
Abstract
Chitin, an insoluble polymer of N-acetylglucosamine, is one of the most abundant biopolymers on Earth. By degrading chitin, chitinolytic bacteria such as Vibrio harveyi are critical for chitin recycling and maintenance of carbon and nitrogen cycles in the world’s oceans. A decisive step in chitin degradation is the uptake of chito-oligosaccharides by an outer membrane protein channel named chitoporin (ChiP). Here, we report X-ray crystal structures of ChiP from V. harveyi in the presence and absence of chito-oligosaccharides. Structures without bound sugar reveal a trimeric assembly with an unprecedented closing of the transport pore by the N-terminus of a neighboring subunit. Substrate binding ejects the pore plug to open the transport channel. Together with molecular dynamics simulations, electrophysiology and in vitro transport assays our data provide an explanation for the exceptional affinity of ChiP for chito-oligosaccharides and point to an important role of the N-terminal gate in substrate transport.
Similar content being viewed by others

Introduction
Chitin, an insoluble polymer of β1,4-linked N-acetylglucosamine (GlcNAc) residues, is one of the most abundant biopolymers on Earth, along with cellulose1,2. Chitin serves as the major structural component of arthropods, the cell walls of some fungi, and the primary exoskeletons of crustaceans and marine zooplankton3. The global estimation of chitin production is ~1010–1011 tons4. Remarkably, while providing a constant rain of polysaccharides to the ocean floor (“marine snow”), no substantial accumulation of chitin in ocean sediments occurs due to the rapid recycling of chitin driven by chitinolytic bacteria, mainly from the family Vibrionaceae5,6. These bacteria utilize only chitin as a sole source of cellular energy, and certain Vibrio species such as Vibrio harveyi and Vibrio parahaemolyticus are extremely fast growing. Hence, these bacteria play critical roles in maintaining the carbon and nitrogen cycles in marine ecosystems.
The chitin utilization pathway of marine Vibrios is highly conserved and incorporates a number of characterized and uncharacterized enzymes, chitin binding proteins, and transport proteins7,8. Proteins involved in the initial processes include chemotaxis proteins responsive to chitin oligosaccharides and extracellular chitinases that degrade the insoluble polymer into water-soluble chitooligosaccharides that are imported across the outer membrane (OM) through a dedicated, chitooligosaccharide-specific channel9,10,11. In the periplasmic space, chitin dextrinase12 and β-N-acetylglucosaminidase13,14 generate the mono- (GlcNAc) and disaccharide (GlcNAc2) that are subsequently transported into the cytoplasm via several ABC transporter complexes in the inner membrane. Finally, at least six cytoplasmic enzymes convert the transport products to fructose-6-P, NH3, and acetate7.
Cloning of a number of chitin utilization genes has been reported from Vibrio furnissii, Vibrio cholera, and V. harveyi and the corresponding proteins have been characterized9,10,12,15,16,17,18,19,20. A key step in chitin utilization represents the cellular acquisition of soluble chitin oligosaccharides (GlcNAc2–6) produced by the action of extracellular chitinases20. This uptake process is carried out by an OM diffusion channel, termed chitoporin (ChiP)9,10,11. ChiP was first identified in V. furnissii and was proposed to be a chitooligosaccharide-uptake channel based on expression profiles in the presence of various sugar substrates21. DNA microarray expression profiles further demonstrated that expression of the chiP gene in V. cholerae is induced by chitin oligosaccharides and that the genes responsible for chitin degradation are under the stringent control of the chiS regulon21. The gene encoding for ChiP is found in most marine members of the Vibrionaceae, emphasizing the general importance of this OM diffusion channel for chitin utilization.
On a molecular level, it is so far unclear how ChiP mediates the specific uptake of chitooligosaccharides. The protein from V. harveyi (denoted VhChiP) has recently been cloned and expressed at high levels in Escherichia coli and purified to homogeneity. VhChiP was purified as a stable trimer, suggesting a structural similarity to the general diffusion porins OmpF and OmpC from E. coli. In single-channel electrophysiology, VhChiP displays a trimeric conductance of 1.8 nS in 1 M KCl, which is half of the trimeric conductance of E. coli OmpF (4.2 nS)22 but substantially larger than its well-characterized sugar-specific analog LamB (maltoporin) from E. coli (160 pS) and Salmonella typhimurium (90 pS)23,24. The addition of micromolar concentrations of chitooligosaccharides such as chitohexaose (GlcNAc6) causes a complete block of ionic current, demonstrating tight binding. Interestingly, closely related sugars such as maltohexaose do not interact with the channel, suggesting remarkable substrate specificity of VhChiP. The binding constant (K) for the most potent substrate chitohexaose is about 5 × 106 M−1, which is at least an order of magnitude higher compared to other OM sugar-specific diffusion channels11. In vitro liposome swelling assays confirm the highly specific nature of the channel for chitooligosaccharides10. Statistical analysis on stochastic fluctuations of ion current through VhChiP in the presence of chitohexaose indicated that VhChiP has multiple binding sites for sugar, and the trapping properties of chitoporin exhibit memory effects, given that the average binding rate of an unblocked monomer is larger when its neighboring monomers are blocked. Such results suggested a possible design strategy to enhance the rate of sugar uptake by the bacterium25,26. A homology model based on the X-ray crystal structure of Delftia acidovorans Omp32 (pdb ID: 2FGR)27 predicted Trp136, located at the mouth of the channel constriction, to be an important residue for chitin transport28. This notion was confirmed by single-channel electrophysiology and liposome swelling experiments, showing that for the preferred substrate chitohexaose, mutation of Trp136 led to decreased binding affinity and reduced rates of uptake respectively.
Here, we report X-ray crystal structures of VhChiP in the absence and presence of chitooligosaccharides. Together with single-channel electrophysiology and molecular dynamics (MD) simulations the results reveal the structural basis for the exquisite substrate specificity of chitoporin and clarify the mechanism of facilitated diffusion of chitooligosaccharides across the OM of Vibrionaceae.
Results
Purification and crystallization of VhChiP
We expressed VhChiP without a His-tag in the OM of the porin-deficient E. coli Bl21 omp8 strain and obtained reasonable yields (~3 mg from 12 l of culture) even though VhChiP overexpression appeared to be highly toxic for E. coli. The protein was expressed without a tag based on the predicted similarity to general porins in which the N- and C-termini of the monomers interact to form a salt bridge. Figure 1a shows that boiled and non-boiled SDS-PAGE samples have different mobilities, with trimers observed in non-boiled samples and monomers after boiling, i.e., VhChiP is heat-modifiable. The purified protein was crystallized in the presence of 0.4% C8E4 as detergent (Methods). The obtained crystals of VhChiP in space group C2 diffracted to reasonable resolutions of ~2.5 Å (Supplementary Table 1). Despite this, structure solution using molecular replacement (MR) failed, presumably due to the lack of a good homology model given that sequence identities of likely search models with VhChiP are 20% or less. Soaking of native crystals with different heavy atoms was also not successful. Next, we attempted to express VhChiP in minimal media in the presence of seleno-methionine (SeMet) for phasing using single/multiple anomalous dispersion (SAD/MAD) approaches, but the cells failed to grow in accordance with the protein toxicity that was observed in rich medium. Subsequently we cloned VhChiP without a signal sequence and with a C-terminal His6-tag into pET28a (Methods) to express the protein into inclusion bodies (IBs) and avoid toxicity issues. As expected, the cells grew now very well in minimum medium in the presence of SeMet. VhChiP from IBs was folded in vitro from 8 M urea and purified as the native protein (Methods). Well-diffracting crystals (~ 2 Å resolution) in space group P21 were obtained and a SAD data set was collected at the absorption edge of Selenium. The structure was solved using Autosol in Phenix and refined in Refmac (Methods).

