
Abstract
In Drosophila, the JNK pathway eliminates by apoptosis aberrant cells that appear in development. It also performs other functions associated with cell proliferation, but analysis of the latter is hindered by the pro-apoptotic activity. We report the response of apoptosis-deficient cells to transient activation of JNK and show that it causes persistent JNK function during the rest of the development. As a consequence, there is continuous activity of the downstream pathways JAK/STAT, Wg and Dpp, which results in tumour overgrowths. We also show that the oncogenic potential of the Ras-MAPK pathway resides largely on its ability to suppress apoptosis. It has been proposed that a hallmark of tumour cells is that they can evade apoptosis. In reverse, we propose that, in Drosophila, apoptosis-deficient cells become tumorigenic due to their property of acquiring persistent JNK activity after stress events that are inconsequential in tissues in which cells are open to apoptosis.
Similar content being viewed by others


Introduction
Apoptosis, i.e. programmed cell death, is a physiological process by which cells trigger their own destruction. Although there is developmentally programmed apoptosis, i.e. the removal of the tail of tadpoles or the articulations of Drosophila legs1,2, a major function of apoptosis is the elimination of aberrant cells that may compromise the viability or the fitness of the organism3,4,5. Apoptosis also functions to remove, through a process known as cell competition3,6,7, oncogenic cells that may arise during development4,8.
The study of apoptosis in Drosophila is facilitated by the simplicity of the genetic program, the availability of mutants affecting apoptosis and of methods to manipulate gene activity. Within Drosophila, the wing imaginal disc (the precursor cells of the adult wing and thorax) is a convenient experimental system, for while it normally does not contain cells in apoptosis, it exhibits a strong apoptotic response to ionising radiation or other forms of stress9. A simplified version of the apoptosis program is shown in Fig. 1a.

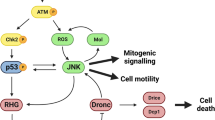
JNK persistent activity after irradiation or a short pulse of p53 and JNK in apoptosis-deficient cells. a Simplified representation of the apoptotic pathway in Drosophila. JNK/p53 are activated after irradiation and induce the activation of the pro-apoptotic genes that block DIAP1, inducing the activation of the apical caspase Dronc and effector caspases Dcp1 and Drice. Besides, Dronc reinforces JNK activity generating an amplification loop that augments the apoptotic response26. As a side event, JNK activates the Dpp, Wg and JAK/STAT pathways. b-g Wing discs showing the effects of X-rays at different time points. JNK activity is monitored by the expression of a LacZ insert at the JNK target puckered (puc, red). The blue background reflects TOPRO staining. b, c puc expression in non-irradiated dronc+ control (n = 10) and null dronc− mutant (n = 17) discs. Note that the label is restricted in both cases to a small region that corresponds to the most proximal zone of the disc. d, e Twenty-four hours after irradiation, both dronc+ (n = 22) and dronc− (n = 18) discs show high expression levels of puc, especially in the region corresponding to the wing pouch. f, g puc expression 96 h after irradiation in dronc+ (n = 28) and dronc− (n = 31) discs. Whereas puc levels have returned to normal in dronc+ (f), they remain high in many zones of the dronc− discs (g). Wing discs of genotype: h UAS-p53/UAS-GFPtubGal80ts;hh-Gal4dronci29/+ (n = 10), i UAS-p53/UAS-GFPtubGal80ts;hh-Gal4dronci29/dronci29 (n = 14), j UAS-hepCA/UAS-GFPtubGal80ts;hhGal4dronci29/+ (n = 10) and k UAS-hepCA/UAS-GFPtubGal80ts;hh-Gal4dronci29/dronci24 (n = 11). Discs are stained with Cubitus interruptus, Ci (green) to mark the anterior compartment and Mmp1 (red) to monitor JNK activity. The short pulse of p53 (h, i) or of hepCA (j, k), at the posterior compartment is administered by temperature shift, detailed in Methods section; 96 h after the change to 17 °C, no activity of JNK can be detected by Mmp1 staining (red) in the posterior compartment (lack of green) in p53 dronc+ (h) and hepCA dronc+ discs (j). In contrast, there is JNK activity (red) at the posterior compartment (lack of green) of p53 dronc− (i) and in hepCA dronc− discs (k). Scale bars are 100 μm
A prime event in the apoptotic response in vertebrates and Drosophila10,11,12 is the activation of the Jun N-terminal kinase (JNK) signalling pathway, a member of the MAPKs family involved in various cellular processes13. In the Drosophila wing disc, JNK is not normally active—except in a small proximal region (Fig. 1b, c)—but after irradiation, there is overall induction of JNK and subsequent apoptosis10,11 (Fig. 1d, e), aimed to remove cells damaged by the irradiation. The apoptotic role of JNK also mediates the elimination of oncogenic cells by cell competition, thus revealing its anti-tumour role4,14.
In addition to its apoptosis-inducing function, JNK is known to play non-apoptotic roles connected with cell proliferation15,16,17. For example, sustained expression of JNK is associated with cancer in vertebrates18,19, and in Drosophila we have shown that forcing JNK activity causes overgrowths in apoptosis-defective wing discs17. Thus, both in flies and in vertebrates, JNK may function as anti-tumorigenic or pro-tumorigenic, depending on the context.
Another factor associated with tumorigenesis in vertebrates is the inappropriate expression of the Ras-MAP kinase pathway20,21. In Drosophila, Ras overexpression can also cause massive overgrowths through interactions with mutations at tumour suppressing genes22,23. Significantly, the development of those overgrowths requires JNK activity3,23. One noteworthy feature of the overexpression of Ras-MAPK in Drosophila is that it makes cells refractory to apoptosis24,25, and a similar observation has been made in mammalian cells26.
