
Abstract
The major challenge facing lithium–oxygen batteries is the insulating and bulk lithium peroxide discharge product, which causes sluggish decomposition and increasing overpotential during recharge. Here, we demonstrate an improved round-trip efficiency of ~80% by means of a mesoporous carbon electrode, which directs the growth of one-dimensional and amorphous lithium peroxide. Morphologically, the one-dimensional nanostructures with small volume and high surface show improved charge transport and promote delithiation (lithium ion dissolution) during recharge and thus plays a critical role in the facile decomposition of lithium peroxide. Thermodynamically, density functional calculations reveal that disordered geometric arrangements of the surface atoms in the amorphous structure lead to weaker binding of the key reaction intermediate lithium superoxide, yielding smaller oxygen reduction and evolution overpotentials compared to the crystalline surface. This study suggests a strategy to enhance the decomposition rate of lithium peroxide by exploiting the size and shape of one-dimensional nanostructured lithium peroxide.
Similar content being viewed by others
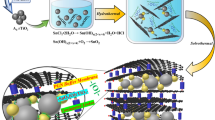


Introduction
The limitation of driving distance per charge is one of the core challenges associated with electric vehicles in supplanting fossil fuel-powered and environmentally unfriendly vehicles1,2. Current battery technology supplies far lower gravimetric and volumetric energy densities compared to fossil fuels, which has propelled the development of advanced batteries3. In this regard, the rechargeable lithium–oxygen (Li–O2) battery is one of the most suitable concepts with the essential precondition of high theoretical energy density (~3 kWh kg−1). In non-aqueous Li–O2 cells, the O2 electrochemistry consists of an overall two-electron transfer (2e–) producing solid lithium peroxide (Li2O2) during discharge (DC), which subsequently decomposes in the reverse reaction during recharge (RC), i.e., 2Li+ + O2 + 2e– ⇆ Li2O2(s)4,5. The low molecular weight of reactants, i.e., Li+ and O2 gas, and lightweight carbon electrode results in high specific capacities, which are typically over 1000 mAh g–1electrode. However, the bulk Li2O2 produced from DC leads to sluggish decomposition due to its insulating wide bandgap6, which is reflected in the high overpotential (η, the difference between thermodynamic reversible potential and measured potential) during RC. Moreover, when the RC potential is over 3.75 V, instabilities of the carbon electrode and non-aqueous electrolyte are exacerbated7,8,9. Therefore, promoting Li2O2 decomposition at little expense of overpotential is highly necessary, which has led to the concerted effort in developing catalysts10,11 including heterogeneous12,13,14 and soluble molecular catalysts15,16,17. These catalysts have shown suppressed RC potentials, but they have also caused unintended problems, such as the degradation of electrolyte solution18 and shuttling of soluble molecules that passivate the negative Li electrode19,20.
Alternatively, we have focused on controlling Li2O2 morphology and structure to enable its facile decomposition. We demonstrated the formation of amorphous and film-like Li2O2 in contrast to crystalline and aggregated (or toroidal) Li2O2 particles by tailoring the carbon electrode surface with oxygen moieties21. The significant advantage of amorphous Li2O2 is the far higher ionic conductivity and improved electrical conductivity in comparison to crystalline Li2O222. With amorphous structure, Li2O2 facilely decomposes from the surface, corresponding to the Li2O2/electrolyte solution interface at potentials below 3.5 V21,23,24. However, such behavior is only observed at the initial RC region, up to ~30% state of RC21, and is followed by a sudden increase in potential, signifying the slow decomposition of the subsequent process. The high electrical resistivity of bulk Li2O2 may cause sluggish charge transport and requires over 4.0 V for full decomposition24. Considering all these processes, the short-lived effect of surface decomposition can largely be attributed to the limited surface area of the Li2O2 film that is accessible to the electrolyte solution. Therefore, a significant increase in the surface area of Li2O2 can promote the decomposition at low overpotential and circumvent the sluggish charge transport.
Here, we demonstrate one-dimensional (1-D) nanostructures of amorphous Li2O2 that can remarkably reduce the RC potential and lead to the high round-trip efficiency of 80%. This result could be achieved by the use of a mesoporous carbon electrode, which guides the growth of these 1-D Li2O2 nanostructures. The fast decomposition of the 1-D nanostructures occurs even at the high RC current rate of 2 A g–1carbon, which is notably distinct from typical Li2O2 films and large toroidal particles.
Results
Discharge–recharge profiles of Li–O2 cells
As the mesoporous carbon electrode in Li–O2 cells, CMK-3 was employed without the use of any additive carbon. CMK-3 has a turbostratic structure comprised of three-dimensional (3-D) hexagonal arrays of mesoporous channels and micropores (Supplementary Figure 1)25. The average diameters of the mesoporous channels (dmeso) and micropores (dmicro) are 3.74 nm and 0.71 nm, respectively, which contribute to the high surface area (SAtotal) of 1128 m2 g–1 and large volume (Vtotal) of 0.9 cm3 g–1 (N2 adsorption–desorption isotherm in Supplementary Figure 1 and Supplementary Table 1). To compare the Li–O2 cell performance of the CMK-3 electrode, we also prepared three non-mesoporous carbon electrodes consisting of large porous carbon (LPC, pore diameter of 80 nm), commercial carbon of Ketjen Black (KB), and multi-walled carbon nanotubes (CNTs), respectively. Figure 1a shows the DC–RC profiles of Li–O2 cells with the above-mentioned carbon electrodes under the same operating conditions. At a glance, all carbon electrodes show comparable DC potentials, but exhibit significantly different RC profiles. At ~67% of the RC process (Q/Qtotal, denoted with the notation 0.67RC) corresponding to 1.0 mAh, the potential for CMK-3 is limited to 3.45 V, while in case of all non-mesoporous carbon electrodes, the potential reaches 4.1 V. Furthermore, CMK-3 shows even lower RC potential in comparison to the catalyst-containing electrodes. At 1.0 mAh, the representative catalysts of cobalt oxide (Co3O4) and ruthenium (Ru) nanoparticles-loaded CNT electrodes attain 3.75–3.8 V, and the soluble redox mediator of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) reaches 3.6 V. Overall, the CMK-3 electrode exhibits the highest round-trip efficiency (~80%), which is 10–13% higher than the non-mesoporous carbon electrodes and 4–7% higher than the catalyst-containing electrodes (Fig. 1b). These results underscore that CMK-3 itself is profoundly effective in suppressing the rise of RC potential in Li–O2 cells.

