal-support interface remains challenging. Herein, we report a simple and efficient method for enhancing the photostability of glutathione-protected Au clusters (Au GSH clusters) loaded on the surface of SiO2 sphere by utilizing multifunctional branched poly-ethylenimine (BPEI) as a surface charge modifying, reducing and stabilizing agent. The sequential coating of thickness controlled TiO2 shells can further significantly improve the photocatalytic efficiency, while such structurally designed core-shell SiO2-Au GSH clusters-BPEI@TiO2 composites maintain high photostability during longtime light illumination conditions. This joint strategy via interfacial modification and composition engineering provides a facile guideline for stabilizing ultrasmall Au clusters and rational design of Au clusters-based composites with improved activity toward targeting applications in photoredox catalysis.
Similar content being viewed by others

Introduction
Noble metal nanoparticles (MNPs) are uniquely suited for heterogeneous catalysis because of their relatively high specific surface areas and abundant active centers1,2,3,4,5. The size of MNPs has been shown to be one of the most important factors that dictates the performance of a catalyst5. However, the catalytically active nano-sized MNPs are at a thermodynamic unstable state, which are inclined to sinter—they form fewer, larger particles—under thermal and/or light irradiation conditions2,6,7,8,9,10. For instance, it has been demonstrated that nano-sized gold nanoparticles irradiated by a femtosecond laser-induced white-light supercontinuum can initiate a growth of larger nanoparticles9. Additionally, the shape of silver nanospheres can be converted into triangular nanoprisms under the illumination of light at wavelengths between 350 and 700 nm10. Thus, a variety of solid support strategies have been utilized in practical catalytic systems for stabilization of the small MNPs in heterogeneous catalysis since the size and shape of MNPs provide important control over the catalytic efficiency6,7.
Recently, the ultrasmall MNPs with size regime of 1–3 nm in diameter (often called metal clusters, e.g., gold (Au) clusters) that are composed of a specific number of metal atoms and of ligands have received considerable attention due to their distinctive physicochemical properties11,12,13,14,15,16, which are different from their plasmonic counterpart owing to the quantum size effect, surface geometric effect (e.g., surface atom arrangement and low-coordinated atoms), and high surface-to-volume ratio17,18. These ultrasmall ligands (typically thiolate)-protected Au clusters exhibit discrete electronic structure18,19, which have been regarded as one of the prototypes of highly tunable molecular-sized materials for catalysis and solar energy conversion. In particular, the thiolate-protected Au clusters, typically glutathione-protected Au clusters (Au GSH clusters), are able to serve as both photosensitizer and catalytic center for multifunctional use in photocatalytic redox systems, such as water splitting, selective organic synthesis, and pollutants degradation20,21,22,23,24,25,26,27,28,29. However, such ultrasmall Au GSH clusters suffer from serious instability under light irradiation due to its extremely high surface energy and large surface15. The loading of ultrasmall Au GSH clusters onto different supports is often inefficient to avoid coalescence and agglomeration of these Au GSH clusters15,27,28,29,30, which thereby results in either the difficulty of unambiguous photocatalytic mechanism assignment or a loss of photocatalytic activity15,27,28.
Regarding the Au clusters-based photocatalysts, seminal works have been reported for understanding the observed aggregation of Au GSH clusters at the semiconductor/metal interface. For instance, the irradiation of high-energy electron beams can lead to the aggregation of Au GSH clusters into larger Au NPs (~5 nm diameter) after 2 h continuous visible light illumination (λ > 420 nm) over Au GSH-sensitized Pt/TiO2 photocatalysts28. It has also been reported that the photogenerated electrons from Au GSH clusters and/or TiO2 can be captured by Au (I) component on the outer layer of Au GSH clusters (i.e., Au(0)@Au(I)-GSH) to produce Au NPs and the photocatalytic decomposition of GSH ligands tethered on the Au clusters surface can also lead to formation of metallic Au NPs27. Recently, a diffusion/aggregation mechanism has been proposed to elucidate the photoinduced coalescence of Au GSH clusters to larger metallic Au NPs at the Au/TiO2 interface under light illumination15. Despite various studies on disclosing underlying mechanism of transformation of ultrasmall molecular-like Au GSH clusters to Au NPs, the effective control of Au GSH clusters with long-term stability on the substrates under in situ photo-irradiation conditions still remains a challenge, which becomes the main bottleneck for the development of Au clusters-based catalysts systems for photoredox applications15,27,28.
Herein, we report a simple, combinatorial approach to stabilize ultrasmall Au GSH clusters on the SiO2 sphere support and improve their photocatalytic efficiency by using multifunctional branched poly-ethylenimine (BPEI) for surface modification and coating thickness controlled semiconductor TiO2 shell for interfacial composition engineering. The BPEI is shown to behave as a surface charge modifying, reducing and stabilizing agent for interfacial modification of SiO2-Au GSH clusters composites. Specifically, the size and structure of Au GSH clusters can be well retained over 10 h under continuous visible light irradiation (λ > 420 nm) because of the high reductive ability of BPEI to inhibit the organic ligand oxidation process over Au GSH clusters. In addition, while simultaneously maintaining the photostability of Au GSH clusters, the sequential coating of TiO2 shells for constructing core-shell SiO2-Au GSH clusters-BPEI@TiO2 (SABT) nanostructures has been demonstrated to significantly ameliorate the photoactivity by regulating the photoelectrochemical and adsorption properties of SABT composites synergistically. Therefore, the strategy via interfacial modification and composition manipulation by coating semiconductor shell provides an efficient way for stabilizing Au clusters with improved photocatalytic activity, which is anticipated to enable the broad development of Au clusters-based composite system for photoredox applications in solar energy conversion.
Results
Structural characterizations of Au GSH clusters
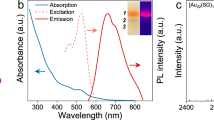
The successful fabrication of Au GSH clusters has been proved by transmission electron microscopy (TEM) analysis and the characteristic emissive properties of Au GSH clusters31. As apparently shown in Supplementary Fig. 1a, TEM image shows the uniform dispersed ultrasmall Au GSH clusters with mean diameter around 1.4 nm (Supplementary Fig. 1b). The Au GSH clusters solution exhibits yellow emissive properties under the blacklight illumination (Supplementary Fig. 1d and e), which is different from that of plasmonic Au counterparts15,27,28. UV-vis absorption spectrum of Au GSH clusters in Supplementary Fig. 1c suggests that the Au GSH clusters show an absorption onset at ∼520 nm with a distinct shoulder around 400 nm, which is attributed to the highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) transition originated from the ligand-to-metal charge transfer15, indicating that Au GSH clusters could be used as visible light photosensitizers. The photoluminescence (PL) excitation spectrum exhibits a maximum at 400 nm, which coincides well with the absorption shoulder observed in the absorption spectrum. The PL emissive spectra of Au GSH clusters with different excitation wavelengths in Supplementary Fig. 1f show a low energy emission band with the peak maximum at 605 nm, which is ascribed to the triplet metal-centered state32, and the shape of the PL spectra is independent of the excitation wavelength15. The large Stokes shift in the emission, where the absorption band shoulder appears at around 400 nm and the emission maximum is seen at 605 nm, is consistent with the excited state being a ligand-metal charge transfer type15,33. In addition, the electrospray ionization mass spectrometric characterization has been employed to investigate the as-synthesized Au GSH clusters. However, because the spray is orthogonal to ion extraction in our utilized instrument, the clusters are unable to be detected in their native state and only certain fragments such as [Au(SG)2-H]−1 (m/z 808) and [Au2(SG)2-H]−1 (m/z 1005) are detectable, as shown in Supplementary Fig. 2, which are in good agreement with the previous reports34,35.