Our results indicate that cells refractory to apoptosis, they may lack the apoptosis machinery or overexpress the Ras pathway, become tumorigenic due to their property of acquiring sustained JNK activity after its transient activation due to stress or other events.
Results
JNK sustained activity in apoptosis-defective cells
In our experiments, we generated entire discs or disc domains that cannot initiate apoptosis. They may be mutant for the caspase Dronc, or may lack activity of pro-apoptotic genes (see Fig. 1 and Supplementary Fig. 1 for details), or may contain constitutive expression of the Ras-MAPK pathway24,25. Then, we analysed the response to events leading to JNK activation such as X-rays, a pulse of p53, or a pulse of JNK itself.
We first analysed JNK response to ionising radiation (3-4000R) using the targets puckered (puc) and metalloprotease 1 (mmp1) to monitor JNK function. Except for the resident JNK expression in a small proximal region (Fig. 1b, c), any additional activity has to be caused by the experimental intervention. We compared the response of discs in which apoptosis is not prevented (controls) with that of discs in which cells cannot undergo apoptosis.
In irradiated discs open to apoptosis, we observe high puc levels 24 h after the irradiation (Fig. 1d), but afterwards those levels decay and eventually disappear by 96 h (Fig. 1f). Cells expressing puc also exhibit Dcp1 effector caspase activity indicating that they are in apoptosis. These results confirm previous work and illustrate the JNK role of removing cells damaged by irradiation10,11.
The JNK response to irradiation of apoptosis-deficient discs is very different.
Although the apoptotic response is suppressed, there is a strong activation of the JNK marker puc, especially in the pouch 24 h after X-rays (Fig. 1e). However, unlike the response of dronc+, mutant dronc− discs maintain high puc levels for the rest of the larval development, including pre-pupal stages (Fig. 1g).
We find similar results in discs in which apoptosis is suppressed only in the posterior compartment: 96 h after irradiation, there are high levels of JNK in the posterior but not in the anterior compartment (Supplementary Fig. 1a, b). Further evidence comes from clones homozygous for the H99 deletion (lacking major pro-apoptotic genes), many of which express JNK markers 96 h after the irradiation, whereas surrounding cells do not (Supplementary Fig. 1c).
We also performed experiments to follow the lineage of the cells that acquire JNK activity after irradiation, both in cells open to apoptosis (dronc+) and in apoptosis-defective (dronc−) cells.
Irradiation of dronc+ discs of genotype puc-Gal4 UAS-Flp UAS-GFPDbox act<stop>lacZ activates puc-Gal4 in many of their cells. These cells become labelled with GFPDbox, an unstable form of GFP27, to avoid an effect of perdurance, and also produce high levels of the recombinase Flippase. In the majority of those cells, Flippase induces recombination in the act<stop>lacZ cassette, what labels them and their progeny indelibly with LacZ. The puc-expressing cells that do not die generate clones marked with LacZ expression (see Supplementary Fig. 1 for details). Twenty-four hours after X-rays, we find a large number of cells expressing GFPDbox and the majority of them are also labelled with lacZ (Supplementary Fig. 1d). Seventy-two hours after irradiation, we observed a smaller number of large LacZ patches, but none of them expresses GFPDbox (Supplementary Fig. 1e), indicating that JNK has been turned off, consistent with the results above (Fig. 1f). The large size of those patches suggests that the mature disc has been reconstituted by few cells, many of which are descendants from those in which JNK was activated by the irradiation. This is proof that X-rays can activate JNK to sub-lethal levels.
The same lineage analysis after irradiation of dronc− discs yields different results. The principal difference is that 72 h after the irradiation, we still observed many groups of cells expressing GFPDbox (Supplementary Fig. 1g), again indicating the persistence of JNK expression in apoptotic defective cells and reinforcing the results above (Fig. 1g). In these discs, the size of the lacZ patches is smaller than in the dronc+ discs, which is expected because the number of cells is greater as there is no cell death after the irradiation.
To explore the possibility that just an initiation event is sufficient to generate persistent JNK activity in apoptosis-defective cells, we carried out two experiments in which we checked JNK expression several days after a short pulse of induction (see Methods for details). In the first experiment, a 16 h pulse of the JNK activator p5328 in the posterior compartment causes persistent JNK activity, visualised 96 h after the pulse (Fig. 1i). In the second experiment, we obtain a similar result after a 16 h pulse of JNK itself (Fig. 1k), achieved by the expression of a form of hemipterous (hepCA, a JNKK4), which causes constitutive activity of the pathway. In control discs, in which apoptosis is not prevented, the brief expression of JNK (Supplementary Fig. 1h) rapidly subsides (Fig. 1h, j).
ROS and moladietz are required for JNK persistent activity
The preceding experiments establish that events leading to JNK activation trigger a genetic operation that causes its sustained adventitious function. This phenomenon is of little consequence when cells are open to apoptosis because they die shortly after JNK induction or recover and the JNK pathway is turned off, but becomes prominent when cells are refractory to apoptosis. This persistent activity cannot be due to the apoptotic amplification loop28 because in the absence of the function of dronc or the pro-apoptotic genes hid/rpr/grim (DfH99 deletion), the loop cannot be completed.
We sought to identify the mechanism(s) responsible for the persistence of JNK activity after a transient initiation event. It has been shown that cellular damage to the wing disc induces a burst of reactive oxygen species (ROS), which in turn activate JNK29. Furthermore, recent work on wing disc regeneration30 has identified the gene moladietz (mol), transcriptionally activated by JNK, which encodes a Duox maturation factor necessary for production of ROS, with subsequent induction of JNK. JNK and mol establish a positive amplification loop that ensures prolonged JNK function during the regeneration process30.