Galvanostatic cycle. a Discharge (DC)–recharge (RC) profiles for the first cycle with mesoporous carbon (CMK-3, red), ketjen black nanoparticles (KB, dark yellow), carbon nanotube (CNT, black), and large porous carbon (LPC, pore diameter ≈80 nm, orange). All electrodes have same carbon mass of ~1.0 mg and galvanostatic tests were performed at a same current rate of 50 mA g–1carbon in 0.5 M LiTFSI in tetraglyme to the fixed capacity of 1.5 mAh. These controlled conditions allow us to assess the round-trip efficiencies. The dashed line indicates a reversible potential of Li–O2 electrochemistry (2.96 V). Along with various carbon electrodes, DC–RC curves with heterogeneous catalysts such as Ru and Co3O4 nanoparticles with CNT (Ru/CNT, blue and Co3O4/CNT, green) and homogeneous catalyst of 10 mM TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) (dark cyan) with CNT were performed. b Comparison of round-trip efficiency calculated from the ratio of the integrated areas under the DC and RC curves
Morphological and chemical characterizations of DC product
We can infer that the differences in the RC potential profiles are correlated with the morphological and structural characteristics of the DC products. Scanning electron microscopy (SEM) and X-ray diffraction (XRD) reveal film or large particle-shaped products and crystalline Li2O2 reflections present on the non-mesoporous carbon electrodes of LPC, KB, and CNT following DC (Supplementary Figures 2–3). In the presence of Co3O4 and Ru nanoparticles (NPs), the crystalline reflections of Li2O2 are suppressed (Supplementary Figure 3), and the resulting poorly crystalline Li2O2 is one of the key reasons for the lower RC potential in comparison to the catalyst-free CNT electrodes (Fig. 1a), as we demonstrated in previous reports13,24. In stark contrast, flake-like products appear on the CMK-3 electrode surface from SEM images (Supplementary Figure 4). The higher-resolution imaging tool of transmission electron microscopy (TEM) addresses the detailed morphology as ultrathin 1-D nanostructures with a width of 6–15 nm (Fig. 2a, b). They grow upward by loosely entangling with one another during DC (from 0.25DC to 1.0DC, Fig. 2b–e) and their contours display flake-like shapes at 1.0DC (Fig. 2e). There is absence of crystalline Li2O2 reflections from the XRD analysis (Supplementary Figure 3), revealing its amorphous character, while the DC products can be identified as Li2O2 from X-ray absorption near-edge structure (XANES) spectroscopy. The O K-edge XANES spectra demonstrate the notable σ*(O–O) bands at 530.5 eV arising from Li2O226 in both total electron yield (TEY) and partial fluorescence yield (PFY) modes (Fig. 3a), which has the escape depth of tens and hundreds of nanometers, respectively. This result indicates the presence of Li2O2 on both the surface and bulk (interior) of mesoporous CMK-3. Fourier transform infrared (FTIR) spectra also reveal the Li–O stretching mode at 535 cm–1 from Li2O2 (Supplementary Figure 5)27. Although additional vibration bands of side products such as lithium carboxylates and carbonates are also present in the FTIR spectra, the predominant DC product is Li2O2, and is further confirmed from the average electrons per oxygen molecule of 2.06 e−/O2, acquired by monitoring the pressure change in the Li–O2 cell during DC (Fig. 3b, c). Taken together, the DC product formed with CMK-3 has 1-D shape at the nanometer scale and amorphous structure, which is considerably different from typical Li2O2 films.

Transmission electron microscopy (TEM) images. a As-prepared, b–e discharging, and f–i recharging CMK-3 surfaces. The scale bars are 50 nm. The top label of the image denotes the depth of DC or RC, denoted by Q/Qtotal at a fixed Qtotal of 1.0 mAh and the current rate of 50 mA g–1carbon. The yellow dashed lines indicate the surface of CMK-3. j Schematic illustration of initial growth of Li2O2, blue poducts, from CMK-3 electrode. From over 0.5DC, the one-dimensional structure and ultrathin Li2O2 grow upward by loosely entangling with one another, which display flake-like shapes (see SEM images in Supplementary Figure 4)

Monitoring DC and RC processes. a O K-edge XANES spectra for CMK-3 electrode (red) at different depths of DC/RC and standard references of Li2O2, Li2CO3, and HCO2Li (black). The dark and light colors indicate bulk-sensitive partial fluorescence yield (PFY) and surface-sensitive total electron yield (TEY) modes, respectively. The different DC and RC states are denoted from Q/Qtotal at a fixed Qtotal of 1.0 mAh and a current rate of 50 mA g–1carbon. b–d In situ pressure and gas analyses during DC and RC, respectively, using online electrochemical mass spectrometry (OEMS): b DC–RC curve at a current rate of 50 mA g−1 and a capacity of 1.5 mAh. c The gas-pressure decrease during DC (overall 2.06e─/O2). d O2 gas (overall 3.17e─/O2) and CO2 gas (over 0.9RC) evolution with the corresponding RC. The horizontal dashed line represents the ideal 2e─/O2
Growth mechanism of Li2O2 and density functional theory calculations
The 1-D nanostructured Li2O2 appearing from the electrode mimics the shape of the mesoporous channels of CMK-3, which is not found with the other non-mesoporous electrodes (Supplementary Figure 2). This allows us to infer that the mesoporous structure of CMK-3 determines the growth process and overall features of the 1-D Li2O2. With CMK-3, the O2 gas and electrolyte solution diffuse into the mesoporous channels by capillary force28,29, where the confined O2 is reduced via two-electron transfer as follows:
Here, * represents adsorbed species on the surface. After Eq. (1), Li2O2 can form by either Eq. (2) or by the disproportionation of LiO2 species (2LiO2 ⇆ Li2O2(s) + O2). At the initial stage of DC with CMK-3, we found that the overpotential of oxygen reduction reaction (ORR), which leads to the formation of amorphous Li2O2 in the mesoporous channels, was lower than the crystalline Li2O2 formed with the non-mesoporous electrodes (Supplementary Figure 6). The lower ORR overpotential on the amorphous Li2O2 surface can be understood by using density functional theory (DFT) calculations to examine the key intermediates of the electrochemical reactions.