Enhancing the photostability of Au GSH clusters
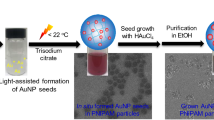
The insulative SiO2 spheres with rather low optical absorption (Supplementary Fig. 3) have been chosen as inert supports to predominantly focus on investigating the photosensitizer role of Au GSH clusters in photocatalytic applications. The schematic synthesis procedure for SiO2-Au GSH clusters composites via interfacial modification process has been displayed in Fig. 1a. The SiO2 spheres become positively charged (Supplementary Fig. 4a) after surface modification with BPEI36, a conjugated reducible dendrimer, which leads to a substantial electrostatic attraction with negatively charged Au GSH clusters (Supplementary Fig. 4c) by Coulombic forces, thereby forming SiO2-Au GSH clusters-BPEI composites (SAB) with strong interfacial interaction between Au GSH clusters and SiO2 supports. In contrast, the common protocol used for loading Au GSH clusters onto the supports involves a pH value adjusted process under acidic conditions15,19,20,22,23,27. As illustrated in Supplementary Fig. 5, SiO2 dispersion is firstly adjusted to acidity (pH value at about 2) by adding dilute hydrochloric acid solution and then the Au GSH clusters solution is added into the above dispersion for fabricating SiO2-Au GSH clusters-pH composites (SAP). Notably, due to the protonation of carboxylic groups in the glutathione ligands under acidic conditions19 and the relatively weak electropositivity of SiO2 spheres (Supplementary Fig. 4b), the as-prepared sample of SAP generally exhibits a weak Au–oxide interaction. The difference in the interaction between SiO2 and Au GSH clusters over SAB and SAP samples may lead to the different loading amounts of Au GSH clusters on SiO2 surface, which has been investigated by the inductively coupled plasma optical emission spectroscopy. As revealed in Supplementary Table 1, the addition amount of Au GSH clusters in SAB (0.86%) is higher than that of SAP with 0.28% Au GSH clusters, which could be attributed to the strong coulombic force between positively charged BPEI-SiO2 spheres and the negatively charged Au GSH clusters. The XRD patterns of SAB and SAP samples in Supplementary Fig. 6 suggest the similar crystal structure of these composites and the diffraction peaks of Au GSH clusters are hardly observed for both of SiO2-Au GSH clusters composites, which may be due to the low loading amount and poor crystallinity of Au GSH clusters15,23,24.

Photostability test and characterizations. a Schematic illustration of synthesis procedure for SiO2-Au GSH clusters-BPEI composites (SAB) and photostability testing of as-prepared SAB; transmission electron microscopy (TEM) images of SAB b before and c after visible light irradiation (λ > 420 nm) for 10 h and SiO2-Au GSH clusters-pH (SAP) e before and f after visible light irradiation for 10 h; the corresponding models of d SAB and g SAP after visible light irradiation for 10 h; high-resolution X-ray photoelectron spectroscopy (XPS) spectra of h S 2p and i Au 4f for SAB before/after visible light irradiation; j UV-vis diffuse reflectance spectrum (DRS) spectrum of SAP after 10 h visible light irradiation. Insets in b and e are the corresponding models of SAB and SAP before visible light irradiation. The histograms in c and f correspond to the particle size distributions of Au GSH clusters in the SAB and SAP after visible light irradiation for 10 h, respectively. The yellow spheres in d represent Au GSH clusters and the purple spheres in g represent Au nanoparticles. Scale bar, 5 nm
To initially investigate the photostability of Au GSH clusters, the composites of SAB and SAP have been exposed to continuous visible light irradiation (λ > 420 nm) for 10 h under ambient conditions. TEM analysis suggests that Au GSH clusters maintain almost the same size on the surface of SiO2 spheres for SAB composites before and after photo-irradiation (Fig. 1b–d), while ~1.4 nm Au GSH clusters aggregate to around 6 nm Au nanoparticles (NPs) for SAP samples after photo-irradiation (Fig. 1e–g), indicating that the aggregation of Au GSH clusters in the SAB samples has been effectively inhibited by BPEI interfacial modification, which is significantly different from that of SAP. The UV-vis diffuse reflectance spectrum (DRS) of SAP after light illumination in Fig. 1j shows a pronounced surface plasmon resonance (SPR) peak located at around 540 nm belonging to the metallic Au NPs15,23,27, which consists with the TEM image of SAP sample after 10 h irradiation of visible light in Supplementary Fig. 7, further suggesting the seriously photoinduced aggregation of Au GSH clusters into Au NPs on the surface of SiO2 spheres over SAP. Additionally, the TEM images in Supplementary Fig. 8 suggest that the size of Au GSH clusters over SAB after 24 h visible light irradiation (λ > 420 nm) is 1.4 nm, which is nearly unchanged as compared to that before light illumination (Fig. 1b), indicating that the interfacial modification of BPEI layer over SAB can provide a long-time protection (24 h) with regard to the stabilization of Au GSH clusters. When the irradiation time over SAB is further prolonged to 36 and 48 h, the size of Au GSH clusters will increase to 2.0 and 2.1 nm, respectively, as illustrated in Supplementary Fig. 9, which indicates the slight aggregation of Au GSH clusters. Such increase in the size of Au GSH clusters may be attributed to the partial depletion of BPEI, since the BPEI layer that serves as a reducing agent would undergo oxidation and/or decomposition in the way to stabilize the Au GSH clusters. To further investigate the role of BPEI in inhibiting the growth of Au GSH clusters, the SAP sample is modified by BPEI (BPEI-SAP) and the obtained BPEI-SAP composite is irradiated under visible light (λ > 420 nm) for 10 h. The HRTEM image of BPEI-SAP after light illumination in Supplementary Fig. 10a suggests that, as compared with SAP samples (Fig. 1e–g), the serious aggregation of Au GSH clusters can be prevented to some extent and the size of Au GSH clusters is calculated to be 2.0 nm (Supplementary Fig. 10b).
To ensure whether the stability enhancement of Au GSH clusters due to the modification of BPEI is universal, we have performed a series of control experiments including the effects of various metal oxide supports and the structure of Au GSH clusters. The surfaces of different metal oxide supports, including rutile TiO2, ZnO, and ZrO2, have been positively charged by the BPEI modification (Supplementary Fig. 11), which can subsequently interact with the negatively charged Au GSH clusters (Supplementary Fig. 4c) via an electrostatic self-assembly method to produce metal oxide-Au GSH clusters-BPEI composites (MABs) and the photostability of MABs has been investigated under the same conditions as that for SAB and SAP. As illustrated in Supplementary Fig. 12, the mean diameter of Au GSH clusters on the surfaces of various metal oxide supports after continuous visible light (λ > 420 nm) for 10 h is determined to be 1.4 nm, indicating the critical role of BPEI on inhibiting the aggregation of Au GSH clusters and excluding the supports effect on the photostability enhancement of Au GSH clusters. Additionally, Supplementary Note 1 elucidates that, even though the Au GSH clusters suffer from serious aggregation on the surface of anatase TiO2 due to the presence of abundance surface hydroxyl group, the presence of BPEI layer can protect ultrasmall Au GSH clusters from being fusion to some extent under visible light illumination, suggesting the universality of the interficial modification strategy on stabilizing Au GSH clusters. To further confirm our inference and achieve a generally valid conclusion, the Au25-glutathione clusters (Au25(SG)18), which are well known as the most stable one among the thiolated clusters of this class15,37,38, have been decorated on the surface of BPEI modified SiO2 spheres to produce SiO2-Au25(SG)18 clusters-BPEI composites (SASB). The photostability test in Supplementary Fig. 13 suggests that the size of Au25(SG)18 clusters can be well maintained, thereby demonstrating that the introduction of BPEI layer for interfacial modification over SASB is an effective strategy to impede the photoinduced coalescence of Au25(SG)18 clusters to larger metallic Au NPs upon visible light illumination (λ > 420 nm).