Therefore, we checked the involvement of ROS and mol on the phenomenon of persistent JNK function observed in our experiments. First, using dihydroethidium (DHE) staining31 to monitor ROS production, we found high DHE levels in dronc− discs 96 h after irradiation (Fig. 2b), whereas DHE is not detected in dronc+ discs (Fig. 2a). Second, mol activity, visualised with a mol-LacZ insert, is also detected in dronc− discs 96 h after irradiation (Fig. 2e), but not in controls (Fig. 2d).

ROS and moladietz are involved in JNK persistent activity. a, b Wing discs stained 96 h after irradiation with DHE to detect ROS, a dronc+ control (n = 10) and b dronc− mutant (n = 30). We observe DHE staining in dronc− mutant disc (b, arrows) in cells that present JNK activity, labelled by the expression of UAS-GFPDbox, but not in (a) dronc+ control. c Wing disc of the genotype: UAS-SOD:UAS-Cat/UAS-GFPtubGal80ts;dronci24/dronci29hh-Gal4 96 h after irradiation (n = 14) (after irradiation, larvae were raised for 24 h at 17 °C to allow ROS activation and then transferred to 29 °C for 72 h to block ROS production). We observe a reduction in the activation of JNK (puc-LacZ, red) only in the posterior compartment (GFP, green), where SOD:Cat are expressed. Wing discs of the genotypes: d (n = 9) TRE:RFP/mol-LacZ and e (n = 22) TRE:RFP/mol-LacZ;dronci24/dronci24 96 h after irradiation where mol-LacZ expression (green) is maintained in dronc− mutants (e) in cells that show JNK activity (TRE:RFP, red). No RFP or ectopic mol-LacZ is observed in (d) dronc+ control. f Wing disc of the genotype: UAS-molRNAi/UAS-GFPtubGal80ts;dronci24/hh-Gal4dronci29 (n = 7) 96 h after irradiation. As in c, the mol RNAi was expressed the last 72 h after irradiation. Reduction of the JNK activity (Mmp1, red) is observed in the posterior compartment (green), but not in the anterior. Scale bars are 100 μm
We checked the functional requirements of ROS and mol activity for the sustenance of JNK function. The reduction of ROS levels in the posterior compartment by overexpression of Superoxide dismutase (SOD) and Catalase (Cat)29, or the suppression of mol function using a molRNAi construct30, resulted in reduced JNK activity in both cases 96 h after the irradiation (Fig. 2c, f).
Sustained JNK induces ectopic expression of target pathways
The preceding results establish the involvement of ROS and mol in the continuity of JNK function after transient activation. Next, we examined whether the persistent JNK function in apoptosis-deficient cells causes continuous mis-expression of pathways downstream JNK like JAK/STAT23,32, Dpp and Wg17,33. Indeed, JAK/STAT, Wg and Dpp are induced after irradiation in territories outside their normal domains, and their expression is associated with the zones of JNK ectopic activity (Fig. 3b, e, g). Moreover, the ectopic expression of JAK/STAT and Wg depends on JNK, for compromising JNK activity with a dominant negative form of Bsk (JNK itself) suppresses their expression (Fig. 3c, h).

JNK persistent activity induces sustained activity of the JAK/STAT, Dpp and Wg pathways. a–c Images of wing discs of genotypes: a STAT-GFP;pucLacZ dronci24/+(n = 15), b STAT-GFP pucLacZ dronci24/dronci29 (n = 29), c UAS-BskDN;nub-Gal4/STAT-GFP;puc-lacZ dronci24/dronci29 (n = 3), 96 h after irradiation stained for STAT-GFP (green) and β-gal (red). Note in b the ectopic STAT expression, especially in the wing pouch, associated with ectopic JNK activity. In c, the suppression of JNK, achieved by forcing bskDN in the wing pouch (the domain of nubbin outlined in yellow), prevents ectopic STAT expression. d, e Images of wing discs of genotypes: d dpp-LacZ;dronci24/+(n = 14) and e dpp-LacZ;dronci24/dronci29 (n = 17) 96 h after irradiation; dpp expression is monitored by a LacZ insert at the dpp locus (dpp-LacZ, green) and JNK function is indicated by Mmp1 staining (red). The dronc+ disc (d) shows wildtype dpp pattern, whereas in dronc− (e) there is ectopic dpp expression (arrows) associated with Mmp1 cells. f, g Images of wing discs of genotypes: f pucLacZ dronci24/+ (n = 9), g puclacZ dronci24/dronci29 (n = 22) 96 h after irradiation, stained with anti-Wg antibody (red) and puc-LacZ (green) to label JNK activity. The wg expression pattern is normal in dronc+ (f), but in dronc−, there is clear ectopic expression (g), especially in the wing pouch (arrow). Note the overall coincidence between the zones of ectopic wg expression and of JNK activity. And h UAS-bskDN;UAS-GFP;puc-lacZ dronci24/hh-Gal4dronci29 (n = 8) 96 h after irradiation, stained with anti-Wg antibody (red), the suppression of JNK activity by bskDN in the posterior compartment (green) prevents ectopic wg expression. Scale bars are 100 μm
Sustained JNK induces tissue overgrowth
We also observed a correlation between the continuous expression of JNK and an increase of cell proliferation, the latter monitored by EdU incorporation, which results in the formation of overgrowths (Fig. 4a, b, d). We also find that the development of the overgrowths depends on JNK activity: in irradiated hh>RHGmiRNA discs the overgrowth and increase of EdU incorporation in the posterior compartment is suppressed if they contain the dominant negative form of Bsk (Fig. 4c, d).