Figure 4a, b shows the calculated free energy diagrams along with the optimized structures of crystalline and amorphous surface, respectively. Three different applied potentials (U) are incorporated into the energetics via –neU, where n is the number of transferred (Li+ + e–). The overpotential (η) for ORR is defined as ηORR = U0 – UDC, where the equilibrium potential U0 is defined as the potential for which the change of free energy for the whole process is zero, and the DC potential UDC is the highest potential that makes the free energy for every step in ORR downhill.30,31 Using these prescriptions, the calculated U0 = 2.77 V for the crystalline structure, reconstructed Li2O2 \((1\bar 100)\) that is the most stable surface30, is in good agreement with the experimental thermodynamic potential of Li2O2 formation (2.96 V).32 For the amorphous Li2O2 surface, we divided the surface into 16 equivalent areas to find the lowest energy adsorption site (Supplementary Figure 7), and the free energy diagram shown in Fig. 4b is based on the most favorable reaction site. We note, as summarized in Supplementary Table 2, that statistically 14 sites out of 16 show lower ηORR compared to that from the crystalline surface, which is in accordance with the experimental results (Supplementary Figure 6). The potential determining step (PDS) for ORR is calculated to be Eq. (2) for both amorphous and crystalline surfaces, indicating that the weaker binding of second *LiO2 adsorption on the amorphous surface is responsible for a lower ηORR. Structurally, this weaker binding of *LiO2 on the amorphous surface is due to the disordered arrangements of the surface Li and O atoms, preventing the newly adsorbing *LiO2 species from forming optimal coordination with the surface. To quantify this, in Fig. 4f, we plotted the binding energies of *LiO2 for the PDS of ORR as a function of the number of newly formed Li–O bonds as a result of *LiO2 adsorption. Overall, the low *LiO2 binding energies on the amorphous surface are indeed associated with the low coordination number that the newly adsorbed *LiO2 forms with the surface, compared to that on the crystalline surface.

Density functional calculations and Bader charge analysis. a, b Calculated free energy diagrams a on the Li2O2 \((1\bar 100)\) and b on amorphous Li2O2, along with the optimized structures. For the amorphous surface, the most favorable adsorption site was considered to construct the diagram. Lithium is colored in light gray (bulk) and light green (adsorbate); oxygen is colored in dark gray (bulk) and red (adsorbate). c, d The variation in electron density upon the first LiO2 adsorption on c the crystalline Li2O2 \((1\bar 100)\) and d amorphous Li2O2 surfaces. The charge density indicated by surface contour is plotted with a threshold value of 0.01e Å−3. The newly adsorbing *LiO2 is denoted explicitly as Li, O1, and O2. O2 which is not shown in c is behind O1. Lithium is in light green and oxygen in red. e, f Binding energies of e 1st LiO2 and f 2nd LiO2 for 16 sites of the amorphous Li2O2 surface plotted versus coordination number. Binding energies on the crystalline surface are also shown for comparison, denoted as c-Li2O2
On the basis of the DFT results, we expect the weakly binding LiO2 to constitute a chelating network within the confined channels33,34, prior to the formation of amorphous Li2O2. As the DC proceeds at low DC current rate, amorphous Li2O2 grows and emerges from the mesoporous channel. The continuous 1-D growth is also observed on the electrode surface as shown in Fig. 2a–e, j, and is similar in shape to the mesoporous channels. Although the prominent vertical growth is apparent, LiO2 species may also bind at the lateral sides of the 1-D Li2O2 protruding from the exterior of the mesoporous channels. This can account for the slightly increased width of the nanostructure than the diameter of mesoporous channel. It is noted that the mesoporous channels are only partially filled with Li2O2 due to slow O2 diffusion and rapid clogging by Li2O229. This is verified from the full DC capacity of ~2300 mAh g−1CMK-3 that is lower than the theoretical capacity of ~2430 mAh g−1 CMK-3 estimated from the full volume of the mesoporous channels, despite the inclusion of Li2O2 formed over the exterior of the channels (Supplementary Figure 8). Therefore, the initial growth occurring in the vicinity of channel entry is critical to lead to the unique shape of Li2O2.
Analysis of RC process
We now turn our attention to the RC process of the 1-D amorphous Li2O2 occuring at low RC potentials. The decreasing height of 1-D nanostructured Li2O2 is followed by its complete depletion from the CMK-3 surface at 0.75RC as shown by TEM images (Fig. 2f–i). The surface-sensitive TEY mode from the O K-edge XANES spectra is also consistent with this result, showing the significant decrease in the σ*(O–O) band by 0.5RC and disappearance at 0.75RC (light solid line in Fig. 3a). However, regarding the interior of CMK-3, the behavior of Li2O2 decomposition is different. The bulk-sensitive PFY mode shows the pronounced σ*(O–O) band at 0.5RC, which decreases at 0.75RC and disappears at 1RC (dark solid line in Fig. 3a). These results clearly demonstrate the preferential decomposition of Li2O2 on the exterior of CMK-3, relative to Li2O2 in the interior of the mesoporous channels, which we will discuss in more detail below. Further insight on the decomposition process is gained from in situ gas analysis using on-line electrochemical mass spectrometry (OEMS). Initially, O2 gas evolves at the significant rate of ~0.25 μmol mA–1 min–1, but gradually decreases throughout the RC process (Fig. 3b, d). As a result, the efficacy of Li2O2 decomposition is determined to be 3.17e–/O2, which is slightly closer to the ideal 2e–/O2 than that from typical Li2O2 film from CNT (3.31e–/O2, Supplementary Figure 9)8,21,24 owing to the lower RC potential. Nevertheless, the deviations from the 2e–/O2 indicates the accompaniment of side reactions as evidenced by the side products of lithium carbonate (Li2CO3, 532.8 eV) and carboxylate-related bands (HCO2Li, 531.6 eV) emerging in the TEY mode of the O K-edge XANES spectra at 0.5RC and PFY mode at 0.75RC, respectively (Fig. 3a). These side products result from the deteriorating carbon electrode and electrolyte due to instabilities with the superoxide intermediate during DC7,9 and singlet oxygen produced during RC35. Decomposition of the side products requires over ~4 V and is reflected in the potential rise at 0.75RC, which directly occurs after the disappearance of Li2O2 from the CMK-3 surface and is met with CO2 gas evolution36 (Fig. 3b, d). The residual side products present after RC can compromise cycling performance (Supplementary Figure 10), which affirms the need for more stable electrolytes and electrode materials.