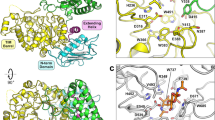
X-ray crystal structure of in vitro-folded VhChiP. a SDS-PAGE gel of OM-expressed (native; 1), in vitro-folded (2) and truncated VhChiP (3) loaded both as non-boiled (left lanes) and boiled samples (right lanes). b Side view and c top view cartoon presentations of the in vitro-folded VhChiP trimer. d, e Top view superpositions of in vitro-folded VhChiP (red) with Neisseria meningitidis PorB (green; d) and Delftia acidovorans Omp32 (blue; e). f Superposition of in vitro-folded VhChiP (red) with OM-expressed VhChiP (green). The N-termini have been labeled
VhChiP forms trimers with N-terminally plugged pores
In vitro-folded VhChiP forms a trimeric assembly of oval β-barrels, each consisting of 16 strands (Fig. 1), like the general diffusion porins OmpF/C from E. coli. Unlike OmpF/C however, the N-terminus of VhChiP does not form an intramolecular salt bridge with the C-terminal carboxyl group. Instead, the first N-terminal 17 amino acids of VhChiP are not part of the barrel and extend into the periplasm where they form a trimerization motif like that observed in the phosphate transporter OprP from Pseudomonas aeruginosa29. The first nine amino acids of the N-terminus are disordered and not visible in the electron density (Fig. 1b). The overall topology of the barrel, including the periplasmic N-terminal region, is correctly predicted by Boctopus and PRED-TMBB30,31. A DALI search32 identifies N. meningitidis PorB, which has 18% sequence identity to VhChiP, as the closest structural homolog in the database (Z = 29; r.m.s.d. 2.7 Å over 301 residues). The second-highest similarity is observed for Omp32 from D. acidovorans (Z = 26; r.m.s.d. 2.7 Å over 295 residues), which was used for previous homology modeling of VhChiP. Overall, the differences between both structural homologs and VhChiP are substantial, especially for most extracellular loops and the functionally important, barrel constricting loop L3 (Fig. 1 d, e).
The crystal structure of the OM-expressed channel is almost identical to that of the in vitro-folded protein. Strikingly however, in OM-expressed VhChiP the N-termini plug the pore of a neighboring β-barrel within the trimeric assembly, effectively blocking all β-barrels for substrate transport (Fig. 2). The blocking of a channel by a neighbor within an oligomeric assembly is, to our knowledge, unprecedented. The reason that the plug is inter- and not intra-molecular might be due to fact that the N-terminal β-strand is pointing away from its own pore towards the neighboring one (Fig. 1f and Supplementary Fig. 1). This is due to the small angle of the strand(s) relative to the OM plane. Thus, to plug its own pore, the N-terminus would have to fold back on itself in a rather elaborate way and would likely need to be longer. All residues of the N-terminus have good electron density in the structure (Supplementary Fig. 1) and a number of polar interactions can be identified between the N-terminal plug and the barrel, most notably between Asp1 (carboxylate)-Arg94/148, Asp1 (α-amino group)-Y118/Asp122, Ala3-Glu53, Asn4-Asn127/Arg312/Glu347, Ser5-Asp122, Asp6-Arg94, and Lys9-Glu76 (Fig. 2). Thus, five out of the first six residues of the N-terminus make polar interactions with the pore of the protein, suggesting that the plug is stably inserted in the pore. It should be noted that for in vitro-folded VhChiP, two residues (MG) were added to the N-terminus of the mature protein for inclusion body expression, and it is possible that the presence of those additional residues has destabilized the plug sufficiently for it be outside the pore.

OM-expressed VhChip has N-terminally plugged channels. a, b Surface representations (top view) of in vitro-folded VhChiP (a) and natively expressed VhChiP (b). The barrel lumen of the natively expressed VhChiP is occupied by the N-terminus of a neighbor barrel. c N-terminal insertion mode of natively expressed VhChiP. The N-terminus of one barrel (blue) plugs the lumen of a neighbor barrel (red). d Polar interactions between the inner barrel wall (red) with the N-terminus of a neighbor barrel (blue)
To explore the stability of the pore plug further, we performed 500 ns equilibrium MD simulations of in vitro-folded and natively expressed VhChiP, with free periplasmic and pore-inserted N-termini respectively. The N-terminal ten residues of in vitro-folded VhChiP are highly flexible and do not insert into the pore (Fig. 3). As expected from the interactions observed in the crystal structure, the plug in natively expressed VhChiP is very stable with root mean square fluctuations (RMSF) of about 1.5 Å. Interestingly, in the presence of the pore plug, residues 143–146 located in loop L3 show enlarged RMSF values (Fig. 3a). The movements in loop L3 are likely to be induced by fluctuations of the tail of the plug within the pore, and we hypothesize that these concerted motions might initiate the N-terminal unbinding process that has to occur in order for substrate to bind.

Behavior of the pore plug in MD simulations. a Average Cα RMSF of the protein dynamics in vitro-folded (red) and OM-expressed (blue) VhChiP. In addition to the differences for the first 14 residues, a difference in the RMSF for residues 143–146 in loop L3 can be clearly observed. b Cumulative total charge as a function of time for a 1 M KCl solution at 200 mV. In addition, the cumulative charges for K+ and Cl− ions separately are shown as well. The data was averaged over the three simulations and the slopes of these curves yield the respective currents. c Free-energy surfaces of unbinding of the N-terminus in wild type and mutant channels. The shaded areas indicate the respective error estimates. The arrow represents the position of the N-terminal plug in the crystal structure of OM-expressed VhChiP
In a next step, MD simulations (3 × 250 ns) were performed to estimate the conductance of the in vitro-folded channel in a 1 M KCl solution in the presence of a 200 mV membrane potential. Consistent with the experiments, the in vitro-folded trimeric channel shows a theoretical conductance value of 2.13 ± 0.15 nS, only slightly higher than the experimental value of 1.8 nS measured at lower voltages22. When the plug is inside the pore, only very few ions can pass the plug and no statistically meaningful current value can be determined in the simulation time (Fig. 3b).