Persistent JNK activity causes overgrowths. a–c Discs of genotypes: a UAS-GFP;hh-Gal4 (n = 10), b UAS-RHGmiRNA/UAS-GFP; hh-Gal4/+(n = 20) and c UAS-BskDN; UAS-RHGmiRNA/UAS-GFP; hh-Gal4/+(n = 11) 72 h after X-rays. EdU incorporation (red) is increased in the posterior compartment (green) of b where apoptosis is suppressed by UAS-RHGmiRNA, but when JNK activity is blocked with BskDN (c), EdU incorporation is similar in both compartments, which is the normal situation (a). d Quantification of the overgrowth in UAS-RHGmiRNA/UAS-GFP;hh-Gal4/+disc (n = 32) after irradiation compared to non-irradiated discs (n = 35) (***p-value < 0.0001). Suppression of JNK by BskDN (n = 14) reverses the posterior compartment to normal size (***p-value < 0.0001), indicating that JNK is responsible for the overgrowth. e, f Discs of genotypes: e UAS-GFPtubGal80ts/+;dronci24/hh-Gal4dronci29 (n = 10) and f UAS-BskDN;UAS-GFPtubGal80ts/+;dronci24/hh-Gal4dronci29 (n = 23) 96 h after X-rays (see details of the experiment in the Methods section). Discs were stained with Mmp1 (red) to monitor JNK activity and GFP (green) to identify the P-compartment; TOPRO staining delimits the discs. The disc in e shows Mmp1 label in both compartments, but expression of the BskDN blocks JNK activity (Mmp1, red) as shown in f. g Quantification of the posterior compartment volume over the total disc volume for the genotypes: UAS-GFPtubGal80ts/+; dronci24/hh-Gal4 dronci29 (column: hh>GFPdronc− X-rays, n = 79), UAS-BskDN; UAS-GFPtubGal80ts/+;dronci24/hh-Gal4dronci29 expressing BskDN 24 h (column: hh>BskDNdronc− 24 h after X-rays, n = 43) or 48 h after irradiation (column: hh>BskDNdronc− 48 h after X-rays, n = 18). There is a significant size decrease when BskDN is expressed 24 h and 48 h after irradiation (***p-value < 0.0001), indicating that inhibiting JNK after irradiation suppresses overgrowth. Quantification for the genotypes: UAS-GFPtubGal80ts/UAS-SOD:UAS-Cat;dronci24/hh-Gal4dronci29 (column: hh>SOD:Cat dronc− 24 h after X-rays, n = 14) inducing the expression of SOD:Cat 24 h after irradiation and UAS-GFPtubGal80ts/UAS-molRNAi; dronci24/hh-Gal4dronci29 (column: hh>molRNAidronc− 24 h after X-rays, n = 7) inducing the expression of molRNAi 24 h after irradiation. There is a significant size decrease after the expression of SOD:Cat and the inhibition of mol (**p-value < 0.001 and *p-value < 0.02, respectively), indicating that ROS levels are important to maintain JNK overgrowth. Columns in the graphs represent mean ± s.d. Statistical test used: Mann–Whitney U-test. Scale bars are 100 μm
To test if the overgrowth observed in the RHGmiRNA experiment is induced by the sustained expression of JNK, we made use of the tub-Gal80ts transgene to suppress the JNK pathway with BskDN 24 and 48 h after the irradiation in the posterior compartment of dronc− mutants. In the 24 h experiment, we already found a significant reduction of the overgrowth in the posterior compartment (Fig. 4f, g), which is also observed in the 48 h experiment (Fig. 4g). Along the same lines, reduction of ROS levels or of mol function 24 h after the irradiation also results in suppression of the overgrowths (Fig. 4g). Overall, these results indicate that the overgrowths observed are dependent on persistent JNK activity.
Lack of apoptosis in ras V12 tissue leads to tumorigenesis
Overexpression of the Ras-MAPK pathway is associated with tumour development in vertebrates and Drosophila34,35,36,37,38. Since it renders cells refractory to stress-induced apoptosis24,25,26, we surmised that this property might confer a tumorigenic potential to cells with elevated levels of Ras activity.
In Drosophila, the overexpression of Ras does not generate significant overgrowth, although it up regulates the oncogene dMyc39,40. In our experiments, the growth of clones or body regions with constitutive Ras function (induced by the rasV12 transgene) is like in the wildtype: the size of the Sal domain (a region covering 15% of the wing disc) is similar in salEpv>GFP (control) and salEpv>rasV12 discs (Supplementary Fig. 2 a–d), and the growth rate of clones containing rasV12 is also similar to that of the surrounding tissue (Supplementary Fig. 2e). We confirmed that rasV12-expressing cells show high levels of dMyc41 (Supplementary Fig. 2d, e). We also confirmed that those cells exhibit a reduced apoptotic response to X-rays: the apoptotic levels after irradiation of the posterior compartment or the Sal domain containing RasV12 are much lower than in controls (Supplementary Fig. 3a–c).
We then checked whether the lack of apoptotic response of rasV12-expressing cells also causes sustained JNK activity after irradiation. Indeed, in salEpv>rasV12 (Supplementary Fig. 3d) and in hh>rasV12 discs (Fig. 5b), JNK remained active in the Sal domain and the posterior compartment, respectively, 72 and 96 h after irradiation. Irradiated (Fig. 5a) and non-irradiated (Fig. 5d, e) controls show no JNK activity. Moreover, Ras activity is associated with ectopic activity of the JAK/STAT and Wg pathways (Fig. 5g, h) and with overgrowth of the posterior compartment (Fig. 5f). These overgrowths are likely the result of the inactivation of the Hippo pathway42 and require JNK activity, for in irradiated hh>rasV12 UAS-puc discs the overgrowth is much reduced (Fig. 5c, f). This observation links the tumorigenic property of the Ras pathway with JNK activity. Interestingly, Ras-induced oncogenic transformations in vertebrates also require c-JNK activity43.