To understand the role of BPEI in improving the stability of Au GSH clusters in SAB, X-ray photoelectron spectroscopy (XPS) analysis has been conducted. Survey XPS spectra in Supplementary Fig. 14d evidence the presence of Au, S, N, C, O and Si elements in the SAB composites before/after visible light irradiation. As mirrored in Fig. 1i, the two doublet 4f peaks in the high-resolution spectrum of Au 4f suggest that the there are two different elemental chemical states of Au species. The Au 4f5/2 and Au 4f7/2 peaks with binding energies of 86.9 and 83.1 eV are ascribed to metallic Au (Au0), and the other doublet at 87.75 and 84.05 eV for Au 4f5/2 and Au 4f7/2 are assigned to Au+ ion39,40,41. The Au0 content determined by XPS is found to constitute ∼90% of all Au atoms. The samples of SAB before/after photo-irradiation exhibit similar Au 4f spectra, demonstrating the well reserved structural properties of Au GSH clusters during the visible light irradiation (λ > 420 nm). Additionally, previous work has verified that the Au GSH clusters aggregation procedure is accompanied with a ligand oxidation process of Au GSH clusters, which results in a new S 2p peak matching the binding energy of R–SO3 species (169.2 eV)15. As for the SAB composites after photo-irradiation of 10 h, only one S 2p3/2 peak located at 161.5 eV (Fig. 1h), corresponding to the ligand features (RS–Au) in various monothiolate Au GSH clusters42,43, is observed, which is similar with that of SAB sample before light irradiation, demonstrating that the organic ligand oxidation process over the Au GSH clusters in SAB has been efficiently inhibited during visible light irradiation. The enhanced photostability of Au GSH clusters in the SAB composites can be attributed to the introduction of BPEI with high reductive ability44,45,46, which could serve as effective reductive agents to hamper the ligand oxidation process over Au GSH clusters during photo-irradiation process. As illustrated in Supplementary Fig. 14a–c, the slight peak shift of the elements of C, O, and N over the SAB sample before/after the visible light irradiation clearly suggests the redox process of BPEI to protect organic ligands from being oxidized and thus enhancing the photostability of Au GSH clusters in the SAB composites. Aiming to in depth explore the underlying mechanism of enhanced photostability of Au GSH clusters over SAB composites, the surface charge modifying agent of 3-aminopropyltriethoxysilane (APTES), which can also positively charge the SiO2 spheres surface47, but with no reductive ability, has been employed to replace BPEI for interfacial modification. After coupling with negatively charged Au GSH clusters, the as-formed SiO2-Au GSH clusters-APTES composites (SAA) have been exposed under visible light irradiation (λ > 420 nm) for 10 h. TEM images of SAA after light irradiation (Supplementary Fig. 15a, b) suggest that the Au GSH clusters (1.4 nm) aggregate into larger Au NPs (3.1 nm) (Supplementary Fig. 15c), indicating that the APTES as a surface charge modifying agent cannot protect the Au GSH clusters from being fusion. The structural formulas of APTES and BPEI in Supplementary Fig. 16 suggest that different from APTES, BPEI contains abundant primary, secondary and tertiary amino groups. It has been well documented that the multiple polymer chains of primary, secondary and tertiary amine groups in the BPEI can encapsulate the as-synthesized Au GSH clusters via crosslinking45, which could fix these clusters on the surface of supports and hamper the migration of clusters, thus stabilizing the ultrasmall Au GSH clusters. Accordingly, based on the above control experiments, we can deduce that it is the high reductive ability and branched structure of BPEI that play a critical role in hampering the ligand oxidation process over Au GSH clusters, thereby stabilizing Au GSH clusters against aggregation under continuous light illumination condition.
The photocatalytic performances of bare SiO2 spheres and SiO2-Au GSH clusters-BPEI (SAB) composites have been tested by degradation of organic dye rhodamine B (RhB), a typical nonselective photocatalytic processes48,49,50, under visible light irradiation (λ > 420 nm). As shown in Supplementary Fig. 17, SiO2 spheres exhibit negligible photoactivity toward RhB degradation during 10 h visible light irradiation, which suggests that SiO2 spheres are not photocatalytically active under visible light irradiation and the dye photosensitization effect of RhB48 on the photoactivity can be ignored. After coupling with Au GSH clusters as visible light photosensitizers, the SAB sample shows obvious activity toward photocatalytic RhB degradation (Supplementary Fig. 17). The observed photoactivity can be attributed to the incorporation of Au GSH clusters, which can generate electron–hole pairs by electron transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) under visible light irradiation23,51,52, thereby driving RhB degradation. However, it should be noted that only 38% of RhB has been degraded after 10 h irradiation of visible light over SAB, indicating the low photosensitization efficiency of Au GSH clusters. Such observed low photoactivity could be attributed to large band gap of SiO2 support47, by which the photogenerated electrons in the LUMO of Au GSH clusters cannot transfer to the conduction band (CB) of SiO2 efficiently, thereby leading to simultaneously serious recombination of charge carriers. Notably, it has been demonstrated that the CB edge potential of metal oxides (e.g., ZnO and rutile TiO2) is more positive than the LUMO potential of Au GSH clusters, which enables the transformation of photoexcited electrons from Au GSH clusters to the metal oxide supports15,24,53. The photoactivities of BPEI modified metal oxides-Au GSH clusters composites (denoted as MAB) have been evaluated toward RhB degradation under visible light irradiation (λ > 420 nm). As shown in Supplementary Fig. 18, the samples of MAB exhibit moderate photoactivity enhancement than bare semiconductors, which suggests that the random loading of ultrasmall Au GSH clusters onto semiconductors without rational structure design is inefficient to achieve high efficient Au GSH clusters-semiconductor composites. Therefore, a thickness tunable TiO2 shell has been coated onto the surface of SAB composites for designing core-shell SiO2-Au GSH clusters-BPEI@TiO2 structures to construct high performance Au GSH clusters-semiconductor composites for solar energy conversion.
Elaboration of SiO2-Au GSH clusters-BPEI@TiO2 samples
The synthesis procedure of core-shell SiO2-Au GSH clusters-BPEI@TiO2 (SABT) composites has been illustrated in Fig. 2a. In brief, the SAB composites are firstly dispersed in ethanol, followed by adding hexadecylamine (HDA) surfactants and ammonia under vigorously stirring at room temperature. The HDA surfactants could segregate the surface of SAB. Then, different amounts of titanium isopropoxide (TIP) have been introduced into the dispersion under stirring. During this process, TIP hydrolysis products (i.e., TiO2) participate in hydrogen-bonding interactions with the amino groups of HDA molecules to form inorganic–organic composites that coat the SAB spheres to produce SABT54. Scanning electron microscopy (SEM) images of SABT composites in Supplementary Fig. 19 show the uniform spherical morphology of these SABT composites. TEM analysis in Fig. 2 shows that the core-shell structures of SABT composites are well defined, and the thickness of TiO2 shell can be well-tuned from few to dozens nanometer by adjusting the addition amounts of TIP from 0.05 to 0.2 mL (marked as SABT-x, where the x represents the volume of added TIP). In particular, the Au GSH clusters embedded into TiO2 shell are distinguishable, especially for the samples of SABT-0.05 (Fig. 2c) and SABT-0.1 (Fig. 2d) with thin TiO2 shell. Noteworthily, the Au GSH clusters in core-shell SABT composites maintain their original sizes, suggesting the stability of Au GSH clusters during such a mild TiO2 coating process at room temperature. The energy-dispersive X-ray (EDX) spectrum in Fig. 2h evidences the presence of Au, O, Si, and Ti elements over SABT-0.05 composites. Additionally, the elemental mapping analysis identifies the spatial distribution of these elements in SABT-0.05. As displayed in Fig. 2b, e, the distribution ranges of Ti and O elements are slightly larger than that of Au, which is itself slightly larger than that of Si. These corroborate the intelligently designed core-shell SABT structure, among which the thin TiO2 layer is located at the outmost surface, while the SiO2 spheres serve as the core and the Au GSH clusters are located between the SiO2 core and TiO2 shell.