Morphological and structural effects
Overall, the 1-D shape with ultrathin amorphous structure plays a pivotal role for facile decomposition. We could confirm this further by modulating the DC current rate to control the product structure. The SEM images in Fig. 5a reveal different Li2O2 morphological shapes with respect to the DC current rate. As the current rate increases from 10 to 100 mA g–1carbon, the flake-like product becomes thinner and less numerous, then finally the conformal film shape is prominent when the current rate is over 100 mA g–1carbon. At higher current rates, there is the swift deposition of Li2O2 over the surface of CMK-337, which nullifies the interior of the electrode by blocking the entry of the mesoporous channels. The XRD results show amorphous characters of both flake and film products (Supplementary Figures 3 and 11), thus their different shapes can be directly correlated to their decomposition behaviors which can be explored with anodic linear sweep voltammetry (LSV) (Fig. 5b). When flake-abundant Li2O2 is present, a significant anodic peak appears at ~3.18 ± 0.04 V, denoted as Ea1, while the peak at higher potential of ~3.40 ± 0.03 V, Ea2, becomes progressively pronounced when increasing proportions of the film are present. Notably, Ea2 is greater than Ea1 at over 100 mA g–1. The corresponding OEMS analysis demonstrates that the amount of O2 evolution is proportional to the peak intensity of Ea1 and Ea2, and decouples this from CO2 evolution at Ea3 (Fig. 5c). Therefore, it can be concluded that there is prior decomposition of the flake-like products comprised of 1-D nanostructured Li2O2 as opposed to the conformal films. As for another control experiment, we induced the partial blockage of the mesoporous channels by annealing CMK-3 at 2000 °C in argon (Ar). This high-temperature annealing brings about disordered and clogged mesoporous channels in some parts of CMK-3 and results in the decrease of the total surface area and volume of the electrode by half, while the turbostratic structure and pore diameter are still preserved (Supplementary Figure 12 and Supplementary Table 1)25. After DC, crystalline and lump-shaped Li2O2 is observed together with flake-like Li2O2 (Supplementary Figure 13), which may deposit on the clogged surface of the electrode. In the subsequent RC, two potential plateaus are observed at ~3.5 and ~4.3 V with the corresponding O2 evolution (Supplementary Figure 14). The distinctly separate plateaus with large potential gap account for the far slower decomposition of the crystalline lumps, which confirms again the critical role of morphology and structure in promoting the facile decomposition of Li2O2.

Dependence of Li2O2 morphology and corresponding RC profiles on DC current rate. a SEM images of CMK-3 electrodes with different current rates of 10, 50, 100, and 200 mA g–1carbon (from bottom to top) at a fixed DC capacity of 500 mAh g–1carbon. The scale bars indicate 1 μm. b Corresponding anodic LSV profiles at a sweeping rate of 0.05 mV s−1. The black open circles represent experimental LSV curve whereas the red lines indicate the sum of the deconvoluted curves. The deconvoluted peaks above 4.0 V have been omitted for simplicity. c Representative in situ gas analysis linked with anodic LSV for Li2O2 formed at a DC current rate of 50 mA g–1carbon
Mechanism of fast RC
The question is then on the mechanism accounting for the facile decomposition. As oxygen evolution reaction (OER) is the reverse process of ORR, the same DFT free energy profile in Fig. 4a, b can be used to understand the enhanced OER mechanism from the amorphous structure. The PDS for OER on both crystalline and amorphous surfaces is the reverse of Eq. (1), LiO2* ⇆ Li+ + e– + O2*. The ηOER for crystalline Li2O2 is calculated to be 0.86 V, which is comparable with the experimental values (~0.9 V, Fig. 1). In contrast, the ηOER for amorphous Li2O2 at the most favorable binding site is calculated to be 0.48 V, almost 50% reduction in overpotential compared to the crystalline case and is consistent with our experiments. With the PDS of OER being LiO2* ⇆ Li+ + e– + O2* for both crystalline and amorphous surfaces, the underlying origin of reduced ηOER is again the weaker binding of *LiO2 on the amorphous surface, as in ORR, which can then be explained with the smaller number of new coordination that the adsorbed *LiO2 creates with the amorphous surface (Fig. 4c). Remarkably, the effect of amorphous structure on the PDS of OER is much more prominent than that of ORR, explaining the great enhancement of the Li2O2 decomposition on the amorphous phase. Bader Population analysis on the crystalline versus amorphous surfaces before and after *LiO2 adsorption demonstrates that the amount of charge transferred from the surface to the adsorbed *LiO2 is indeed much more moderate on the amorphous surface due to disordered surface geometries and associated weaker electronic interactions with the adsorbates (Fig. 4e, f).
Along with the thermodynamic understanding of the structural effect, the morphological benefit of 1-D and ultrathin nanostructure is significant. Considering that the 1-D Li2O2 is continuous from the interior of mesoporous channel to exterior, the interior would be embedded in the conducting electrode while the exterior part is enclosed with the electrolyte solution. One can expect facile charge transport across Li2O2 that is confined in the mesoporous channels. However, Li+ dissolution which is responsible for the electron transfer (see Eqs. (1) and (2)) is less plausible within the closed mesoporous channels due to the violation of charge balance. Therefore, preferential depletion takes place from Li2O2 that is surrounded by the electrolyte solution (Fig. 7a). This is in good agreement with the O K-edge XANES results showing the disappearance of Li2O2 from the exterior of CMK-3 surface (Fig. 3a). The cyclic voltammogram (CV) analyses of both CMK-3 and non-mesoporous CNT electrode also confirm this hypothesis. Figure 6 and Supplementary Figure 15 show significant cathodic responses for both CMK-3 and CNT during ORR in comparison with the featureless response under Ar environment. The subsequent OER reveals the pronounced anodic response exclusive to CMK-3 in the potential range of 3.2–4.2 V (Fig. 6a). When the total charges of ORR versus OER, i.e., (QORR/QOER), are estimated, they are 1.39 and 1.01 for CNT and CMK-3 electrodes, respectively. The charge ratio at unity indicates more facile decomposition of 1-D Li2O2 and also the negligible loss of Li2O2 during OER. This also implies the less favorable depletion of Li2O2 from the interior of channel, which would otherwise no longer anchor the Li2O2 on exterior of the electrode, leading to the physical loss of Li2O2 and increase in the charge ratio.