Constitutive expression of the Ras-MAPK pathway causes overgrowths due to permanent activity of the JNK pathway after irradiation. Images of wing discs of genotypes: a control tubGal80ts/+;hh-Gal4/+(n = 13), b UAS-RasV12/tubGal80ts;hh-Gal4/+(n = 9) and c tubGal80ts/UAS-RasV12;hhGal4/UAS-puc2A (n = 11) 72 h after irradiation. Non-irradiated wing discs of genotypes: d control tubGal80ts/+;hh-Gal4/+(n = 9) and e UAS-RasV12/tubGal80ts;hh-Gal4/+ (n = 10). Anterior compartments are stained with Ci (green). JNK activity is monitored by Mmp1 (red). Staining with TOPRO facilitates delimiting the discs. Note in b the large overgrowth of the posterior compartment that contains UAS-RasV12, associated with ectopic permanent JNK activity. Much of the overgrowth is prevented (c) when JNK function is compromised by overexpressing puc. There is no Mmp1activity (red) in non-irradiated discs (d, e). f Quantification of the posterior compartment area (in percentage) over the total disc area in the genotypes: non-irradiated tubGal80ts/+;hh-Gal4/+control (column: control No X-rays, n = 12), tubGal80ts/UAS-RasV12;hh-Gal4/+(column: RasV12 No X-rays, n = 6), and irradiated tubGal80ts/+;hh-Gal4/+control (column: control X-rays, n = 8), tubGal80ts/UAS-RasV12;hh-Gal4/+(column: RasV12 X-rays, n = 9) and tubGal80ts/UAS-RasV12;hhGal4/UAS-puc2A (column: RasV12 puc X-rays, n = 34). There is a statistically significant size increase of the posterior compartment after irradiation when RasV12 is expressed, compared with non-irradiated discs and with irradiated controls (***p-value < 0.0004, Mann–Whitney U-test). However, if JNK activity is suppressed by overexpressing puc, the size of the posterior compartment significantly decreases (***p-value < 0.0001, Mann–Whitney U-test). Columns in the graph represent mean ± s.d. Images of wing discs of genotypes: g STAT-GFP/tubGal80ts;UAS-RasV12/hh-Gal4 (n = 20) and h UAS-GFP,tubGal80ts/UAS-RasV12;hh-Gal4/+(n = 9) 72 h after irradiation; g shows the ectopic expression of STATGFP (green) only in the posterior compartment (delimited by the lack of Ci in red) where RasV12 is expressed; h shows the ectopic expression of Wg (red) only in the posterior compartment (green) where RasV12 is active. Scale bars are 100 μm
Since in rasV12 cells the apoptotic response is not completely suppressed, we studied the growth of rasV12 tissue also mutant for dronc. After irradiation, salEpv>rasV12 dronc− discs showed very large overgrowths in comparison with non-irradiated discs of the same genotype (Supplementary Fig. 3e, f). In this experiment, we introduced a lineage-tracing cassette (see Methods) to follow the progeny of rasV12-expressing cells, which expand to the surrounding tissue. It is worth emphasising that these tumorigenic features are triggered by a single 3000 R irradiation administered 96 h before. In these discs, we compared the relative growth of the rasV12 tissue with that of the rest of the disc. As shown in Supplementary Fig. 3F, the rasV12 cells generate a disproportionate part of the overgrowth. We believe that the strong tumorigenic character of irradiated rasV12 cells defective in apoptosis is caused by the sum of two factors: (1) persistent tumorigenic JNK activity and (2) the high metabolic activity conferred by the high dMyc levels40,44,45.
Discussion
JNK is associated in vertebrates and in Drosophila both with anti-tumorigenic and pro-tumorigenic roles. The anti-tumour function of the single Jun kinase of Drosophila is achieved by its ability to induce apoptosis in oncogenic cells3,4. In our experiments, we have characterised the pro-tumorigenic function of JNK in apoptosis-suppressed cells by examining the response to a brief induction of JNK caused by X-rays or by a short pulse of p53, or of JNK itself. We find that it causes sustained activity of JNK and of genetic factors, e.g. the JAK/STAT, Wg and Dpp pathways, which function downstream of JNK. Furthermore, it gives rise to the appearance of tumorous overgrowths caused by over-proliferation of the apoptosis-defective cells. We emphasise the differences between these experiments and those reported after blocking cell death in apoptotic cells with the baculovirus protein P359,33. Those “undead” cells persistently express JNK, Dpp and Wg, which generate tissue overgrowths. However, in undead cells, the apoptosis program remains fully active, except for the blocking of the effector caspases, and the continuous expression of Dronc generates persistent JNK function28,46. In contrast, in the present experiments, the apoptosis program is completely suppressed.
The fact that a short-term activation event is sufficient to induce persistent JNK activity in apoptosis-defective tissues defines an important property of the pathway: it includes a self-sustenance mechanism. In normal tissues open to apoptosis, this feature is largely irrelevant because the cells die soon after JNK activation, but in cells refractory to apoptosis, the function of JNK and of subsidiary pathways is maintained for the rest of the development of the disc. The result is the production of tumour outgrowths. A key aspect of this process is the mechanism of JNK sustenance after the initial event. Based on the results of Khan et al.30 and our own, we propose a simple model: the activation of JNK (by X-rays, or P53 or Hep itself) induces mol function, which generated high ROS levels that in turn induce JNK function. As mol is induced downstream of JNK30, it generates an amplification loop that maintains indefinite activity of JNK.