To obtain a more quantitative result concerning the binding energies of the N-termini, we determined one-dimensional free-energy surfaces for wild type and D1A, D6A and K9A N-terminal plug mutants (Methods) using the umbrella sampling technique with two 2.24 µs long simulations. The mutated residues are involved in electrostatic interactions with channel-lining residues (Figs 2 and 3) and are therefore likely to be important for the stability of the plug-inserted state. As can be seen in Fig. 3c, the binding energy of the wild type N-terminus is about 22 kcal mol−1 and the profile shares similar features as that obtained recently for the monomeric cyclodextrin channel CymA33. The D6A and D1A mutants had relatively little effect, while the K9A mutant reduced the binding energy roughly by a factor of two (Fig. 3c). The result suggests that Lys9 plays an important role in the stability of the plug-inserted state. Intriguingly, in CymA it has been verified experimentally that a similar positive residue is involved in the stability of the plug-bound conformation33. It is important to note that all determined free-energy surfaces show only one minimum, which is for the N-terminus located inside the pore. This strongly supports the stability of the closed state of the VhChiP channel as observed in the crystal structure of the natively expressed protein.
Chitohexaose binding to in vitro-folded VhChiP
To obtain a structure of VhChiP in complex with a substrate, we co-crystallized in vitro-folded VhChiP with the substrate chitohexaose. Well-diffracting crystals grew under the same conditions and in the same space group as for the apo protein (Supplementary Table 1). Following MR, density was observed for all six units of the GlcNAc6 oligosaccharide (Fig. 4 and Supplementary Fig. 2). The substrate is bound in an extended conformation, with the density for the GlcNac units at both ends of the substrate (GlcNac-1 and -6) somewhat less clear compared to that for the central GlcNac-2–GlcNac-5 units. However, the density is of sufficient quality for placement of the substrate molecule with its reducing end (denoted GlcNAc-1) on the periplasmic side (Supplementary Fig. 2), stacked against the aromatic ring of Trp123. On the extracellular side of the channel constriction, the last sugar unit (denoted GlcNAc-6) stacks against Trp331 (Fig. 4). The same orientation of substrate (reducing end periplasmic) has been observed for maltoporin, albeit at lower resolution (2.8 Å)34.

Chitohexaose binding to in vitro-folded VhChiP. a Overview from the extracellular side. b Slabbed view from the side, with substrate residues labeled. c Schematic, showing interactions of key VhChiP residues with chitohexaose
Besides the ring stacking, few other interactions are present between GlcNAc-1 and GlcNAc-6 and VhChiP. The aromatic ring of Trp136 stacks against both GlcNAc-4 and GlcNAc-5, providing a clear explanation as to why this residue is important for chitohexaose binding by VhChiP28. Both Trp123 and Trp136 are conserved in VhChiP orthologs, whereas Trp331 is not (Fig. 5). The structure provides a clear rationale for the fact that (GlcNAc6) is the best substrate for VhChiP, since beyond GlcNAc-1 and GlcNAc-6 the channel widens, decreasing the potential for interactions between the substrate and the channel. The central four residues of the sugar chain (GlcNAc-2 to GlcNAc-5) form several polar interactions with the channel interior, some of which are mediated by water molecules (Fig. 4). Importantly, the density for the central part of GlcNAc6 is well-defined and allows unambiguous assignment of the acetamido groups, which for successive GlcNAc units point in opposite directions, as also observed for the chitohexaose molecule bound to the E. coli transglycosidase MltA35. The acetamido carbonyl groups of GlcNAc-2 and GlcNAc-3 are likely to be especially important for binding. The former interacts closely with the amide of Asn127 and to a lesser extent with Arg312, whereas the latter makes strong hydrogen bonds to the side chains of Arg94 and Arg148 (Fig. 4). In addition, the acetamido amides of GlcNAc-2 and GlcNAc-3 interact with the carboxylates of Glu347 and Asp122 respectively.

Alignment of VhChiP orthologs from Vibrio species. Observed secondary structure elements have been indicated (orange cylinders, helices; green arrows, β-sheets). The first helix that is part of the N-terminal plug is presented in red. Key aromatic (purple) and hydrophilic residues (cyan) interacting with chitooligosaccharides are colored. The following orthologs have been aligned: V. harveii (GenBank ID: HF558985.1), V. parahaemolyticus (GenBank ID: CPO12950.1), V. cholera (GenBank ID: DQ774012.1), V. furnissii (GenBank ID: AF129934.1), and V. fischeri (GenBank ID: CP001139.1). Chitoporin from V. harveyi (UniProtKB/TrEMBL entry: L0RVU0) was used as protein template to identify putative chitoporins. The alignment was generated using “CLUSTALW” algorithm in the DNASTAR package and displayed in Genedoc. The secondary structure of VhChiP was generated by ESPript v. 2.2
Chitotetraose binding to natively expressed VhChiP
Given that the channels of the OM-expressed apo-protein are closed due to the insertion of the N-terminus of a neighboring monomer (Fig. 2), it is important to establish whether the incoming substrate from the extracellular space could displace the N-terminus to bind in the pore. In addition, we wanted to establish how chitooligosaccharides of different lengths bind to VhChiP. To answer these questions we co-crystallized OM-expressed VhChiP with chitotetraose (GlcNAc4). OM-expressed VhChiP in the presence of the GlcNAc4 substrate also crystallizes under the same conditions and in the same space group as the closed apo-protein (Supplementary Table 1). Importantly, the oligosaccharide is bound in the channel and has displaced the N-terminus, which is now in the periplasmic space and has the same conformation as that of the in vitro-folded protein.
The GlcNAc4 substrate occupies approximately the positions of GlcNAc-2 to GlcNAc-5 of the GlcNAc6 chain (Fig. 6), suggesting that the central positions of the binding site provide the most binding energy for chitooligosaccharides (note that the subunits of GlcNAc4 are numbered 2–5 to provide consistency with those of chitohexaose). However, there are differences of up to 5 Å for individual atoms between the two structures, especially at both ends of the substrate (GlcNAc-2 and GlcNAc-5). In most cases, however, this does not dramatically change the interactions between the sugar and the channel. For example, the acetamido carbonyl group GlcNAc-2 interacts in both structures with Asn127 and Arg312 whereas that of GlcNAc-3 interacts with Arg94/148 (Fig. 6). It appears therefore that VhChiP accommodates compounds of different length via subtle changes in the way the substrates are bound. For compounds of four GlcNAc units or larger, the central binding sites (2–5) are likely to be always occupied, in a manner as observed in our structures. However, since X-ray crystallography is an averaging technique it is possible that the binding register may vary, i.e., small populations of, e.g., chitohexaose might be bound with GlcNAc-1 at position 2 (with GlcNAc-6 being disordered). As alluded to above, it is also possible that mixed populations are present with regards to the orientation of the substrate, i.e., the reducing end might be extracellular in part of the population. Future MD simulations could shed light on these details.