Cyclic voltammetry. a CMK-3 and b CNT electrodes at a sweeping rate of 1 mV s–1 in bulk electrolysis cell with 0.5 M LiTFSI/tetraglyme. The blue dashed lines indicates input potential curve. The light and dark solid curves indicate Ar (O2-free) and O2 environment, respectively
Discussion
We propose that the critical advantage of 1-D nanostructured Li2O2 on the exterior of the channel is the extensive accessibility of Li+ in all directions to the electrolyte solution (Fig. 7a), unlike the case of bulk Li2O2 where electrolyte accessibility is restricted to the outer top surface38,39 (Fig. 7b). Such large surface area is activated by being in equilibrium with the electrolyte solution (see dark blue area in Fig. 7) and reduces the non-active volume of Li2O2. One of the vital steps of decomposition involve electron and charge (hole, h+, in Li2O2) transport giving Li+ dissolution and O2 evolution, which is determined by the corresponding Li2O2 characteristics. Given the 1-D shape of Li2O2, electron transport occurs at highly conductive sites are expected at the interface where Li2O2, electrode, and electrolyte solution are closely in contact (denoted with the star mark in Fig. 7a). From these conductive sites, the charge carriers of holes may swiftly migrate along to the sidewalls of the Li2O2 where highly mobile Li+ and charge carriers lead to facile decomposition, which can account for the decreasing height of nanostructures during RC (Fig. 2f–i). In all, the decomposition rate and behavior of 1-D nanostructured Li2O2 indicate its higher conductivity than bulk Li2O240,41. Electrochemical impedance spectroscopy (EIS) analysis in Supplementary Figure 16 also demonstrates the lowest resistance of 1-D amorphous Li2O2, ~170 Ω for the sum of interface and charge transport resistance, compared with typical amorphous films (~220 Ω) and crystalline Li2O2 lumps (~330 Ω).

Schematic illustration for decomposition processes. a 1-D nanostructured and b bulk Li2O2 film. The dark blue region that is distinguished from inside of Li2O2 with the light blue color indicates activated Li2O2 surface where free access of Li+ is allowed. The star mark denotes highly conductive sites where Li2O2, electrode, and electrolyte solution closely contact. Once RC commences, facile Li+ dissolution and charge transport takes place along the activated Li2O2 surface, which results in O2 evolution and decomposition of Li2O2 at low potential. The charge carrier hole in Li2O2 and electron are denoted as h+ and e–, respectively. Since Li+ dissolution of Li2O2 enclosed within the mesoporous channel would violate charge balance rule, this part of Li2O2 decomposes later. The bulk Li2O2 film decomposes sluggishly due to the limited surface area of Li2O2.
The conductive Li2O2 should exhibit high rate capability, which could be examined with increasing RC current rates. Figure 8a reveals that the low RC potential is still maintained below 4 V by 0.75RC from CMK-3 electrode even at 2 A g–1. In sharp contrast, the Li2O2 film on CNT leads to the significant rise in potential, reaching ~4.5 V at 0.2RC and 2 A g–1 (Fig. 8b). With increasing current rate, the decomposition rate of 1-D Li2O2 is notably distinct from bulk film, implying the exceedingly rapid Li+ dissolution and charge transport. In summary, we have demonstrated the high round-trip efficiency of ~80% in Li–O2 batteries by forming 1-D nanostructures of amorphous Li2O2. This unique shape of Li2O2 could be formed from the guided growth of the mesoporous carbon channels and retains the ability to suppress RC overpotential even with very high current rates, which is distinct from bulk Li2O2. The initial DC potential profiles correlated with DFT calculations demonstrate that amorphous Li2O2 has lower overpotential for both oxygen reduction and evolution reactions than the crystalline Li2O2 due to disordered geometric arrangements of the surface atoms and associated weaker electronic interactions of the key reaction intermediates, namely lithium superoxide. The 1-D nanostructured shape also has significant benefit for facile decomposition from the highly abundant and facile mobility of Li+ and charge carriers which are present along the surface of Li2O2. This expectation is confirmed from the decreasing height of 1-D structure during the decomposition process and the lowest resistance among other film-shape and bulk Li2O2. This study shows an alternative strategy to surmount the sluggish decomposition of Li2O2 by controlling its shape and structure, and paves the way to promote the facile decomposition of Li2O2 without catalysts.

Galvanostatic rate capability. a CMK-3 and b CNT electrodes after DC at 50 mA g–1 carbon and capacity of 1.0 mAh. The RC was performed at a current rate of 50, 500, 1000, and 2000 mA g–1carbon (from dark to light color)
Methods
Synthesis of CMK-3
CMK-3 was synthesized with reference to the procedure reported by Terasaki and co-workers, but slightly modified42. The SBA-15 silica was used as the template, and the carbon source of phenol–formaldehyde mixture was employed to impregnate the silica template.
The SBA-15 template was prepared by using tetraethyl orthosilicate (TEOS) and triblock co-polymer poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO, Pluronic® P123, Sigma Aldrich, Mn ~5800)43. Briefly, 1.0 g of triblock co-polymer surfactant was dissolved in 30 mL of 2.0 M hydrochloric acid (HCl, Wako Pure Chemicals, 35–37%) and after full dissolution 2.2 g of TEOS (Wako Pure Chemicals, ~95%) was added. The mixture was stirred at 40 °C for 24 h and transferred to a Teflon-lined autoclave for hydrothermal reaction at 100 °C for 24 h. The resulting solid precipitate of SBA-15 was filtrated, thoroughly washed with de-ionized (DI) water, and dried at room temperature. Afterward, calcination was carried out at 600 °C in air for the complete removal of organic residues. The SBA-15 was then impregnated with carbon precursors; 0.5 g of as-prepared SBA-15 was added to aqueous solution (6.0 mL DI water) including 0.6 g of phenol (Wako Pure Chemicals, 99%) and 0.5 g of formaldehyde (Wako Pure Chemicals, 36–38%). After stirring at room temperature for 30 min, 100 μL of neat H2SO4 (Wako Pure Chemicals, 98%) was added and the temperature was increased to 80 °C. The vigorous stirring continued until the mixture became dry and the dry precipitate was heated at 150 °C for 3 h. These steps were repeated one more time but with different amount of carbon precursors: 0.3 g of phenol and 0.25 g of formaldehyde. The carbonization was conducted by pyrolysis at 950 °C for 6 h in Ar. Lastly, SBA-15 template was completely etched by soaking in 10% aqueous HF (hydrofluoric acid, Wako Pure Chemicals, 46–48%) for 10 h with stirring. This etching process was carried out for two times. The silica-free mesoporous carbon, i.e., CMK-3, was collected by centrifugation after washing with DI water for several times and dried at 80 °C under vacuum.