Our results also bear on the tumorigenic properties of the Ras pathway, whose deregulated expression is associated with cancer development in mammals21. We show that the principal tumorigenic feature of overexpressing Ras in Drosophila is that it makes cells refractory to apoptosis; these cells can acquire irrepressible JNK activity after an irradiation event that would have eliminated them but for the overexpression of Ras. It is of interest that Ras activity is known to have anti-apoptotic activity in mammalian cells26, although the apoptotic response to irradiation has not been studied. It is also worth noting that oncogenic transformations associated with de-repressed function of Ras in vertebrates require activity of the c-Jun pathway43; thus both in Drosophila and in vertebrates, tumour development appears to require combined activities of the Ras and JNK pathways.
In their review of Cancer, Hanahan and Weinberg21 state that one principal feature of cancer cells is that they can evade apoptosis. We would argue the reverse: it is the cells that evade apoptosis that become oncogenic; a simple stress to apoptosis-deficient tissue may have tumorigenic consequences.
Methods
Drosophila strains
The Drosophila stocks used in this study were: dronci29, dronci24 (A Bergmann, MD Anderson Center, Houston, TX, USA); puc-lacZ line (pucE69)47, hh-Gal4 and en-Gal4 (gift of T Tabata, IMBC, Tokyo, Japan), nub-Gal4(our own lab), salEpv-Gal4 (gift from J. F. de Celis, CBMSO, Madrid, Spain), tub-Gal80ts48, UAS-RHGmiRNA49, STAT-10xGFP50, act>stop>lacZ (gift of G. Struhl, Harvard Medical School, Boston, MA, USA), UAS-SOD:UAS-Cat29, TRE:RFP51, UAS-GFPDbox27, M(3) 67C FRT2A Ubi-GFP, UAS-Flp, UAS-GFP, Df (3L)H99, UAS-hepCA, UAS-p53.EX, P(dpp-lacZ), UAS-BskDN, UAS-puc2A, UAS-molRNAi, mol-LacZ and UAS-Ras85DV12 (Bloomington Drosophila Stock Center).
Imaginal discs staining
Immunostaining was performed as described previously28. Images were captured with a Leica (Solms, Germany) DB5500 B confocal microscope. The following primary antibodies were used: rabbit anti-Dcp1 (Cell Signalling, antibody #9578) 1:200; mouse anti-β -galactosidase (DSHB 40-1a) 1:50; rabbit anti-β-galactosidase (ICN Biomedicals) 1:2000; mouse anti-Wingless (DSHB 4D4) 1:50; mouse anti-Mmp-1 (DSHB, a combination, 1:1:1, of 3B8D12, 3A6B4 and 5H7B11) 1:50; rat anti-Ci antibody (DSHB 2A1) 1:50; rabbit anti-PH3 (Millipore) 1:100 and guinea pig anti-Myc (our own lab) 1:100. Fluorescently labelled secondary antibodies (Molecular Probes Alexa) were used in a 1:200 dilution. TO-PRO3 (Invitrogen) was used in a 1:600 dilution to label the nuclei.
DHE staining was performed as in Owusu-Ansah et al.31 with modifications. Larvae were dissected in 1× PBS and incubated with DHE (ThermoFisher Scientific, catalogue number D1168) 30 μM for 5 min at room temperature followed by three washes of 5 min in 1× PBS. Discs were mounted in 1× PBS and images were captured with Leica (Solms, Germany) DB5500 B confocal microscope.
All the n numbers stated in the text represent individual discs of the mentioned genotypes. All experiments are replicated at least three times.
p53/JNK short pulse experiments
Larvae of the genotypes: UAS-hepCA /UAS-GFP tubGal80ts;hh-Gal4 dronci29/+, UAS-hepCA /UAS-GFP tub-Gal80ts; hh-Gal4 dronci29/dronci24, UAS-p53 /UAS-GFP tub-Gal80ts; hh-Gal4 dronci29/+ and UAS-p53 /UAS-GFP tub-Gal80ts; hh-Gal4 dronci29/dronci29 were raised at 17 °C for 7 days, then changed to 29 °C for 16 h to activate the corresponding transgene and subsequently changed back to 17 °C. After 96 h, they were dissected and the wing imaginal discs stained with the appropriate antibodies.
Experiments to suppress JNK
To block JNK activity, the UAS-BskDN transgene was expressed in larvae of the following genotypes: UAS-BskDN; tub-Gal80ts, UAS-GFP; dronci29 hh-Gal4/dronci24 and UAS-BskDN; nub-Gal4/STATGFP; dronci29/ dronci24pucLacZ. Larvae were raised at 25 °C and changed to 29 °C 24 h before 4000 X-rays dose to enable the expression of BskDN. Irradiation was performed with an X-ray machine Phillips MG102. Larvae were kept at 29 °C for 72 h and then dissected and stained with the appropriate antibodies.
We have also used the UAS-puc2A transgene in the following genotype: tub-Gal80ts/UAS-RasV12; hh-Gal4/UAS-puc2A. The high levels of the negative regulator puc suppress most of JNK function. Larvae were raised at 25 °C and changed to 29 °C 24 h before 3000 X-rays dose to enable the expression of RasV12 and puc2A. Larvae were kept at 29 °C for 72 h and then dissected and stained with the appropriate antibodies.
Quantification of the overgrowths
No statistical methods were used to estimate the sample size, as we use Drosophila melanogaster, and because it has a high fecundity, we can use a large number of animals for our experiments. No randomisation or blind experiments were performed.
For quantification in the RHGmiRNA experiments, we used larvae of the genotypes: UAS-RHG-miRNA/+; hh-Gal4 UAS-GFP/+and UAS-BskDN; UAS-RHG-miRNA/+; hh-Gal4 UAS-GFP/+. Larvae were raised at 25 °C and changed to 29 °C 24 h before X-rays (4000 R) to induce expression of the BskDN and RHG-miRNA transgenes. Control non-irradiated UAS-RHG-miRNA/+; hh-Gal4 UAS-GFP/+larvae were raised in the same conditions. All larvae were kept at 29 °C for 72 h and then dissected and stained with GFP and TO-PRO3. Stacks were captured with a Leica (Solms, Germany) DB5500 B confocal microscope, and analysed with Fiji52 voxel counter plugin to obtain the volume of the samples.