Chitotetraose binding to VhChiP displaces the N-terminal plug. Side views in the same orientations, showing the bound chitotetraose in natively expressed VhChiP (left panel) and chitohexaose in in vitro-folded VhChiP (right panel). Residues interacting with the sugars are labeled
Ejection of the pore plug in electrophysiology
To probe the role of the N-terminus in substrate binding and transport in more detail we carried out single-channel electrophysiological experiments for in vitro-folded and OM-expressed VhChiP. Based on the crystal structure we also generated a VhChiP variant with the first 19 N-terminal residues removed, denoted truncated VhChiP. At standard electrophysiological conditions with applied potentials of −100 and +100 mV, the single-channel conductance is essentially the same (~2.0 nS) for all three VhChiP proteins (Fig. 7a–c), and are fully consistent with data for the native protein obtained in previous studies10,11. Surprisingly, there is no clear evidence of a closed channel for any of the three VhChiP proteins. However, the OM-expressed protein and (to a lesser extent) in vitro-folded ChiP show short-lived channel closures that are absent in the N-terminally truncated channel (Fig. 7c), suggesting infrequent insertions of the plug inside the pore in the presence of a high and non-physiological membrane potential. To probe this further, we first induced closing of the OM-expressed VhChiP channels by applying a high voltage (+199 V; Fig. 7d). We then decreased the voltage to obtain very low membrane potentials (±2.5 mV; Fig. 7e). Under these conditions we observe long-lived channel closures that are fully consistent with the plug-inserted state observed in the crystal structure. Interestingly, three subconductance levels are observed corresponding to trimers with one, two or three open pores, indicating that the pore plugs move in and out of the channels independently. The channels open permanently when the voltage is increased to ±10 mV (Fig. 7f). The low voltages required for channel opening are somewhat surprising given the large free energy of plug binding derived from MD simulations (Fig. 3).

Voltage-induced ejection of the pore plug in VhChiP. a–c Single-channel electrophysiology of OM-folded VhChiP (a), in vitro-folded VhChiP (b) and truncated VhChiP (c). Left panels show typical traces at −100 mV, whereas the center and right panels show the current–voltage profiles. Data represent mean ± s.d., n = 3. d Stepwise voltage-induced closure of OM-folded VhChiP channels at 200 mV. e, f Typical traces observed at ±2.5 mV (e) and at ±10 mV (f). The data shown in d–f are for the same experiment, with protein only added at the start (d). Traces shown are representative of three independent experiments
A plug peptide blocks the pore of plug-less VhChiP
To probe the role of the pore plug further, we added a peptide corresponding to the first nine residues of VhChiP (DGANSDAAK) to the N-terminally truncated protein. At 100 μM peptide, frequent current blockages corresponding to the complete closure of a single channel are observed (Fig. 8). No blockages are observed for the control OmpF in the presence of 200 μM peptide. Interestingly, blockage of VhChiP is asymmetric, i.e., it occurs only upon peptide addition from one side (trans), regardless of the sign of the voltage (Supplementary Fig. 3). Given that proteins are added cis and preferentially insert into the lipid bilayer without translocating the long extracellular loops (i.e., these remain on the cis side), trans most likely corresponds to the periplasmic side of the channel. This experiment therefore recapitulates the physiological insertion of the pore plug. To further test specificity we also tested two unrelated peptides of similar length for VhChiP pore blockage. Neither of the control peptides affect the VhChiP-mediated currents (Supplementary Fig. 4), indicating that pore blockage is specific.

Channel blockage by an N-terminal plug peptide. Single-channel current traces at −100 mV (1 M KCl) of truncated VhChiP in the absence (a) and presence of 100 μM (b) of the plug peptide DGANSDAAK. c E. coli OmpF traces in the presence of 200 μM peptide. Traces shown are representative of three independent experiments
Effect of the N-terminus on substrate binding and transport
Next, we tested the three proteins in the presence of increasing concentrations of chitohexaose (Fig. 9 and Supplementary Figs. 5–8). Under the conditions employed (−100 mV voltage) all three channels are permanently open (previous section). The results show that one subunit of the full-length channels was frequently blocked even at very low concentrations of chitohexaose (0.5 µM), while under the same conditions blocking events were rarely seen with the truncated channel. At 2.5 µM, occlusion of all three subunits was observed for the full-length channels, but not for truncated VhChiP. The data therefore suggest that the truncated channel interacts weaker with the sugar compared to the full-length channels. When the equilibrium binding constant K is evaluated, the full-length channels show relatively similar binding constants of ~2–5 × 105 M−1 in agreement with previous results (Supplementary Table 2)11,28. The binding constants vary somewhat with different membrane potentials and are generally two- to threefold higher for the in vitro-folded protein for reasons that are not clear. However, removal of the first nine residues affects substrate binding significantly, with binding constants 10–15-fold lower for truncated VhChiP compared to OM-expressed, full-length protein (Supplementary Table 2).

Chitoxexaose binds to VhChiP with high affinity. Single-channel electrophysiology experiments for natively expressed VhChiP (a), refolded VhChiP (b), and N-terminally truncated VhChiP (c). The fully open VhChiP trimeric channels were exposed to different concentrations of chitohexaose. Ion current traces were acquired at −100 mV with sugar added on the cis side. For full trace recordings of 2 min duration and analyses see Supplementary Figs. 3–6
For most systems, a limitation of single-channel electrophysiology is the inability to distinguish substrate binding from substrate translocation through the channel, i.e., a compound can be released on the same side as where it binds. We therefore also assayed in vitro transport of various sugars via proteoliposome swelling assays. Various monomeric sugars translocate efficiently through VhChiP in the presence and absence of the N-terminus (Fig. 10). The truncated variant exhibited a more non-specific channel behavior and was approximately twofold more efficient in transport of various types of small monosaccharides compared to the full-length channels. By contrast, translocation of oligomeric sugars is exquisitely specific in accordance with previous results10,11, i.e., only chito-oligosaccharide substrates are translocated efficiently. Interestingly, for the oligosaccharides the absence of the N-terminus leads to a substantial decrease in transport rates, which is in qualitative agreement with the decreased binding constants observed in electrophysiology. Collectively the results suggest that the N-terminal segment plays an important role in chito-oligosaccharide substrate translocation through the VhChiP channel (Fig. 10).
Discussion
The X-ray crystal structure of trimeric OM-purified VhChiP shows an unprecedented closing of the transport pore by the N-terminus of a neighboring monomer. Potential of mean force calculations suggest that this structural feature is highly stable in the absence of substrate. However, the applied-field MD simulations do not explain why the N-terminal plug is ejected at very low voltages in the experiments. These ejections might be rather slow processes involving details of the ion distributions and electro-osmotic forces on the plug that are currently out of scope for the present MD simulations due to the limited simulation time scales. In addition, the apparent discrepancy between the high computed binding free-energy of the plug and the measured low voltage of plug ejection might be due to differences in salt conditions, constant protonation states in the simulations and force-field approximations.