Synthesis of LPC
A macroporous carbon, LPC (pore diameter (d) ~80 nm) was synthesized by using a hard template of silica NP array that was synthesized with reference to Stöber process44. Briefly, a mixture of 4.0 mL DI water and 4.0 mL of 25% (v/v) aqueous NH3 (Wako Pure Chemicals) was slowly added to the TEOS solution comprised of 6.0 g of TEOS in 100.0 mL ethanol. The mixture was stirred in a sealed bottle for 20 h at room temperature, showing that the transparent solution became turbid due to formation of silica NPs. After drying the solvent at 70 °C, the collected silica NPs (diameter (d) ~80 nm) were sintered at 800 °C for 20 min under air, during which an array of silica NPs was formed45. The following processes for impregnation of carbon source, carbonization, and removal of silica NP template were the same as the one described for synthesis of CMK-3.
Synthesis of Co3O4 NPs on CNT (Co3O4/CNT)
Co3O4 NPs (d = 8–10 nm) were synthesized according to the procedure reported by Yamada et al.46 but slightly modified; 0.3 g of cobalt acetate tetrahydrate (Co(C2H5O2)2‧4H2O, Wako Pure Chemicals, 99%) was dissolved in 30 mL DI water and 2 mL of 25% (v/v) aqueous NH3 was added. After 20 min stirring at room temperature, 100 μL of H2O2 (Wako Pure Chemicals, 30–35.5%) solution was added. By continuing stirring for another 12 h, Co3O4 NPs were formed, which were collected by centrifugation followed by drying at 80 °C in air. To prepare the Co3O4/CNT electrodes, the Co3O4 NPs were mixed with multi-walled CNT (MWCNT, Sigma Aldrich, outer diameter versus length is 7–15 nm versus 0.5–10 μm) with ~40 wt% by grinding using mortar and pestle.
Synthesis of Ru/CNT
To synthesize Ru NPs, 0.3 g of RuCl3‧nH2O (Wako Pure Chemicals) was dissolved in 20 mL ethylene glycol (EG) and the pH was adjusted to 11 by adding 0.1 M NaOH. After stirring for 30 min, temperature was increased to 200 °C and solvothermal reaction was carried out for 4 h in N2 atmosphere. The Ru NPs were incorporated onto MWCNT by dispersing 45 mg of MWCNT in 80 mL ethanol and adding as-prepared Ru colloidal solution to be ~40 wt% loading. A 0.1 M HCl was added to adjust pH to ~5 and the solution was kept under stirring for 6 h. Subsequently, Ru/CNT was centrifuged, washed with DI water–ethanol mixture (1:1 v/v) for several times, and finally dried at 80 °C under vacuum.
Fabrication of various electrodes
Slurries of the carbon materials CMK-3 and LPC were prepared by mixing with LITHionTM (Ion Power) binder (4:1 ratio (w/w) for carbon/LITHion) in N-methyl-2-pyrrolidone (NMP). They were tape casted to porous carbon paper (Toray, TGP-H-030) using doctor blade, dried slowly at room temperature followed by at 80 °C under vacuum, and finally punched to disks (d = 12 mm). KB (EC-600JD) carbon was mixed with LITHionTM(Ion Power, 4:1 ratio (w/w) for carbon/LITHion) in isopropanol (IPA) by 10 min sonication. The resulting slurry was sprayed on glass fiber (GF/C, Whatman) and dried at room temperature. This carbon-coated glass fiber was punched to disks (d = 12 mm). Free-standing CNT electrodes were prepared by dispersing MWCNT powder in IPA followed by tip sonication for 10 min. The Co3O4/CNT and Ru/CNT were also prepared as a slurry by dispersing in IPA using tip sonication. The slurry for CNT, Co3O4/CNT, and Ru/CNT was filtered through glass fiber (GF/C) under vacuum, dried at room temperature, punched to disks (d = 12 mm), and peeled off from the glass fiber. All electrodes were completely dried at 200 °C for 5 h using a Büchi oven equipped with vacuum and transferred to Ar-filled glovebox without any exposure to air. The carbon loading mass for all electrodes was 1.0 ± 0.2 mg.
Assembly of Li–O2 cells and electrochemical measurements
All assembly processes were carried out in Ar-filled glove box (O2, H2O < 1 ppm). The cell components were dried at 80 °C under vacuum for 8 h. The Li–O2 cell was assembled with the negative electrode of metallic Li (Honjo) rolled on a stainless steel current collector (d = 12 mm), two separators of Celgard® 2500 (thickness (t) = 25 μm, d = 26 mm) and GF/C (d = 16 mm), and the positive electrode of as-prepared carbon samples. The non-aqueous electrolyte solution of 0.5 M LiTFSI (Lithium bis(trifluoromethanesulfonyl)imide, Kanto Chemical, >99.7%) in tetraethylene glycol dimethyl ether (tetraglyme, battery grade, UBE) was added with each 70 μL to three interfaces of Li|Celgard|GF/C|carbon electrode. The as-received tetraglyme was dried using molecular sieves to reduce H2O content <10 ppm (measured from Karl Fischer titration) before cell assembly. The assembled cell was taken out from the glove box and filled with O2 gas (>99.99995% Tomoe Shokai). The Li–O2 cells were examined with galvanostatic electrochemical testers (WonATech WBCS3000L) in a constant-temperature incubator (25 °C).
Characterizations
All post-mortem electrode samples were transferred using hermetic transfer vessel and loaded into characterizing instruments without any exposure to air except microscopy observations. The morphological features of carbon electrodes and DC products were observed using field-emission scanning electron microscope (FE SEM, Hitachi S 4800) and transmission electron microscopes (TEM) with JEOL JEM-1230 (accelerating voltage 80 kV) and JEOL JEM-ARM200F (accelerating voltages 200 and 80 kV). The loading time for samples was minimized to avoid sample contamination from air. During TEM analysis of the cycled electrodes, the electron beam intensity was carefully adjusted by controlling the aperture size to avoid any damage to the thin Li2O2 due to incident beam. The N2 adsorption–desorption isotherm was obtained from BEL Japan Inc. BELSORP-mini II and the surface area was calculated by Brunauer–Emmett–Teller (BET) method, pore size distribution and pore volume were estimated by Barrett–Joyner–Halenda (BJH) method. Powder XRD pattern (PXRD) was collected from RigakuSmartLab X-ray diffractometer. The XANES spectra were acquired at the beamline 11 of the Synchrotron Radiation (SR) center of Ritsumeikan University in Japan. FTIR spectroscopy was also performed for chemical analysis of products by using an FTIR spectrometer (Thermo Fisher Scientific Nicolet iS50) equipped with Ar-filled glovebox. For in situ gas analysis, OEMS was used during RC and pressure change was recorded during DC. This custom-built gas analysis system was programmed to automatically detect the collecting gas evolving during RC and the pressure change was concurrently measured for quantitative gas analysis. The detailed description of the OEMS setup can be found in our previous report24.