To test if the sustained activity of JNK is responsible for the overgrowths, we blocked JNK activity after irradiation as follows. Larvae of the following genotypes: tub-Gal80ts, UAS-GFP; dronci29 hh-Gal4/dronci24 and UAS-BskDN; tub-Gal80ts, UAS-GFP; dronci29 hh-Gal4/dronci24 were raised at 17 °C and transferred to 29 °C 24 h before irradiation (4000 R) or transferred to 29 °C 24 h or 48 h after irradiation. All larvae were kept at 29 °C for 72 h and then dissected and stained with GFP and TO-PRO3. Stacks were captured with a Leica confocal microscope, and analysed with Fiji52 voxel counter plugin to obtain the volume of the samples. Some samples were also stained with Mmp1 to check the JNK activity.
To test if ROS and mol are involved in promoting overgrowth, we also quantified the volumes of discs overexpressing SOD:Cat and molRNAi, as in the previous experiment for the BskDN.
In the RasV12 experiments, we used larvae of the genotypes: tub-Gal80ts/+; hh-Gal4/+, tub-Gal80ts/UAS-RasV12 hh-Gal4/+ and tub-Gal80ts/UAS-RasV12; hh-Gal4/UAS-puc2A. Larvae were raised at 17 °C and changed to 29 °C 24 h before irradiation (3000 R) to allow transgene expression. Non-irradiated tub-Gal80ts/+; hh-Gal4/+and tub-Gal80ts/UAS-RasV12 hh-Gal4/+larvae were raised in the same conditions. All larvae were kept at 29 °C for 72 h and then dissected and stained with rat anti-Ci 1:50 and TO-PRO3. Stacks were captured with a Leica confocal microscope, and analysed with Fiji to obtain the area of the samples.
Statistical analysis was performed with the software Prism5 (Graphpad).
EdU incorporation
Wing discs were cultured in 1 mL of EdU labelling solution for 20 min at room temperature and subsequently fixed in 4% paraformaldehyde for 30 min at room temperature. Rabbit anti-GFP (Invitrogen) 1:200 antibody was used overnight at 4 °C before EdU detection to protect GFP fluorescence. EdU detection was performed according to the manufacturer instructions (Click-iT EdU Alexa Fluor 555 Imaging Kit, ThermoFisher Scientific).
Lineage tracing experiments
To follow the lineage of cells after irradiation, we performed the following experiment. Larvae of the genotypes: UAS-GFPDbox/act>stop>lacZ; puc-Gal4/UAS-flp dronci24 and UAS-GFPDbox/act>stop>lacZ; dronci29 puc-Gal4/UAS-flp dronci24 were irradiated (4000 R) and kept at 25 °C for 24 h or 72 h before dissection and staining.
In the experiments in which we overexpressed the Ras pathway in the Sal domain of dronc mutant larvae, e.g. salEpv-Gal4, UAS-GFP/act>stop>lacZ UAS-Flp; dronci29 UAS-RasV12/dronci24, the flippase activity directed by the salEpv-Gal4 driver induces recombination of the act>stop>lacZ cassette in all the cells of the Sal domain. These cells become irreversibly labelled with lacZ, thus allowing the identification of the cellular progeny of the domain.
Data availability
The authors declare that the data supporting the findings reported in this manuscript are available within the article and the Supplementary Information files or from the corresponding author upon request.
References
Kerr, J. F., Harmon, B. & Searle, J. An electron-microscope study of cell deletion in the anuran tadpole tail during spontaneous metamorphosis with special reference to apoptosis of striated muscle fibers. J. Cell. Sci. 14, 571–585 (1974).
Manjon, C., Sanchez-Herrero, E. & Suzanne, M. Sharp boundaries of Dpp signalling trigger local cell death required for Drosophila leg morphogenesis. Nat. Cell. Biol. 9, 57–63 (2007).
Ballesteros-Arias, L., Saavedra, V. & Morata, G. Cell competition may function either as tumour-suppressing or as tumour-stimulating factor in Drosophila. Oncogene 33, 4377–4384 (2014).
Igaki, T. Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis 14, 1021–1028 (2009).
Lowe, S. W., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432, 307–315 (2004).
Morata, G. & Ripoll, P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42, 211–221 (1975).
Moreno, E., Basler, K. & Morata, G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416, 755–759 (2002).
Menendez, J., Perez-Garijo, A., Calleja, M. & Morata, G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl Acad. Sci. USA 107, 14651–14656 (2010).
Perez-Garijo, A., Martin, F. A. & Morata, G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 131, 5591–5598 (2004).
Luo, X., Puig, O., Hyun, J., Bohmann, D. & Jasper, H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 26, 380–390 (2007).
McEwen, D. G. & Peifer, M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132, 3935–3946 (2005).
Tournier, C. et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 (2000).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Morata, G. & Ballesteros-Arias, L. Cell competition, apoptosis and tumour development. Int. J. Dev. Biol. 59, 79–86 (2015).
Bosch, M., Serras, F., Martin-Blanco, E. & Baguna, J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev. Biol. 280, 73–86 (2005).
Noselli, S. & Agnes, F. Roles of the JNK signaling pathway in Drosophila morphogenesis. Curr. Opin. Genet. Dev. 9, 466–472 (1999).
Perez-Garijo, A., Shlevkov, E. & Morata, G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development 136, 1169–1177 (2009).