Interestingly, the single-channel experiments on truncated VhChiP in the presence of chitohexaose suggest that the absence of the N-terminus results in a weaker binding affinity of the substrate for the channel. Likewise, the proteoliposome swelling experiments show lower rates of chitooligosaccharide transport in the VhChiP variant lacking the N-terminus. These results have several implications. First, they would be difficult to reconcile with a scenario where the N-terminus would always be disordered and located outside the barrel, as observed in the crystal structure obtained from in vitro-folded VhChiP. Thus, the results suggest that N-terminal gating also occurs in the lipid bilayer, which is confirmed by single-channel experiments at very low membrane potential, approaching physiological conditions where the Donnan potential across the OM will be close to zero36. Second, the N-terminal gate is most likely not a passive structural feature but instead promotes substrate translocation. How this occurs is still unclear, but one possibility is that the N-terminal plug generates the optimal configuration of the substrate binding site. This hypothesis is given some support by the MD simulations, which show that the presence of the N-terminus results in increased dynamics of a region in loop L3 close to several substrate-interacting residues (Fig. 3a). This could be beneficial to substrate binding/orientation and consequently would make transport more efficient, providing support for the electrophysiology and in vitro transport data.
Is the putative gating mechanism conserved and why would there be a need for gating? Inspection of the sequence alignment (Fig. 5) shows that the N-terminal extension forming the pore plug is present only in a subset of VhChiP orthologs from Vibrio spp., indicating that other VhChiP orthologs may not be gated, or perhaps are gated in a different way. Regarding the need for a gating mechanism, in other enteric bacteria such as E. coli it is well established that the general porins OmpF and OmpC are not gated, but differently expressed depending on the osmolarity of the medium. At high osmolarity, expression of the smaller-diameter OmpC is upregulated relative to that of OmpF, presumably restricting the salt concentrations in the periplasmic space. Vibrio spp. also have orthologs of the general porins (named OmpU and OmpT in V. cholerae). While the structures of OmpU and OmpT have not yet been solved it is known that their expression is also regulated by osmolarity, with the larger-pore OmpU protein downregulated under conditions of high osmolarity37. Thus, a general picture emerges where under conditions of high osmolarity the entry of excess ions is restricted via upregulation of smaller-diameter channels and/or downregulation of larger ones. VhChiP is highly upregulated in marine environments in the presence of chitin21; for such a highly expressed channel, gating would be an effective means to restrict the entry of ions in a high-osmolarity environment. As mentioned above, how VhChiP orthologs with a shorter N-terminus (Fig. 5) would restrict ion access to the periplasmic space is not yet clear.
In conclusion, our studies on an important OM channel from marine Vibrio spp. highlight an emerging theme that substrate transport by OM channels is often more complicated than simple diffusion through a large, static pore. Ligand gating has been observed before in monomeric 14-stranded OM proteins33,38, but gating by a neighbor within an oligomeric assembly has, to our knowledge, not been reported in any membrane protein. Moreover, our results demonstrate that gating, rather than being a passive switch between open and closed states, might play a more active role in substrate transport. Future studies will be required to illuminate how gating improves substrate translocation and how ligands cause the necessary conformational changes that open the gate.
Methods
Cloning and expression of OM-expressed VhChiP
A BlastP search with chitoporin from V. furnissii as input (UniProtKB/TrEMBL entry: Q9KK91) identified VIBHAR_01269 as a putative chitoporin ortholog (GenBANK accession number YP_001444474) from the V. harveyi type strain ATCC BAA-1116 (also named V. campbellii ATCC BAA-1116). Therefore, oligonucleotides were designed for the gene from the BAA-1116 BB120 strain to clone the chitoporin gene from our laboratory strain (V. harveyi type strain 650). Genomic DNA was prepared using the PureLink Genomic DNA Kit (Invitrogen, Gibthai Company Ltd., Bangkok, Thailand) and used as the template for PCR. The oligonucleotides used for PCR are listed in Supplementary Table 3. The PCR product was of the expected size (1.1 kbp) and was cloned into pET23d(+) using Nco I and Xho I restriction sites, following the protocol of the manufacturer. Nucleotide sequences of both strands of the PCR product were determined by automated sequencing (First BASE Laboratories Sdn Bhd, Selangor Darul Ehsan, Malaysia). The recombinant plasmid harboring the chiP gene (pET23d(+)/chiP) was transformed into E. coli BL21(DE3) omp8 rosetta (ΔlamB ompF::Tn5ΔompAΔompC39) (a gift from Prof. Roland Benz, Jacob University Bremen, Germany). The transformed cells were grown at 37 °C in Luria-Bertani (LB) liquid medium containing 100 μg mL−1 ampicillin, 25 μg mL−1 kanamycin and 1% (w/v) glucose. The expression was induced by adding 0.4 mM IPTG at an OD600 ~ 0.6 and the cells were incubated for 6 h at 37 °C. After cell disruption, the membranes were spun down and incubated with 2% (w/v) SDS for 1 h at 50 °C. Afterwards the membranes were washed with 0.125% (v/v) octyl-POE and finally extracted with 3% (v/v) octyl-POE. Insoluble particles were centrifuged for 30 min at 235,000 × g and the supernatant was loaded onto a HiPrep Mono Q anion exchange column. The column was washed with 10 column volumes (CV) 20 mM (Phosphate buffer) sodium phosphate and 0.2% (v/v) LDAO at pH 7.4. The protein was eluted with a linear gradient of 0–1 M KCl. Fractions containing VhChiP were pooled and subjected to size exclusion chromatography using a HiLoad 16/600 superdex 200 (GE Healthcare) using 10 mM HEPES, 100 mM LiCl, 0.4% (v/v) C8E4, pH 7.5. The purified protein was concentrated to 10–12 mg mL−1 and directly flash-frozen into liquid nitrogen prior to setting up crystallization trials.
The gene encoding for truncated VhChiP, lacking the first nineteen residues of the mature sequence (EVYGII--), was synthesized by GenScript and cloned into pET23a (Novagen). Subsequent protein expression and purification was carried out as described above for the full-length protein.
Inclusion body expression of SeMet-labeled VhChiP
For expression of VhChiP into IBs, the gene without signal sequence and His-tag was cloned into pET28a (Novagen) via NcoI and XhoI (Supplementary Table 3). This procedure added the sequence “MG” to the N-terminus of the mature protein (MGDGANS--). The plasmid was transformed into BL21(DE3) cells (New England Biolabs). The cells were grown in minimal media (LeMasters-Richards) to OD600 ~ 0.6 at 37 °C before SeMet in combination with lysine, phenylalanine, threonine, leucine, isoleucine, and valine were added. Half an hour later the cells were induced with 1 mM IPTG at 37 °C for 3 h. Cells were harvested by centrifugation at 4500 × g for 30 min (Beckman Coulter). After cell disruption, IBs were harvested by centrifugation at 12,000 × g for 20 min and washed once in TBS with 1% Triton X-100 for 30 min at room temperature. This was followed by two washes without Triton X-100, followed by centrifugation for 20 min at 12,000 × g after each step. IBs were then solubilized at room temperature by stirring in ~25 mL TBS with 8 M Urea for 2 h. Non-solubilized pellet was removed by centrifugation at 200,000 × g for 30 min. For in vitro folding, the supernatant (~20 mL) was added dropwise to 200 mL TBS including 3% Elugent (Calbiochem) and the folding reaction was allowed to proceed at room temperature overnight with slow stirring. Subsequently, the folded protein was applied to a 10 mL anion exchange column and purified as described above for the OM-expressed protein.