Morphology and chemical composition of SABT samples. a Schematic illustration of fabrication process for core-shell SiO2-Au GSH clusters-BPEI@TiO2 (SABT) composites; b high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image (scale bar, 50 nm); e elemental mapping results; and h energy-dispersive X-ray (EDX) spectrum for the boxed area in the main image of SABT-0.05; TEM images of c SABT-0.05, d SABT-0.1, f SABT-0.15, and g SABT-0.2, scale bar, 20 nm. Insets in the c, d, f, and g are the corresponding models of SABT with different TiO2 shell thickness
The phase composition and optical properties of the SABT composites have been characterized by XRD and DRS analysis, respectively. As reflected in Supplementary Fig. 20, all of the samples possess analogous XRD patterns with a broad peak located at ca. 23°, corresponding to amorphous silica55, and the intensities of SiO2 diffraction peaks become weak with increasing the thickness of TiO2 shells. However, no typical diffraction peaks of TiO2 have been observed in XRD spectra because TiO2 is amorphous by TIP hydrolysis54. Due to the relatively low content and weak diffraction intensity of Au GSH clusters, the diffraction peak of Au GSH clusters in the SABT has also not been perceived15,23,24,25,26,27,28. The DRS spectra of SABT composites in Supplementary Fig. 21 show that the introduction of TiO2 shell can greatly increase the UV absorption intensities of the samples, while the visible light absorptions in the range of 400–520 nm caused by Au GSH clusters almost maintain their intensities.
Photocatalytic activity and stability of SABT composites
The photocatalytic performances of SABT composites have been evaluated toward rhodamine B (RhB) photodegradation under visible light illumination (λ > 420 nm). As delineated in Fig. 3a, the photoactivity over SAB sample can be significantly enhanced by coating TiO2 shell and the dependence of RhB degradation efficiency on the thickness of the TiO2 shell shows a clear volcano behavior. The photocatalytic degradation of RhB firstly increases with increasing thickness of TiO2 layer, and the maximum RhB degradation efficiency has been achieved over SABT-0.15 and then decreases. Specifically, 99.1% conversion of RhB is achieved over SABT-0.15 after visible light irradiation for 0.5 h, and the photocatalytic efficiency of SABT-0.15 is about 19 times higher than that of SAB with only 4.9% conversion of RhB under identical reaction conditions. Furthermore, the sample of TiO2-Au GSH cluster-BPEI (TAB) exhibits worse catalytic activity than the SABT composites, which indicates that the intelligently designed core-shell structure of SABT is favorable for enhancing the photocatalytic performance of Au GSH clusters-semiconductor composites. The pseudo-first order kinetic of the degradation of RhB based on the above data is shown in Supplementary Table 2. It is clear that all of the SABT samples show higher reaction rate than the SAB and TAB samples, among which SABT-0.15 exhibits the highest reaction rate of 0.119 min−1 among these SABT composites. Additionally, the photocatalytic degradation of RhB over SiO2-Au GSH clusters-pH@TiO2 composites (SAPT) with different TiO2 shell thickness has also been investigated. Due to the addition amount of Au GSH clusters in SAP is different from that of SAB (Supplementary Table 1), the photocatalytic activities of RhB degradation over various SAPT composites with different TiO2 shell thickness have been normalized with respect to the loading amount of Au GSH clusters in SAB for a fair comparison. As displayed in Supplementary Fig. 22, the coating of TiO2 shell greatly improves the photocatalytic efficiency of SAP sample and the SAPT-0.15 composites exhibit the best photoactivity among these samples. However, the RhB degradation efficiency over SAPT-0.15 composites is worse than that of SABT-0.15 (Fig. 3a) under identical reaction conditions, which suggests that the presence of BPEI layer is beneficial for the photoactivity enhancement since the photostability of Au GSH clusters in the SABT-0.15 is significantly ameliorated.

Photocatalytic activity and the underlying mechanism Photocatalytic degradation of a RhB over blank SiO2 spheres, TiO2, SAB, TAB, and SABT composites with different TiO2 shell thickness under visible light irradiation (λ > 420 nm) for 0.5 h; photocatalytic reduction of b p-methoxy nitrobenzene to p-methoxy aniline over blank SiO2 spheres, TiO2, SAB, TAB, and SABT composites with different TiO2 shell thickness under visible light irradiation for 5 h; c recycling photocatalytic degradation of RhB over optimal SABT-0.15 composite under visible light irradiation (λ > 420 nm); d transient photocurrent densities of SiO2 spheres, SAB and SABT composites with different TiO2 shell thickness under visible light irradiation (λ > 420 nm); e scheme illustrating the photogenerated electron transport pathways between Au GSH clusters and TiO2 shell (the blue arrow represents the photoexcitation process of electron–hole pairs; the orange dash line means the recombination of electron-hole pairs; e and h in the blue cycles correspond to the photogenerated electron and hole, respectively); f cyclic voltammograms of SiO2 spheres, SAB and SABT composites with different TiO2 shell thickness; g bar plots showing the remaining RhB in reaction solutions after being kept in dark for 3 h to achieve the adsorption–desorption equilibrium over SiO2 spheres, SAB and SABT composites with different TiO2 shell thickness; h nitrogen adsorption–desorption isotherms of SiO2 spheres, SAB and SABT composites with different TiO2 shell thickness; i surface area of SiO2 spheres, SAB and SABT composites with different TiO2 shell thickness. Note that the error bars represent the photoactivity s.d. values calculated from triplicate experiments
To investigate the effect of surface coating of TiO2 shell on the photocatalytic performance and exclude the possible dye photosensitization effect of RhB on the activity56, the visible light photoactivities of SiO2 spheres, TAB, SAB, and SABT composites toward photocatalytic reduction of p-methoxy nitrobenzene to p-methoxy aniline have been evaluated, which is a typical six-electron reduction reaction (Supplementary Fig. 23)24,27,47,57. As presented in Fig. 3b, the higher photocatalytic activity of SABT composites than that of SiO2 spheres and SAB sample verifies the positive role of TiO2 shell in improving the photoactivity of SiO2-Au GSH clusters composites. And the SABT-0.15 shows the best photoactivity among these samples, elucidating that the thickness of TiO2 shell plays a critical role in determining the photocatalytic performance of SABT toward p-methoxy nitrobenzene reduction. Moreover, due to the intelligently designed core-shell structure, all of the SABT composites show higher catalytic activity than the TAB composite.
In addition to the TiO2 shell thickness, the crystallization of TiO2 layer in composites is also a factor that affects the photocatalytic performance of SABT composites and it has been reported that the amorphous TiO2 usually exhibits poor photocatalytic activity58,59,60,61,62. We have paid our endeavors to crystallize TiO2 shell in SABT-0.15, including calcination and hydrothermal treatment, since the high temperature is generally required to transform amorphous TiO2 particle into an anatase one. Even though the crystallization of TiO2 can be improved by calcination and hydrothermal treatment, the Au GSH clusters in SABT-0.15 suffer from serious instability under high temperature and aggregate into metallic Au nanoparticles with large size, as displayed in Supplementary Fig. 24, 25, thereby deteriorating the photocatalytic performance of SABT-0.15 sample toward various photocatalytic reactions under visible light illumination (λ > 420 nm) (Supplementary Fig. 26).