Calculation details
Spin polarized DFT calculations were performed using the Vienna ab initio simulation package (VASP)47,48 code with the revised Perdew–Burke–Ernzerhof (RPBE)49 exchange-correlation functional. The potentials of the atoms were described by projector-augmented wave (PAW)50. Throughout this study, we used a cut-off energy of 400 eV. The DFT calculations included two cycles of Li2O2 formation since the calculation unit cell contains two formula units of Li2O2 product. The free energy of O2 was indirectly calculated according to the water-splitting reaction, and the energy of (Li+ + e–) is assumed to be in equilibrium with the bulk Li, in a way similar to the Computational Hydrogen Electrode method51. For the crystalline structure, the reconstructed Li2O2 \((1\bar 100)\) surface reported was used30. The Brillouin zone was sampled with a 2 × 2 × 1 Monkhorst-Pack mesh. The bottom two layers were fixed to their bulk positions, with the top two layers fully relaxed. For the amorphous Li2O2 model, we adopted the amorphous structure predicted by Tian et al.,23 which described precisely the Li–O2 DC product in recent isotopic labeling expreiments. To reduce the computational cost, 144 atoms (72 Li, 72 O) from the top surface out of 256 atoms were used to make a supercell and half of the atoms from the bottom were fixed. To minimize the calculation artifacts on the amorphous case, we systematically scanned all possible adsorption sites with a 4 × 4 grid. Gamma k-points were used. The free energies of intermediates at zero potential and pH = 0, ΔG = ΔE + ΔZPE – TΔS, was evaluated as follows: The reaction energy ΔE is calculated using DFT. The zero point energy and entropy terms were neglected considering very small changes in those terms along the reaction in the solid phases. The Bader charge analysis was performed to estimate the amount of charge transfer before and after the adsorption of LiO2 in which the charges of isolated LiO2 were used as a reference52. For both crystalline and amorphous models, the 15 Å vacuum space was used in the z-direction. The convergence of energy and forces were set to 1 × 10–5 eV and 0.05 eV Å–1, respectively.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author on request.
References
Larcher, D. & Tarascon, J. M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 7, 19–29 (2015).
Abraham, K. M. Prospects and limits of energy storage in batteries. J. Phys. Chem. Lett. 6, 830–844 (2015).
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012).
McCloskey, B. D., Scheffler, R., Speidel, A., Girishkumar, G. & Luntz, A. C. On the mechanism of nonaqueous Li–O2 electrochemistry on C and its kinetic overpotentials: some implications for Li–air batteries. J. Phys. Chem. C. 116, 23897–23905 (2012).
Lu, J. et al. Aprotic and aqueous Li–O2 batteries. Chem. Rev. 114, 5611–5640 (2014).
Radin, M. D. & Siegel, D. J. Charge transport in lithium peroxide: relevance for rechargeable metal-air batteries. Energy Environ. Sci. 6, 2370–2379 (2013).
McCloskey, B. D. et al. Twin problems of interfacial carbonate formation in nonaqueous Li–O2 batteries. J. Phys. Chem. Lett. 3, 997–1001 (2012).
McCloskey, B. D. et al. Limitations in rechargeability of Li-O2 batteries and possible origins. J. Phys. Chem. Lett. 3, 3043–3047 (2012).
Ottakam Thotiyl, M. M., Freunberger, S. A., Peng, Z. & Bruce, P. G. The carbon electrode in nonaqueous Li–O2 cells. J. Am. Chem. Soc. 135, 494–500 (2013).
Oh, S. H., Black, R., Pomerantseva, E., Lee, J.-H. & Nazar, L. F. Synthesis of a metallic mesoporous pyrochlore as a catalyst for lithium–O2 batteries. Nat. Chem. 4, 1004–1010 (2012).
Xu, J.-J., Wang, Z.-L., Xu, D., Zhang, L.-L. & Zhang, X.-B. Tailoring deposition and morphology of discharge products towards high-rate and long-life lithium-oxygen batteries. Nat. Commun. 4, 2438 (2013).
Li, F. et al. Ru/ITO: a carbon-free cathode for nonaqueous Li–O2 battery. Nano Lett. 13, 4702–4707 (2013).
Yilmaz, E., Yogi, C., Yamanaka, K., Ohta, T. & Byon, H. R. Promoting formation of noncrystalline Li2O2 in the Li–O2 battery with RuO2 nanoparticles. Nano Lett. 13, 4679–4684 (2013).
Black, R., Lee, J.-H., Adams, B., Mims, C. A. & Nazar, L. F. The role of catalysts and peroxide oxidation in lithium–oxygen batteries. Angew. Chem. Int. Ed. 52, 392–396 (2013).
Bergner, B. J., Schürmann, A., Peppler, K., Garsuch, A. & Janek, J. TEMPO: a mobile catalyst for rechargeable Li-O2 batteries. J. Am. Chem. Soc. 136, 15054–15064 (2014).
Lim, H.-D. et al. Rational design of redox mediators for advanced Li–O2 batteries. Nat. Energy 1, 16066 (2016).
Chen, Y., Freunberger, S. A., Peng, Z., Fontaine, O. & Bruce, P. G. Charging a Li–O2 battery using a redox mediator. Nat. Chem. 5, 489–494 (2013).
McCloskey, B. D. et al. On the efficacy of electrocatalysis in nonaqueous Li–O2 batteries. J. Am. Chem. Soc. 133, 18038–18041 (2011).
Aurbach, D., McCloskey, B. D., Nazar, L. F. & Bruce, P. G. Advances in understanding mechanisms underpinning lithium–air batteries. Nat. Energy 1, 16128 (2016).
Ha, S. et al. Investigation into the stability of Li metal anodes in Li-O2 batteries with a redox mediator. J. Mater. Chem. A 5, 10609–10621 (2017).
Wong, R. A. et al. Structurally tuning Li2O2 by controlling the surface properties of carbon electrodes: implications for Li–O2 batteries. Chem. Mater. 28, 8006–8015 (2016).
Zhang, Y. et al. Amorphous Li2O2: chemical synthesis and electrochemical properties. Angew. Chem. Int. Ed. 55, 10717–10721 (2016).
Tian, F., Radin, M. D. & Siegel, D. J. Enhanced charge transport in amorphous Li2O2. Chem. Mater. 26, 2952–2959 (2014).