Crystallization and structure solution
Initial crystallization trials for OM-expressed VhChiP (10 mg mL−1) were performed at 295 K by sitting-drop vapor diffusion using MemGold1- and MemGold2-Screen from Molecular Dimensions with a mosquito robot (TTP Labtech). The initial hits were optimized by fine-screening with larger drops by hanging drop vapor diffusion. Crystals in space group C2 were grown in 28% (w/v) PEG 400, 0.2 M sodium acetate, 0.1 M MES pH 6.5. Crystals were directly flash-frozen in liquid nitrogen. A data set was collected at IO2 at the Diamond Light Source (DLS), UK. Se-Met crystals, derived from in vitro-folded VhChiP (~12 mg mL−1), were obtained in two different crystal forms. Crystal form I in space group P21 was crystallized in 30% (w/v) PEG 400, 0.05 M NaCl, 0.1 M sodium citrate pH 5.5. Crystals in crystal form II were grown in 28% (w/v) PEG 400, 0.5 M potassium iodide, 0.1 M Tris pH 8.5. A SAD data set for crystal form I was collected to 1.95 Å at beamline IO2 at the Diamond Light Source (DLS), UK. Data were integrated and scaled with XDS40. Initial phasing and modeling was done using AUTOSOL within PHENIX41. Further model building was performed using the program COOT42. The protein model was refined with REFMAC43. Phases for OM-expressed VhChiP and in vitro-folded VhChiP in crystal form II were obtained by MR using MOLREP44 with the refined structure of SeMet-VhChiP as a search model. Model building was performed using COOT42 and the structure was refined with REFMAC43. The program MolProbity45 was used to evaluate the final model and PyMOL46 (Schrödinger, LLC) for the visualization of the protein structures.
For VhChiP complex structures, in vitro-folded protein (12 mg mL−1) was co-crystallized with 2.5 mM GlcNAc6 in 34% (w/v) PEG 400, 0.05 M NaCl, 0.1 M sodium citrate pH 5.5. Accordingly, OM-expressed protein (10 mg mL−1) was co-crystallized with 10 mM GlcNAc4 in 28% (w/v) PEG 400, 0.2 M sodium acetate, 0.1 M MES pH 6.5. The structures were solved via MR as described above for the apo proteins.
MD simulations
The starting structures of the MD simulations were prepared as follows. The crystal structures of the VhChiP trimer in closed and open conformations were embedded in a 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) bilayer together with TIP3P water and 1 M KCl salt. Subsequently, an energy minimization was performed using the steepest descent algorithm for 5000 steps followed by a 10 ns equilibration in the NVT and NPT ensembles with positional restraints on the heavy atoms of the proteins and lipids. The final unbiased simulations of VhChiP systems in open and closed conformations were performed for 500 ns. Furthermore, we carried out applied-field simulations for the in vitro-folded channel (200 mV: 3 × 250 ns each) and OM-expressed channel (200 mV: 3 × 250 ns each). For the free-energy calculations, we built an in silico VhChiP trimer variant with one mutation per monomer (D1A, D6A, and K9A) and a wild type system, both in the plug-inserted conformation, and including the necessary number of potassium ions to neutralize the system.
All MD simulations were performed with the GROMACS 4.6.5 program47 using the CHARMM36 all-atom force field48,49. The particle-mesh Ewald approach was used to calculate the long-range electrostatic interactions with a cutoff of 12 Å. Short-range Coulomb and Lennard Jones interactions were explicitly calculated up to the cutoff distance. The LINCS algorithm was applied to constraint the lengths of all bonds containing hydrogen atoms50. Moreover, the unbiased simulations discussed in this study were carried out in the NPT ensemble achieved by a semi-isotropic Parrinello-Rahman barostat51 at 1 bar with a coupling constant of 5 ps and the Nosé–Hoover thermostat52,53 with a coupling constant of 1 ps. The applied-field simulations were performed in a NVT ensemble with an electric field corresponding to transmembrane potential of 200 mV towards the periplasmic side of the channel. The field strength is proportional to the voltage V, i.e., E = V/L z , where L z denotes the system length in z direction normal to the membrane. During the MD simulations snapshots were collected at every 10 ps and the MD results were analyzed using the tools available in GROMACS 4.6.5 package.
To estimate the energetics for unbinding of the N-terminus, two umbrella sampling calculations were performed for the wild type and the above-described trimer variant system. To save computational resources, we took advantage of the trimeric organization of the channel and simultaneously simulated the free energy profiles of the three mutant plugs (D1A, D6A, and K9A) along a single reaction coordinate. The one-dimensional reaction coordinate corresponded to the z-coordinate distance between the centers of mass (COM) of the N-terminal plug (Cα atoms of residues 1–10) and the corresponding barrel (Cα atoms of residues 25–350). The reaction coordinate was divided into 28 windows and we simulated each window for 90 ns with a harmonic potential of 1000 kJ mol−1 nm−2 on the reaction coordinate. The unbiased free energy profiles were calculated from the last 80 ns of each window trajectory using the g_wham54 implementation of the weighted histogram analysis method55.
Single-channel electrophysiology
Planar lipid bilayer reconstitution was carried out in electrolyte containing 1 M KCl and 20 mM HEPES pH 7.5, at 25 °C28,56. Montal–Mueller type solvent-free bilayer formation was carried out using 1,2-diphytanoyl-sn-glycero-3-phosphatidylcholine (DPhPC; Avanti Polar Lipids, Alabaster, AL). First, the aperture was pre-painted with a few microliters of 1%(v/v) hexadecane in hexane, after which a planar bilayer was formed across the aperture by lowering and raising the liquid level. Ion currents were detected using Ag/AgCl electrodes with a patch-clamp amplifier connected to a two-electrode bilayer head-stage (PC-ONE plus PC-ONE-50; Dagan Corp., Minneapolis, MN, USA). The bilayer setup was operated within a Faraday cage on a vibration-dampened table, with an A/D converter (LIH 1600, HEKA Elektronik, Lambrecht, Germany) and was operated using the PULSE software program (HEKA Elektronik, Lambrecht, Germany). One of the electrodes, immersed in 1 M KCl electrolyte on the cis side of the cuvette, was connected to ground, while the electrode on the trans side was connected to the amplifier head-stage. VhChiP was always added to the cis side of the cuvette. Conductance values were determined from the I–V curves obtained from single-channel insertions at different voltages.
To investigate sugar translocation, single channels of the VhChiP variants were reconstituted in the artificial lipid membrane. To prevent multiple insertions during data acquisition, the protein solution in the chamber was gently diluted after the first insertion by sequential additions of working electrolyte. Subsequently the fully open channel was titrated with chitohexaose with concentrations ranging from 0.1 to 10 µM. Sugars were added to the cis side of the chamber. Ion flow was usually recorded for 2 min at different transmembrane potentials. The equilibrium binding constant K (M−1) was derived from the decrease in the ion conductance resulting from increasing concentrations of sugar using the following Equation (1) (ref. 57):
Gmax is the average conductance of the fully open VhChiP channel and Gc is the average conductance at a given concentration [c] of a chitooligosaccharide. Imax is the initial current through the fully open channel without sugar and Ic is the current at a particular sugar concentration.