The photostability of the optimal SABT-0.15 has been evaluated via recycling experiments, as shown in Fig. 3c. The apparent activity loss has not been observed for RhB photodegradation during ten successive recycle runs, indicating that SABT-0.15 is able to serve as a stable visible light photocatalyst. Unfortunately, the size information of Au GSH clusters in SABT-0.15 after visible light illumination cannot be directly obtained due to the thick TiO2 shell. Alternatively, the SABT-0.05 composite with a thin layer of TiO2 has been selected as a representative sample for observing the size of Au GSH clusters after photocatalytic reactions. The TEM image (Supplementary Fig. 27) and EDX spectrum (Supplementary Fig. 28) of SABT-0.05 irradiated in RhB solution under visible light for 4 h suggest that the Au GSH clusters maintain their particle size, verifying the excellent photostability of Au GSH clusters in SABT-0.05.
Mechanism of the photoactivity improvement
To understand the origin of photoactivity enhancement of SABT compared to SAB sample and the effects of TiO2 shell thickness on photoactivities of SABT composites, we have investigated the structure-photoactivity relationship in terms of joint analysis on the charge separation efficiency and surface area. The photoelectrochemical (PEC) analysis has been performed to investigate the charge carrier transfer process over SiO2 spheres, SAB and SABT with different TiO2 shell thickness. Figure 3d shows the periodic on/off transient photocurrent response of these samples under the intermittent visible light photo-irradiation. Bare SiO2 spheres show no photocurrent response under visible light irradiation (λ > 420 nm), which is consistent with its large band gap47. After coupling with Au GSH clusters, the obvious photocurrent density over SAB verifies the substantial photosensitization effect of Au GSH clusters on the PEC performance of SAB in the range of visible light15,63,64. With the coating of TiO2 thin layer, the SABT-0.05 sample exhibits higher photocurrent density than SAB, which suggests that the separation of photoinduced charge carriers over SABT-0.05 has been efficiently facilitated as compared to that of SAB. However, a thicker TiO2 shell leads to the decreased photocurrent densities of SABT composites, which may be attributed to the fact that the thicker layer of TiO2 will block the transport of photogenerated electrons from Au GSH clusters to the back contact, thus reducing the overall photocurrent response. The cyclic voltammograms (CV) curves of bare SiO2 spheres, SAB and SABT composites in Fig. 3f reveal that the introduction of thin TiO2 layer can enhance the current densities of SABT composites as compared to that of SAB, while the thick TiO2 layer results in decreased current densities, which is consistent with the transient photocurrent results in Fig. 3d. Electrochemical impedance spectroscopy (EIS), as a method to monitor charge transfer process on the electrode and at the contact interface between electrode and electrolyte64, has further confirmed that the SABT with thin shell of TiO2 layer improves the charge carrier migration of SABT composites efficiently as compared to that with thick one (Supplementary Fig. 29). The above PEC experiments together confirm that the coating of thin TiO2 layer is beneficial for efficiently separating and transferring the photogenerated electrons from Au GSH clusters (Fig. 3e), hampering the recombination of charge carriers, and thereby resulting in the higher photocatalytic efficiency of SABT composites than that of SiO2 spheres and SAB under visible light illumination (λ > 420 nm).
The nitrogen (N2) adsorption–desorption isotherms have been performed to investigate the surface area of blank SiO2 spheres, SAB and SABT composites. As illustrated in Fig. 3h, i and Supplementary Table 3, the loading of Au GSH clusters leads to little variation of surface area of bare SiO2 spheres, while the surface area of the SABT is gradually improved by increasing the thickness of TiO2 shell. Adsorption experiments toward RhB (Fig. 3g) and p-methoxy nitrobenzene (Supplementary Fig. 30) in the dark suggest that the SABT composites exhibit improved adsorption capacity as compared to SiO2 spheres and SAB sample. In addition, with the increased thickness of TiO2 shell, the adsorption capacity of SABT composites increases, which is consistent with the surface area of SABT composites (Fig. 3i). The above characterizations suggest that the primary role of coating TiO2 semiconductor layer onto SAB for ameliorating the photocatalytic efficiency of SABT composites is two-fold. One is to improve the separation and migration efficiency of photoinduced charge carriers from excited Au GSH clusters. The other is to enhance the adsorption capacity of the catalysts toward the reactants. By adjusting the thickness of TiO2 shells, the photoactivity of SABT can be tuned due to cooperative adsorption and PEC properties. Therefore, the optimal SABT-0.15 composites exhibit the highest activity toward photocatalytic degradation of RhB among these SABT samples.
Control experiments using different radical scavengers help us to further understand the underlying reaction mechanism of the degradation of RhB over SABT-0.15. As shown in Supplementary Fig. 31, the addition of benzoquinone (BQ) scavenger for superoxide radicals65 almost terminates the photocatalytic reaction of RhB degradation. A similar and obvious inhibition phenomenon for photocatalytic reaction is also observed, when tert-butyl alcohol (TBA) scavenger for hydroxyl radicals66, ammonium formate (AF) scavenger for holes67, and potassium persulfate (K2S2O8) scavenger for electrons68 are added, indicating that the hydroxyl radicals, holes, electrons, and superoxide radicals all show a significant impact on the degradation of RhB. Nevertheless, the inhibition degree of photoactivity induced by the addition of TBA, AF, and K2S2O8 is smaller than the case of BQ, suggesting that the superoxide radicals play a more important role than other reactive species toward photodegradation of RhB over SABT-0.15. Taking K2S2O8 as scavenger for photogenerated electrons68, the control experiments for photoreduction of p-methoxy nitrobenzene under visible light illumination (λ > 420 nm) have been performed (Supplementary Fig. 32). The addition of K2S2O8 greatly inhibits the reduction of p-methoxy nitrobenzene, which confirms that the reduction reactions are driven by the photoexcited electrons. Supplementary Fig. 33 shows the electron spin resonance (ESR) spectra of superoxide radicals detected over the samples, which suggests that the coating of thin TiO2 shells contributes to improving the amounts of such reactive oxidative species (ROSs) generated over SABT. This is consistent with the more efficient separation and transfer of charge carriers over SABT than that over SiO2 spheres and SAB. Although the onslaught of ROSs on organic ligands of Au GSH clusters can lead to the instability of Au GSH clusters and their aggregation into large Au nanoparticles15, the following factors are considered to be the decisive role for achieving high photostability of Au GSH clusters in SABT composites. On one hand, as discussed above, BPEI has high reductive capability44,45, which can primitively protect the organic ligands of Au GSH clusters from being oxidized during the photo-irradiation process. On the other hand, the intelligent design of core-shell SABT composites by coating TiO2 shell can enhance the adsorption capacity of catalysts toward reactants, which would consume the ROSs timely and effectively reduce the possibility of the reactions between ROSs with the organic ligands of Au GSH clusters, thus leading to the improvement of the photostability of Au GSH clusters in SABT composites.
Discussion
Although prior literatures have already elucidated the coalescence and agglomeration mechanism of ultrasmall Au GSH clusters, it still remains a challenging task to achieve highly stable Au GSH clusters on the substrates under in situ photo-irradiation conditions, which significantly impedes the practical implementations of Au clusters-based catalysts systems. In this work, the utilization of multifunctional BPEI as a surface charge modifying, reducing and stabilizing agent for interfacial modification of SiO2-Au GSH clusters composites has demonstrated to be an efficient and universal strategy for protecting the Au GSH clusters from being fusion during longtime light illumination. Such photostability enhancement is ascribed to the high reductive ability of BPEI, by which the organic ligand oxidation process over Au GSH clusters can be inhibited, and this in turn results in the improvement of durability. Additionally, while simultaneously maintaining the photostability of Au GSH clusters, the sequential coating of thickness tunable TiO2 shells for constructing core-shell SiO2-Au GSH clusters-BPEI@TiO2 (SABT) nanostructures has been demonstrated to significantly ameliorate the photoactivity by regulating the photoelectrochemical and adsorption properties of SABT composites synergistically.
In summary, we have reported a combined strategy via interfacial modification and composition engineering for enhancing the long-term stability of ultrasmall Au GSH clusters with improved photocatalytic performance, which is expected to provide a facile guideline for rationally structural design of Au clusters-based composites toward various targeting applications in solar energy conversion.