Chang, Q. et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J. Hepatol. 50, 323–333 (2009).
Sakurai, T., Maeda, S., Chang, L. & Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl Acad. Sci. USA 103, 10544–10551 (2006).
Fernandez-Medarde, A. & Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2, 344–358 (2011).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Brumby, A. M. & Richardson, H. E. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779 (2003).
Wu, M., Pastor-Pareja, J. C. & Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 463, 545–548 (2010).
Bergmann, A., Agapite, J., McCall, K. & Steller, H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95, 331–341 (1998).
Kurada, P. & White, K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95, 319–329 (1998).
Cox, A. D. & Der, C. J. The dark side of Ras: regulation of apoptosis. Oncogene 22, 8999–9006 (2003).
Chen, H. M., Huang, Y., Pfeiffer, B. D., Yao, X. & Lee, T. An enhanced gene targeting toolkit for Drosophila: Golic+. Genetics 199, 683–694 (2015).
Shlevkov, E. & Morata, G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 19, 451–460 (2012).
Santabarbara-Ruiz, P. et al. ROS-induced JNK and p38 signaling is required for unpaired cytokine activation during Drosophila regeneration. PLoS Genet. 11, e1005595 (2015).
Khan, S. J., Abidi, S. N. F., Skinner, A., Tian, Y. & Smith-Bolton, R. K. The Drosophila Duox maturation factor is a key component of a positive feedback loop that sustains regeneration signaling. PLoS Genet. 13, e1006937 (2017).
Owusu-Ansah, E., Yavari, A. & Banerjee, U. A protocol for in vivo detection of reactive oxygen species. Protocol Exchange doi: 10.1038/nprot.2008.23 (2008).
La Fortezza, M. et al. JAK/STAT signalling mediates cell survival in response to tissue stress. Development 143, 2907–2919 (2016).
Ryoo, H. D., Gorenc, T. & Steller, H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7, 491–501 (2004).
Der, C. J., Krontiris, T. G. & Cooper, G. M. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc. Natl Acad. Sci. USA 79, 3637–3640 (1982).
Parada, L. F., Tabin, C. J., Shih, C. & Weinberg, R. A. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 297, 474–478 (1982).
Santos, E., Tronick, S. R., Aaronson, S. A., Pulciani, S. & Barbacid, M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature 298, 343–347 (1982).
Stephen, A. G., Esposito, D., Bagni, R. K. & McCormick, F. Dragging ras back in the ring. Cancer Cell. 25, 272–281 (2014).
Taparowsky, E. et al. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature 300, 762–765 (1982).
Karim, F. D. & Rubin, G. M. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125, 1–9 (1998).
Prober, D. A. & Edgar, B. A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 16, 2286–2299 (2002).
Patel, J. H., Loboda, A. P., Showe, M. K., Showe, L. C. & McMahon, S. B. Analysis of genomic targets reveals complex functions of MYC. Nat. Rev. Cancer 4, 562–568 (2004).
Ohsawa, S. et al. Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490, 547–551 (2012).
Behrens, A., Jochum, W., Sibilia, M. & Wagner, E. F. Oncogenic transformation by ras and fos is mediated by c-Jun N-terminal phosphorylation. Oncogene 19, 2657–2663 (2000).
Dejure, F. R. & Eilers, M. MYC and tumor metabolism: chicken and egg. EMBO J. 36, 3409–3420 (2017).
Johnston, L. A., Prober, D. A., Edgar, B. A., Eisenman, R. N. & Gallant, P. Drosophila myc regulates cellular growth during development. Cell 98, 779–790 (1999).
Wells, B. S., Yoshida, E. & Johnston, L. A. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr. Biol. 16, 1606–1615 (2006).
Martin-Blanco, E. et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557–570 (1998).
McGuire, S. E., Le, P. T., Osborn, A. J., Matsumoto, K. & Davis, R. L. Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765–1768 (2003).
Siegrist, S. E., Haque, N. S., Chen, C. H., Hay, B. A. & Hariharan, I. K. Inactivation of both Foxo and reaper promotes long-term adult neurogenesis in Drosophila. Curr. Biol. 20, 643–648 (2010).
Bach, E. A. et al. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene. Expr. Patterns 7, 323–331 (2007).
Chatterjee, N. & Bohmann, D. A versatile PhiC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS ONE 7, e34063 (2012).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
We thank the members of our lab for comments and discussions and Angélica Cantarero and Rosa Gonzalez for technical help. We also thank Jose F. de Celis for comments and suggestions. This work has been supported by grants BFU-2015-67839-P of the Ministerio de Economia y Competitividad and the XVII Concurso Nacional of the Fundación Ramón Areces.
Author information
Authors and Affiliations
Contributions
N.P., M.M. and G.M. conceived the project, designed the study and analysed the data. N.P., M.M. and I.M. performed the experiments. N.P. and G.M. wrote the manuscript. All authors edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pinal, N., Martín, M., Medina, I. et al. Short-term activation of the Jun N-terminal kinase pathway in apoptosis-deficient cells of Drosophila induces tumorigenesis. Nat Commun 9, 1541 (2018). https://doi.org/10.1038/s41467-018-04000-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-04000-6
This article is cited by
-
Transient loss of Polycomb components induces an epigenetic cancer fate
Nature (2024)
-
Lack of apoptosis leads to cellular senescence and tumorigenesis in Drosophila epithelial cells
Cell Death Discovery (2023)
-
A 50 Hz magnetic field influences the viability of breast cancer cells 96 h after exposure
Molecular Biology Reports (2023)
-
Toll signaling promotes JNK-dependent apoptosis in Drosophila
Cell Division (2020)
-
Outcompeting cancer
Nature Reviews Cancer (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