Liposome swelling experiments
VhChiP-reconstituted proteoliposomes were prepared using soybean l-α-phosphatidylchloline (20 mg mL−1, freshly prepared in chloroform) (Sigma-Aldrich) to form multi-lamellar liposomes10. Following this, 200 ng of VhChiP was reconstituted into 200 μL of liposome suspension by sonication, and 17% (w/v) dextran (40 kDa) was entrapped in the proteoliposomes by drying under vacuum and resuspending in 10 mM phosphate buffer pH 7.5. For determination of the isotonic solute concentration, d-Raffinose solutions were prepared in phosphate buffer with concentrations of 40, 50, 60, and 70 mM. The obtained isotonic concentration was then used for the adjustment of the isotonic concentration for other solutes. All small sugars (Fig. 10a) were prepared at 60 mM concentration. For chitooligosaccharides (Fig. 10b) we prepared the solutions differently, and used 1 mM of each chitosugar in 59 mM raffinose. The osmolarity of each sugar solution was checked to be at the isotonic concentration using a Genotec Osmometer 300. The reasons for using low concentrations of the chitosugars are twofold: (i) GlcNAc4–6 are not soluble at 60 mM and (ii) GlcNAc4–6 interact with VhChiP at low µM concentration (not mM). The liposome-swelling assays were carried out by adding 25 µL of proteoliposome suspension to 600 µL of sugar solution and the changes in absorbance at 500 nm were monitored immediately. The absorbance change over the first 60 s was used to estimate the swelling rate (s−1) according to: φ = (1/A i )dA/dt, in which φ is the swelling rate, A i the initial absorbance, and dA/dt the rate of absorbance change during the first 60 s. The swelling rate for each sugar was normalized by setting the rate of l-arabinose (150 Da), the smallest sugar, to 100%. The values presented are averages from three to five independent experiments. The sugars tested were d-glucose (180 Da), d-mannose (180 Da), d-galactose (180 Da), N-acetylglucosamine (GlcNAc) (221 Da), d-sucrose (342 Da), d-melezitose (522 Da), GlcNAc2 (424 Da), GlcNAc3 (628 Da), GlcNAc4 (830 Da), GlcNAc5 (1034 Da), GlcNAc6 (1237 Da), and maltodextrins. Protein-free liposomes and proteoliposomes without sugars were used as negative controls.

The N-terminus is important for chito-oligosaccharide substrate transport. Proteoliposome swelling assays in the presence of various mono- and oligosaccharides (a) as well as chito-oligosacharides of different lengths (b). No permeation of any sugars was observed when tested with control liposomes without VhChiP. “Folded” denotes in vitro-folded VhChiP. Data represent mean ± s.d., n = 3. For details see Methods
Data availability
The atomic coordinates and the associated structure factors have been deposited in the Protein Data Bank (http://www.pdbe.org) with accession codes 5MDO (In vitro-folded VhChiP, crystal form 1), 5MDP (In vitro-folded VhChiP, crystal form 2), 5MDQ (OM-expressed VhChiP), 5MDR (In vitro-folded VhChiP with chito-hexaose), and 5MDS (OM-expressed VhChiP with chito-tetraose). Other data supporting the findings of this study are available from the corresponding authors upon reasonable request.
References
Klemm, D. et al. Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 44, 3358–3393 (2005).
Muzzarelli, R. A. A. Chitin (Pergamon Press, UK, 1977).
Gooday, G. W. The ecology of chitin degradation. Adv. Microb. Ecol. 11, 387–430 (1990).
Tracey, M. A. Chitin. Rev. Pure Appl. Chem. 7, 1–13 (1957).
Hirono, I., Yamashita, M. & Aoki, T. Note: molecular cloning of chitinasegenes from Vibrio anguillarum and V. parahaemolyticus. J. Appl. Microbiol. 84, 1175–1178 (1998).
Zobell, C. E. & Rittenberg, S. C. The occurrence and characteristics of chitinoclastic bacteria in the sea. J. Bacteriol. 35, 275–287 (1938).
Hunt, D. E. et al. Conservation of the chitin utilization pathway in the Vibrionaceae. Appl. Environ. Microbiol. 74, 44–51 (2008).
Jung, B. O., Roseman, S. & Park, J. K. The central concept for chitin catabolic cascade in marine bacterium, Vibrios. Macromol. Res. 16, 1–15 (2008).
Keyhani, N. O., Li, X. B. & Roseman, S. Chitin catabolism in the marine bacterium Vibrio furnissii. Identification and molecular cloning of a chitoporin. J. Biol. Chem. 275, 33068–33076 (2000).
Suginta, W. et al. Molecular uptake of chitooligosaccharides through chitoporin from the marine bacterium Vibrio harveyi. PLoS ONE 8, e55126 (2013).
Suginta, W. et al. Chitoporin from Vibrio harveyi, a channel with exceptional sugar specificity. J. Biol. Chem. 288, 11038–11046 (2013).
Keyhani, N. O. & Roseman, S. The chitin catabolic cascade in the marine bacterium Vibrio furnissii. Molecular cloning, isolation, and characterization of a periplasmic chitodextrinase. J. Biol. Chem. 271, 33414–33424 (1996).
Suginta, W. et al. Novel β-N-acetylglucosaminidases from Vibrio harveyi 650: cloning, expression, enzymatic properties, and subsite identification. BMC Biochem. 11, 40 (2010).
Keyhani, N. O. & Roseman, S. The chitin catabolic cascade in the marine bacterium Vibrio furnissii. Molecular cloning, isolation, and characterization of a periplasmic beta-N-acetylglucosaminidase. J. Biol. Chem. 271, 33425–33432 (1996).
Bassler, B. L. et al. Chitin utilization by marine bacteria. Chemotaxis to chitin oligosaccharides by Vibrio furnissii. J. Biol. Chem. 266, 24268–24275 (1991).
Keyhani, N. O. et al. The chitin catabolic cascade in the marine bacterium Vibrio furnissii. Characterization of an N,N′-diacetyl-chitobiose transport system. J. Biol. Chem. 271, 33409–33413 (1996).
Bouma, C. L. & Roseman, S. Sugar transport by the marine chitinolytic bacterium Vibrio furnissii. Molecular cloning and analysis of the glucose and N-acetylglucosamine permeases. J. Biol. Chem. 271, 33457–33467 (1996).
Park, J. K., Keyhani, N. O. & Roseman, S. Chitin catabolism in the marine bacterium Vibrio furnissii. Identification, molecular cloning, and characterization of A N,N′-diacetylchitobiose phosphorylase. J. Biol. Chem. 275, 33077–33083 (2000).
Li, X. & Roseman, S. The chitinolytic cascade in Vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc. Natl Acad. Sci. USA 101, 627–631 (2004).
Suginta, W. et al. An endochitinase A from Vibrio carchariae: cloning, expression, mass and sequence analyses, and chitin hydrolysis. Arch. Biochem. Biophys. 424, 171–180 (2004).