Methods
Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O), acetonitrile (C2H3N, HPLC grade), sodium hydroxide (NaOH), hydrochloric acid (HCl), tetraethyl orthosilicate (C8H20O4Si, TEOS), isopropanol (C3H8O), ethanol (C2H6O), ammonia (NH3·H2O), p-methoxy nitrobenzene (C7H7NO3), rhodamine B (C28H31ClN2O3, RhB); ammonium formate (CH5NO2), potassium persulfate (K2S2O8), tert-butyl alcohol (C4H10O), p-benzoquinone (C6H4O2) were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). l-glutathione reduced (C10H17N3O6S, GSH), hexadecylamine (C16H35N, HDA), branched poly-ethylenimine (Mw ~25,000, BPEI) and titanium isopropoxide (C12H28O4Ti, TIP), 5,5-dimethyl-1-pyrroline-N-oxide (C6H11NO, DMPO) were obtained from Sigma-Aldrich. All materials were analytical grade and used as received without further purification. Deionized (DI) water used in the synthesis was from local sources.
Catalyst preparation
Synthesis of Au GSH clusters: Au GSH clusters were fabricated by procedures reported previously with modification31. Typically, 0.24 g of gold (III) chloride trihydrate were first dissolved in 200 mL DI water. Then, 100 mL DI water contained 0.276 g of l-glutathione were added into above solution under stirring. A colorless solution was obtained after 2 h stirring and the mixture was heated at 343 K in an oil bath for 24 h. After removing the heat source, the Au GSH clusters were recrystallized in acetonitrile and then thoroughly washed with DI water and acetonitrile (1: 3 in volume) for three times by centrifuging at 10,500×g for 5 min. A desired concentration of Au GSH clusters solution was obtained by redisperse the purified Au GSH clusters in DI water with assistance of few drops of 0.5 M NaOH and stored at 277 K for further use.
Synthesis of SiO2 spheres: In a typical reaction, a mixture contained 13 mL of ammonia (25–28%), 63.3 mL of isopropanol and 23.5 mL of DI water was heated to 308 K in an oil bath. A volume of 0.6 mL of TEOS (99%) was introduced into this solution dropwise and the mixture was kept at 308 K for 30 min. Then, 5 mL of TEOS were added dropwise into the reaction system. After 2 h reaction, the SiO2 spheres were separated by centrifugation, washed with ethanol and DI water repeatedly, and finally dried in air.
Synthesis of BPEI-modified SiO2 spheres: A volume of 4 mL BPEI solution (86 mg mL−1) was added into 200 mL ethanol with SiO2 spheres concentration of 2 mg mL−1. The mixtures were refluxed at 333 K in an oil bath for 4 h. The BPEI-modified SiO2 spheres were washed with ethanol and DI water repeatedly, and finally dried in air.
Synthesis of SiO2-Au GSH clusters-BPEI composite (SAB): 0.1 g positively charged BPEI-modified SiO2 spheres were dispersed in 50 mL DI water by ultrasonication. Then, 5 mL negative charged Au GSH clusters (0.2 mg mL−1) were added dropwise into this dispersion. After stirring for 1 h, SAB were isolated by centrifugation, washed with ethanol and DI water repeatedly, and finally dried in air.
Synthesis of core-shell SiO2-Au GSH clusters-BPEI@TiO2 nanostructures: 0.08 g of as-prepared SAB composites and 0.08 g of HDA (90%) were ultrasonically dispersed in 9.74 mL of ethanol. Then, 0.2 mL of ammonia was introduced into the solution under stirring. After 1 min, different amounts of TIP (97%) were added into the dispersion. After reaction for 10 min, the core-shell SiO2-Au GSH clusters-BPEI@TiO2 composites were collected by centrifugation and washed several times with DI water and ethanol54. The details for synthesis of Au25(SG)18 clusters, SiO2-Au25(SG)18 clusters-BPEI composite (SASB), BPEI modified-metal oxide nanoparticles (ZnO and ZrO2), metal oxide nanoparticles (ZnO and ZrO2)-Au GSH clusters-BPEI composites (MABs) and SiO2-Au GSH clusters-pH@TiO2 nanostructures by hydrolysis of TIP are presented in Supplementary Methods.
Characterization
Zeta sizer 3000HSA was employed to determine the Zeta potentials (ξ) values of the samples at room temperature. The optical properties of the samples were measured by UV-vis spectrophotometer (Perkin Elmer Lambda 950 UV-Vis-NIR). The XRD patterns of different samples were collected by a Bruker D8 Advance X-ray diffractometer (40 kV, 40 mA) using Ni-filtered Cu Kα radiation. The SEM images were taken on FEI Nova NANOSEM 230 spectrophotometer. TEM, EDX, and elemental mapping results were obtained by a JEOL model JEM 2010 EX instrument. PL spectra of the samples were recorded on an Edinburgh FL/FS920 spectrophotometer. XPS measurement was performed on a Thermo Scientific ESCA Lab 250 spectrometer with monochromatic Al Kα as the X-ray source. The C1s peak at 284.6 eV was used to calibrate binding energies. The loading amounts of Au GSH clusters in the SAB and SAP samples were quantified by an inductively coupled plasma optical emission spectroscopy instrument (ICP-OES, PerkinElmer Optima 2000DV). The surface areas of various samples were obtained by the Micromeritics ASAP 3020 equipment. Mass spectra were recorded on an Agilent Technologies ESI-TOF-MS. ESR signal was recorded using a Bruker EPR A300 spectrometer.
Photoelectrochemical analysis was carried out in a three-electrode quartz cell, among which Ag/AgCl electrode and Pt plate were used as reference electrode and counter electrode, respectively. FTO glass was used as support to prepare working electrode. Specifically, the slurry prepared by dispersing 5 mg sample in 0.5 mL of DMF was spread onto FTO glass with exposed area of 0.25 cm2. After air drying, the working electrodes were dried at 393 K for 2 h to enhance adhesion. BAS Epsilon workstation was used to measure the photocurrent density of samples in the electrolyte of 0.2 M Na2SO4 aqueous solution (pH = 6.8). CV and EIS experiments were carried out on Autolab, PGSTATM204 in the electrolyte of 0.5 M KCl aqueous solution containing 0.01 M K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) under open circuit potential conditions.
Photoactivity testing
Degradation of organic dye: Ten milligrams of catalyst were suspended in 40 mL of 10 mg L–1 RhB solution. The mixture was stirred in dark for 3 h and then irradiated with a 300 W Xe arc lamp (λ > 420 nm) under ambient conditions. After photocatalytic reaction, 4 mL of sample solution were taken from reaction system and analyzed on a Varian UV-vis spectrophotometer (Cary 50, Varian Co.).
Reduction of aromatic nitro compound: Fifty milligrams of photocatalyst and 100 mg ammonium formate were dispersed in 30 mL of 5 mg L−1 p-methoxy nitrobenzene solution. After stirring in dark for 3 h, the mixture was irradiated by visible light (λ > 420 nm) using a 300 W Xe arc lamp. After photocatalytic reaction, 4 mL of sample solution were taken from reaction system and analyzed on a Varian UV–vis spectrophotometer (Cary 50, Varian Co.).
Data availability
The data that support the findings of this study are available from the figshare repository with DOI number of 10.6084/m9.figshare.5938669.
References
Bell, A. T. The impact of nanoscience on heterogeneous catalysis. Science 299, 1688–1691 (2003).
Chen, T. & Rodionov, V. O. Controllable catalysis with nanoparticles: bimetallic alloy systems and surface adsorbates. ACS Catal. 6, 4025–4033 (2016).
Van Santen, R. A. Complementary structure sensitive and insensitive catalytic relationships. Acc. Chem. Res. 42, 57–66 (2009).