Yang, C. et al. Unexpected Li2O2 film growth on carbon nanotube electrodes with CeO2 nanoparticles in Li–O2 batteries. Nano Lett. 16, 2969–2974 (2016).
Inagaki, M., Toyoda, M. & Tsumura, T. Control of crystalline structure of porous carbons. RSC Adv. 4, 41411–41424 (2014).
Gallant, B. M. et al. Influence of Li2O2 morphology on oxygen reduction and evolution kinetics in Li-O2 batteries. Energy Environ. Sci. 6, 2518–2528 (2013).
Xiao, J. et al. Investigation of the rechargeability of Li–O2 batteries in non-aqueous electrolyte. J. Power Sources 196, 5674–5678 (2011).
Choi, M. & Ryoo, R. Ordered nanoporous polymer-carbon composites. Nat. Mater. 2, 473–476 (2003).
Luo, X. et al. Mass and charge transport relevant to the formation of toroidal lithium peroxide nanoparticles in an aprotic lithium-oxygen battery: an experimental and theoretical modeling study. Nano Res. 10, 4327–4336 (2017).
Hummelshøj, J. S. et al. Communications: elementary oxygen electrode reactions in the aprotic Li-air battery. J. Chem. Phys. 132, 071101 (2010).
Viswanathan, V. et al. Electrical conductivity in Li2O2 and its role in determining capacity limitations in non-aqueous Li-O2 batteries. J. Chem. Phys. 135, 214704 (2011).
Girishkumar, G., McCloskey, B., Luntz, A. C., Swanson, S. & Wilcke, W. Lithium−air battery: promise and challenges. J. Phys. Chem. Lett. 1, 2193–2203 (2010).
Doyle, A. D., Montoya, J. H. & Vojvodic, A. Improving oxygen electrochemistry through nanoscopic confinement. ChemCatChem 7, 738–742 (2015).
Zhai, D. et al. Disproportionation in Li–O2 batteries based on a large surface area carbon cathode. J. Am. Chem. Soc. 135, 15364–15372 (2013).
Mahne, N. et al. Singlet oxygen generation as a major cause for parasitic reactions during cycling of aprotic lithium–oxygen batteries. Nat. Energy 2, 17036 (2017).
Gallant, B. M. et al. Chemical and morphological changes of Li–O2 battery electrodes upon cycling. J. Phys. Chem. C 116, 20800–20805 (2012).
Adams, B. D. et al. Current density dependence of peroxide formation in the Li-O2 battery and its effect on charge. Energy Environ. Sci. 6, 1772–1778 (2013).
Ganapathy, S. et al. Nature of Li2O2 oxidation in a Li–O2 battery revealed by operando x-ray diffraction. J. Am. Chem. Soc. 136, 16335–16344 (2014).
Ganapathy, S. et al. Operando nanobeam diffraction to follow the decomposition of individual Li2O2 grains in a nonaqueous Li–O2 battery. J. Phys. Chem. Lett. 7, 3388–3394 (2016).
Xia, C. et al. Evolution of Li2O2 growth and its effect on kinetics of Li–O2 batteries. ACS Appl. Mater. Interfaces 6, 12083–12092 (2014).
Radin, M. D., Rodriguez, J. F., Tian, F. & Siegel, D. J. Lithium peroxide surfaces are metallic, while lithium oxide surfaces are not. J. Am. Chem. Soc. 134, 1093–1103 (2012).
Jun, S. et al. Synthesis of new, nanoporous carbon with hexagonally ordered mesostructure. J. Am. Chem. Soc. 122, 10712–10713 (2000).
Zhao, D. et al. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 279, 548 (1998).
Stöber, W., Fink, A. & Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 26, 62–69 (1968).
Kang, S., Yu, J.-S., Kruk, M. & Jaroniec, M. Synthesis of an ordered macroporous carbon with 62 nm spherical pores that exhibit unique gas adsorption properties. Chem. Commun. 1670–1671 (2002).
Yamada, Y., Yano, K., Xu, Q. & Fukuzumi, S. Cu/Co3O4 nanoparticles as catalysts for hydrogen evolution from ammonia borane by hydrolysis. J. Phys. Chem. C 114, 16456–16462 (2010).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007).
Tang, W., Sanville, E. & Henkelman, G. A grid-based bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 21, 084204 (2009).
Acknowledgements
This work is financially supported by RIKEN, JST ALCA-SPRING, the National Research Foundation (NRF) of Korea (Grant NRF-2016R1C1B2008690, NRF-2016M3D1A1021147, and NRF-2017R1A2B3010176), and the Nano Material Technology Development Program through the NRF funded by the Ministry of Science, ICT and Future Planning (Grant 2009-0082580). A.D. is grateful for financial support from BK 21 plus fellowship through NRF funded by the Ministry of Education of Korea. The synchrotron XANES experiments at the SR center, Ritsumeikan University, were performed with the approval of “project for creation of research platforms and sharing of advanced research infrastructure of MEXT” (Proposal No. R1432). The authors thank Prof. Seong-Ho Yoon from Kyushu University for high-temperature annealing of CMK-3 and National Institute for Materials Science (NIMS) Battery Research Platform for assistance with TEM observation.
Author information
Authors and Affiliations
Contributions
A.D. conceived the research and carried out the syntheses, electrochemical tests, and characterizations. R.A.W. performed the OEMS experiments and analyses. W.P. and Y.J. performed and interpreted the DFT calculations. K.Y. and T.O. performed the XANES measurements and analyses. H.R.B. supervised the overall research. All authors discussed the experiments and final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dutta, A., Wong, R.A., Park, W. et al. Nanostructuring one-dimensional and amorphous lithium peroxide for high round-trip efficiency in lithium-oxygen batteries. Nat Commun 9, 680 (2018). https://doi.org/10.1038/s41467-017-02727-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-02727-2
This article is cited by
-
Selectively anchoring Pd single atoms on specific sites in defective cobalt oxides for efficient lithium-oxygen batteries
Science China Materials (2024)
-
A bifunctional nitrile additive for high-performance lithiumoxygen batteries
Nano Research (2024)
-
Addressing Transport Issues in Non-Aqueous Li–air Batteries to Achieving High Electrochemical Performance
Electrochemical Energy Reviews (2023)
-
Improving the electrophilicity of nitrogen on nitrogen-doped carbon triggers oxygen reduction by introducing covalent vanadium nitride
Science China Materials (2023)
-
Ag doped urchin-like α-MnO2 toward efficient and bifunctional electrocatalysts for Li-O2 batteries
Nano Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