Meibom, K. L. et al. The Vibrio cholerae chitin utilization program. Proc. Natl Acad. Sci. USA 101, 2524–2529 (2004).
Schmitt, E. K., Vrouenraets, M. & Steinem, C. Channel activity of OmpF monitored in nano-BLMs. Biophys. J. 91, 2163–2171 (2006).
Benz, R. et al. Pore formation by LamB of Escherichia coli in lipid bilayer membranes. J. Bacteriol. 165, 978–986 (1986).
Schülein, K. & Benz, R. LamB (maltoporin) of Salmonella typhimurium: isolation, purification and comparison of sugar binding with LamB of Escherichia coli. Mol. Microbiol. 4, 625–632 (1990).
Suginta, W. & Smith, M. F. Single-molecule trapping dynamics of sugar-uptake channels in marine bacteria. Phys. Rev. Lett. 110, 238102 (2013).
Suginta, W., Winterhalter, M. & Smith, M. F. Correlated trapping of sugar molecules by the trimeric protein channel chitoporin. Biochim. Biophys. Acta-Biomembr. 1858, 3032–3040 (2016).
Zachariae, U. et al. High resolution crystal structures and molecular dynamics studies reveal substrate binding in the porin Omp32. J. Biol. Chem. 281, 7413–7420 (2006).
Chumjan, W. et al. Chitoporin from the marine bacterium Vibrio harveyi: probing the essential roles of Trp136 at the surface of the constriction zone. J. Biol. Chem. 290, 19184–19196 (2015).
Moraes, T. F. et al. An arginine ladder in OprP mediates phosphate-specific transfer across the outer membrane. Nat. Struct. Mol. Biol. 14, 85–87 (2007).
Hayat, S. et al. Inclusion of dyad-repeat pattern improves topology prediction of transmembrane β-barrel proteins. Bioinformatics 32, 1571–1573 (2016).
Tsirigos, K. D., Elofsson, A. & Bagos, P. G. PRED-TMBB2: improved topology prediction and detection of beta-barrel outer membrane proteins. Bioinformatics 32, i665–i671 (2016).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
van den Berg, B. et al. Outer-membrane translocation of bulky small molecules by passive diffusion. Proc. Natl Acad. Sci. USA 112, E2991–E2999 (2015).
Dutzler, R., Wang, Y. F., Rizkallah, P., Rosenbusch, J. P. & Schirmer, T. Crystal structures of various maltooligosaccharides bound to maltoporin reveal a specific sugar translocation pathway. Structure 4, 127–134 (1996).
van Straaten, K. E. et al. Structure of Escherichia coli lytic transglycosylase MltA with bound chitohexaose: implications for peptidoglycan binding and cleavage. J. Biol. Chem. 282, 21197–21205 (2007).
Sen, K., Hellman, J. & Nikaido, H. Porin channels in intact cells of Escherichia coli are not affected by Donnan potentials across the outer membrane. J. Biol. Chem. 263, 1182–1187 (1988).
Chakrabarti, S. R. et al. Porins of Vibrio cholerae: purification and characterization of OmpU. J. Bacteriol. 178, 524–530 (1996).
Lepore, B. W. et al. Ligand-gated diffusion across the bacterial outer membrane. Proc. Natl Acad. Sci. USA 108, 10121–10126 (2011).
Prilipov, A., Phale, P. S., Van Gelder, P., Rosenbusch, J. P. & Koebnik, R. Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol. Lett. 163, 65–72 (1998).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Vagin, A. & Teplyakov, A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 (1997).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
The PyMOL Molecular Graphics System, v.1.8 (Schrödinger, LLC., 2017).
Pronk, S. et al. Gromacs 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Best, R. B. et al. Optimization of the additive CHARMM All-atom protein force field targeting improved sampling of the backbone φ, ψ and Side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Hess, B., Bekker, H., Berendsen, H. J. C. & Johannes, G. E. M. F. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Nosé, S. A. Molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 (1984).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695 (1985).
Hub, Jochen S., de Groot, B. L. & van der Spoel, D. g_whams: a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 (2010).
Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A. & Rosenberg, J. M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13, 1011–1021 (1992).
Schulte, A. et al. The outer membrane protein VhOmp of Vibrio harveyi: pore-forming properties in black lipid membranes. J. Membr. Biol. 230, 101–111 (2009).
Benz, R. & Hancock, R. E. Mechanism of ion transport through the anion-selective channel of the Pseudomonas aeruginosa outer membrane. J. Gen. Physiol. 89, 275–295 (1987).
Acknowledgements
B.v.d.B. would like to acknowledge the Royal Society for salary support. We are also indebted to the staff at beam lines I02, I04, and I04-1 of the Diamond Light Source UK for beam time (proposal mx9948) and assistance with data collection. Finally, we thank Prof. Jirawat Yongsawatdigul (Suranaree University of Technology, Thailand), for control peptides. The work of W.S. was financially supported by Suranaree University of Technology, the Office of the Higher Education Commission under NRU project of Thailand, and the Thailand Research Fund (Grant no. BRG578001, SUT1-102-57-36-18 and SUT1-102-59-12-17). A.A. received a Ph.D. scholarship through the Royal Golden Jubilee Ph.D. Program (Grant no. PHD53K0022). The research leading to these results was conducted as part of the Translocation consortium (www.translocation.eu) and has received support from the Innovative Medicines Initiatives Joint Undertaking under Grant Agreement No. 115525, resources that are composed of financial contributions from the European Union’s seventh framework program (FP7/2007–2013) and European Federation of Pharmaceutical Industries and Associations companies in-kind contribution.
Author information
Authors and Affiliations
Contributions
A.A. and M.Z. cloned, expressed, and purified VhChiP. M.Z. crystallized VhChiP and determined crystal structures. A.A. and W.S. performed single-channel electrophysiology experiments. A.S. assisted with low-voltage electrophysiological experiments. A.A. carried out liposome swelling assays. A.K. carried out MD simulations, K.R.P. assisted with the free energy calculations, and U.K. planned and supervised the simulations. A.B. collected crystallography data and maintained the Newcastle Structural Laboratory. B.v.d.B. and W.S. designed research. B.v.d.B. wrote the paper, assisted by W.S. and M.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aunkham, A., Zahn, M., Kesireddy, A. et al. Structural basis for chitin acquisition by marine Vibrio species. Nat Commun 9, 220 (2018). https://doi.org/10.1038/s41467-017-02523-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-02523-y
This article is cited by
-
Inherited chitinases enable sustained growth and rapid dispersal of bacteria from chitin particles
Nature Microbiology (2023)
-
Periplasmic chitooligosaccharide-binding protein requires a three-domain organization for substrate translocation
Scientific Reports (2023)
-
Genome sequence of Vibrio fluvialis 362.3 isolated from coral Mussismilia braziliensis reveals genes related to marine environment adaptation
Archives of Microbiology (2021)
-
Understanding microbial community dynamics to improve optimal microbiome selection
Microbiome (2019)
-
Membrane directed expression in Escherichia coli of BBA57 and other virulence factors from the Lyme disease agent Borrelia burgdorferi
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