Roucoux, A., Schulz, J. & Patin, H. Reduced transition metal colloids: a novel family of reusable catalysts? Chem. Rev. 102, 3757–3778 (2002).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Corma, A. & Garcia, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 37, 2096–2126 (2008).
White, R. J., Luque, R., Budarin, V. L., Clark, J. H. & Macquarrie, D. J. Supported metal nanoparticles on porous materials. Methods and applications. Chem. Soc. Rev. 38, 481–494 (2009).
Cavicchi, R. E., Meier, D. C., Presser, C., Prabhu, V. M. & Guha, S. Single laser pulse effects on suspended-Au-nanoparticle size distributions and morphology. J. Phys. Chem. C 117, 10866–10875 (2013).
Besner, S., Kabashin, A. V., Winnik, F. M. & Meunier, M. Synthesis of size-tunable polymer-protected gold nanoparticles by femtosecond laser-based ablation and seed growth. J. Phys. Chem. C 113, 9526–9531 (2009).
Jin, R. et al. Photoinduced conversion of silver nanospheres to nanoprisms. Science 294, 1901–1903 (2001).
Qian, H., Zhu, M., Wu, Z. & Jin, R. Quantum sized gold nanoclusters with atomic precision. Acc. Chem. Res. 45, 1470–1479 (2012).
Jin, R. Atomically precise metal nanoclusters: stable sizes and optical properties. Nanoscale 7, 1549–1565 (2015).
Jin, R. et al. Size focusing: a methodology for synthesizing atomically precise gold nanoclusters. J. Phys. Chem. Lett. 1, 2903–2910 (2010).
Maity, P., Xie, S., Yamauchi, M. & Tsukuda, T. Stabilized gold clusters: from isolation toward controlled synthesis. Nanoscale 4, 4027–4037 (2012).
Liu, S. & Xu, Y.-J. Photo-induced transformation process at gold clusters-semiconductor interface: implications for the complexity of gold clusters-based photocatalysis. Sci. Rep. 6, 22742 (2016).
Habeeb Muhammed, M. A. & Pradeep, T. Au25@SiO2: quantum clusters of gold embedded in silica. Small 7, 204–208 (2011).
Negishi, Y., Kurashige, W., Niihori, Y. & Nobusada, K. Toward the creation of stable, functionalized metal clusters. Phys. Chem. Chem. Phys. 15, 18736–18751 (2013).
Jin, R., Zeng, C., Zhou, M. & Chen, Y. Atomically precise colloidal metal nanoclusters and nanoparticles: fundamentals and opportunities. Chem. Rev. 116, 10346–10413 (2016).
Abbas, M. A., Kim, T.-Y., Lee, S. U., Kang, Y. S. & Bang, J. H. Exploring interfacial events in gold-nanocluster-sensitized solar cells: insights into the effects of the cluster size and electrolyte on solar cell performance. J. Am. Chem. Soc. 138, 390–401 (2016).
Yu, C., Li, G., Kumar, S., Kawasaki, H. & Jin, R. Stable Au25(SR)18/TiO2 composite nanostructure with enhanced visible light photocatalytic activity. J. Phys. Chem. Lett. 4, 2847–2852 (2013).
Han, N. S. et al. Unexpected size effect observed in ZnO-Au composite photocatalysts. ACS Appl. Mater. Interfaces 8, 1067–1072 (2016).
Stamplecoskie, K. G., Chen, Y.-S. & Kamat, P. V. Excited-state behavior of luminescent glutathione-protected gold clusters. J. Phys. Chem. C 118, 1370–1376 (2014).
Xiao, F.-X. et al. Metal-cluster-decorated TiO2 nanotube arrays: a composite heterostructure toward versatile photocatalytic and photoelectrochemical applications. Small 11, 554–567 (2015).
Xiao, F.-X., Zeng, Z. & Liu, B. Bridging the gap: electron relay and plasmonic sensitization of metal nanocrystals for metal clusters. J. Am. Chem. Soc. 137, 10735–10744 (2015).
Negishi, Y. et al. Enhanced photocatalytic water splitting by BaLa4Ti4O15 loaded with ~ 1 nm gold nanoclusters using glutathione-protected Au25 clusters. Nanoscale 5, 7188 (2013).
Stamplecoskie, K. G. & Kamat, P. V. Size-dependent excited state behavior of glutathione-capped gold clusters and their light-harvesting capacity. J. Am. Chem. Soc. 136, 11093–11099 (2014).
Xiao, F.-X. et al. Light-induced in situ transformation of metal clusters to metal nanocrystals for photocatalysis. ACS Appl. Mater. Interfaces 7, 28105–28109 (2015).
Chen, Y.-S. & Kamat, P. V. Glutathione-capped gold nanoclusters as photosensitizers. Visible light-induced hydrogen generation in neutral water. J. Am. Chem. Soc. 136, 6075–6082 (2014).
Kogo, A., Sakai, N. & Tatsuma, T. Photocatalysis of Au25-modified TiO2 under visible and near infrared light. Electrochem. Commun. 12, 996–999 (2010).
Ramasamy, P. et al. Size tuning of Au nanoparticles formed by electron beam irradiation of Au25 quantum clusters anchored within and outside of dipeptide nanotubes. J. Mater. Chem. 19, 8456–8462 (2009).
Luo, Z. et al. From aggregation-induced emission of Au(I)–thiolate complexes to ultrabright Au(0)@Au(I)–thiolate core–shell nanoclusters. J. Am. Chem. Soc. 134, 16662–16670 (2012).
Cha, S.-H., Kim, J.-U., Kim, K.-H. & Lee, J.-C. Preparation and photoluminescent properties of gold(I)–alkanethiolate complexes having highly ordered supramolecular structures. Chem. Mater. 19, 6297–6303 (2007).
Wu, Z. & Jin, R. On the ligand’s role in the fluorescence of gold nanoclusters. Nano. Lett. 10, 2568–2573 (2010).
Shibu, E. S., Muhammed, M. A. H., Tsukuda, T. & Pradeep, T. Ligand exchange of Au25SG18 leading to functionalized gold clusters: spectroscopy, kinetics, and luminescence. J. Phys. Chem. C 112, 12168–12176 (2008).
Negishi, Y., Nobusada, K. & Tsukuda, T. Glutathione-protected gold clusters revisited: bridging the gap between gold(I)–thiolate complexes and thiolate-protected gold nanocrystals. J. Am. Chem. Soc. 127, 5261–5270 (2005).
Sapsford, K. E. et al. Functionalizing nanoparticles with biological molecules: developing chemistries that facilitate nanotechnology. Chem. Rev. 113, 1904–2074 (2013).
Sakai, N. & Tatsuma, T. Photovoltaic properties of glutathione-protected gold clusters adsorbed on TiO2 electrodes. Adv. Mater. 22, 3185–3188 (2010).
Negishi, Y., Chaki, N. K., Shichibu, Y., Whetten, R. L. & Tsukuda, T. Origin of magic stability of thiolated gold clusters: a case study on Au25(SC6H13)18. J. Am. Chem. Soc. 129, 11322–11323 (2007).
Li, H. et al. Mesoporous Au/TiO2 nanocomposites with enhanced photocatalytic activity. J. Am. Chem. Soc. 129, 4538–4539 (2007).
Jiang, J. et al. Oxidation at the core–ligand interface of Au lipoic acid nanoclusters that enhances the near-IR luminescence. J. Phys. Chem. C. 118, 20680–20687 (2014).
Huang, J., Dai, W.-L. & Fan, K. Remarkable support crystal phase effect in Au/FeOx catalyzed oxidation of 1,4-butanediol to γ-butyrolactone. J. Catal. 266, 228–235 (2009).
Battocchio, C. et al. Gold nanoparticles stabilized with aromatic thiols: interaction at the molecule–metal interface and ligand arrangement in the molecular shell investigated by SR-XPS and NEXAFS. J. Phys. Chem. C 118, 8159–8168 (2014).
Odio, O. F., Lartundo-Rojas, L., Santiago-Jacinto, P., Martínez, R. & Reguera, E. Sorption of gold by naked and thiol-capped magnetite nanoparticles: an XPS approach. J. Phys. Chem. C 118, 2776–2791 (2014).
Tan, S., Erol, M., Attygalle, A., Du, H. & Sukhishvili, S. Synthesis of positively charged silver nanoparticles via photoreduction of AgNO3 in branched polyethyleneimine/HEPES solutions. Langmuir 23, 9836–9843 (2007).
Kretschmer, F., Mansfeld, U., Hoeppener, S., Hager, M. D. & Schubert, U. S. Tunable synthesis of poly-(ethylene imine)-gold nanoparticle clusters. Chem. Commun. 50, 88–90 (2014).
Rau, S. et al. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem., Int. Ed. 45, 6215–6218 (2006).
Zhang, N. et al. Near-field dielectric scattering promotes optical absorption by platinum nanoparticles. Nat. Photon. 10, 473–482 (2016).
Zhang, N., Liu, S. & Xu, Y.-J. Recent progress on metal core@semiconductor shell nanocomposites as a promising type of photocatalyst. Nanoscale 4, 2227–2238 (2012).
Zhang, N., Zhang, Y. & Xu, Y.-J. Recent progress on graphene-based photocatalysts: current status and future perspectives. Nanoscale 4, 5792–5813 (2012).
Yuan, L., Yang, M.-Q. & Xu, Y.-J. Tuning the surface charge of graphene for self-assembly synthesis of a SnNb2O6 nanosheet-graphene (2D-2D) nanocomposite with enhanced visible light photoactivity. Nanoscale 6, 6335–6345 (2014).
Zhu, M., Aikens, C. M., Hollander, F. J., Schatz, G. C. & Jin, R. Correlating the crystal structure of a thiol-protected Au25 cluster and optical properties. J. Am. Chem. Soc. 130, 5883–5885 (2008).
Zhu, M., Eckenhoff, W. T., Pintauer, T. & Jin, R. Conversion of anionic [Au25(SCH2CH2Ph)18]–cluster to charge neutral cluster via air oxidation. J. Phys. Chem. C 112, 14221–14224 (2008).
Negishi, Y. Toward the creation of functionalized metal nanoclusters and highly active photocatalytic materials using thiolate-protected magic gold clusters. Bull. Chem. Soc. Jpn. 87, 375–389 (2013).
Guan, B. Y., Yu, L., Li, J. & Lou, X. W. A universal cooperative assembly-directed method for coating of mesoporous TiO2 nanoshells with enhanced lithium storage properties. Sci. Adv. 2, 1501554 (2016).
Lee, J., Park, J. C. & Song, H. A nanoreactor framework of a Au@SiO2 yolk/shell structure for catalytic reduction of p-nitrophenol. Adv. Mater. 20, 1523–1528 (2008).
Zhong, J., Chen, F. & Zhang, J. Carbon-deposited TiO2: synthesis, characterization, and visible photocatalytic performance. J. Phys. Chem. C 114, 933–939 (2010).
Weng, B., Liu, S., Zhang, N., Tang, Z.-R. & Xu, Y.-J. A simple yet efficient visible-light-driven CdS nanowires-carbon nanotube 1D–1D nanocomposite photocatalyst. J. Catal. 309, 146–155 (2014).
Sakatani, Y. et al. Optimised photocatalytic activity of grid-like mesoporous TiO2 films: effect of crystallinity, pore size distribution, and pore accessibility. J. Mater. Chem. 16, 77–82 (2006).
Liu, H. et al. Crystallinity control of TiO2 hollow shells through resin-protected calcination for enhanced photocatalytic activity. Energy Environ. Sci. 8, 286–296 (2015).
Zhang, Q., Gao, L. & Guo, J. Effects of calcination on the photocatalytic properties of nanosized TiO2 powders prepared by TiCl4 hydrolysis. Appl. Catal. B 26, 207–215 (2000).
Ohtani, B. & Ogawa, Y. & Nishimoto, S-i. Photocatalytic activity of amorphous–anatase mixture of titanium(IV) oxide particles suspended in aqueous solutions. J. Phys. Chem. B 101, 3746–3752 (1997).
Tian, G., Fu, H., Jing, L., Xin, B. & Pan, K. Preparation and characterization of stable biphase TiO2 photocatalyst with high crystallinity, large surface area, and enhanced photoactivity. J. Phys. Chem. C. 112, 3083–3089 (2008).
Xiao, F.-X., Miao, J. & Liu, B. Layer-by-layer self-assembly of CdS quantum dots/graphene nanosheets hybrid films for photoelectrochemical and photocatalytic applications. J. Am. Chem. Soc. 136, 1559–1569 (2014).
Weng, B., Quan, Q. & Xu, Y.-J. Decorating geometry- and size-controlled sub-20 nm Pd nanocubes onto 2D TiO2 nanosheets for simultaneous H2 evolution and 1,1-diethoxyethane production. J. Mater. Chem. A 4, 18366–18377 (2016).
Weng, B., Wu, J., Zhang, N. & Xu, Y.-J. Observing the role of graphene in boosting the two-electron reduction of oxygen in graphene–WO3 nanorod photocatalysts. Langmuir 30, 5574–5584 (2014).
Zhang, Y., Zhang, N., Tang, Z.-R. & Xu, Y.-J. Graphene transforms wide band gap ZnS to a visible light photocatalyst. The new role of graphene as a macromolecular photosensitizer. ACS Nano 6, 9777–9789 (2012).
Pan, X. & Xu, Y.-J. Efficient thermal- and photocatalyst of Pd nanoparticles on TiO2 achieved by an oxygen vacancies promoted synthesis strategy. ACS Appl. Mater. Interfaces 6, 1879–1886 (2014).
Yuan, L., Yang, M.-Q. & Xu, Y.-J. A low-temperature and one-step method for fabricating ZnIn2S4-GR nanocomposites with enhanced visible light photoactivity. J. Mater. Chem. A 2, 14401–14412 (2014).
Acknowledgements
Y.-J.X. acknowledges the support from the National Natural Science Foundation of China (U1463204, 21173045), the Award Program for Minjiang Scholar Professorship, the NSF of Fujian Province for Distinguished Young Investigator Rolling Grant (2017J07002), the Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment (No. 2014A05), the 1st Program of Fujian Province for Top Creative Young Talents, and the Program for Returned High-Level Overseas Chinese Scholars of Fujian province. H.M.C. appreciates the support from the Ministry of Science and Technology, Taiwan (Contracts No. MOST 106-2119-M-002-031).
Author information
Authors and Affiliations
Contributions
Y.-J.X. proposed the research direction and supervised the project. B.W. designed and preformed the experiments. Z.C.T. performed MS measurement and H.M.C. provided helpful suggestions in conducting the study. K.Q.L. carried out TEM characterization and analysis. Y.-J.X. and B.W. wrote and revised the manuscript. All authors participated in discussion and reviewed the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weng, B., Lu, KQ., Tang, Z. et al. Stabilizing ultrasmall Au clusters for enhanced photoredox catalysis. Nat Commun 9, 1543 (2018). https://doi.org/10.1038/s41467-018-04020-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-04020-2
This article is cited by
-
Atomically precise photothermal nanomachines
Nature Materials (2024)
-
Molecular motion cools off gold nanoclusters
Nature Materials (2024)
-
Co-introduction of oxygen vacancies and cocatalysts into protonic titanate derived TiO2 nanoparticles for enhanced photocatalytic hydrogen production
Carbon Letters (2024)
-
Facet-engineered TiO2 drives photocatalytic activity and stability of supported noble metal clusters during H2 evolution
Nature Communications (2023)
-
Hot spots engineering by dielectric support for enhanced photocatalytic redox reactions
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
