
Abstract
Hydrosilylation of allenes is the addition of a hydrogen atom and a silyl group to a carbon–carbon double bond of an allene molecule and represents a straightforward and atom-economical approach to prepare synthetically versatile allylsilanes and vinylsilanes. However, this reaction generally produces six possible isomeric organosilanes, and the biggest challenge in developing this reaction is to control both regioselectivity and stereoselectivity. The majorities of the developed allene hydrosilylation reactions show high selectivity towards the production of vinylsilanes or branched allylsilanes. By employing a cobalt catalyst generated from readily available and bench-stable cobalt precursor and phosphine-based ligands, here we show that this reaction proceeds under mild conditions in a regioselective and stereoselective manner, and affords synthetically challenging, but valuable linear cis-allylsilanes with excellent stereoselectivity (generally cis to trans ratios: >98:2). This cobalt-catalyzed (Z)-selective allene hydrosilylation provides a general approach to access molecules containing stereodefined (Z)-alkene units.
Similar content being viewed by others
Introduction
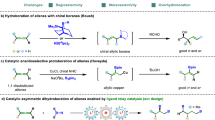
Allylsilanes are a type of organosilanes with an allyl group on a silicon atom, and the stereochemistry around the allylic double bond may be E (cis) or Z (trans). Allylsilanes are synthetically valuable building blocks because of their non-toxicity, high stability and versatile applications in organic synthesis and material science1,2,3. They have been employed as monomers for syntheses of silicon-containing polymers and undergo a variety of organic transformations4,5,6. As such, various methods have been developed to prepare allylsilanes and the majority of these approaches produce thermodynamically more stable (E)-allylsilanes7,8,9. However, the stereoselective synthesis of a wide range of (Z)-allylsilanes still remains challenging and rare10,11,12,13,14. Catalytic hydrosilylation of allenes (Fig. 1a) is one of the most straightforward and atom-economical approaches to synthesize (Z)-allylsilanes, provided that catalysts and reaction conditions favoring the formation of (Z)-allylsilanes can be identified.

Hydrosilylation of allenes. a Overview of hydrosilylation of allenes. b Metal-catalyzed β,γ-hydrosilylation of allenes. c Metal-catalyzed α,β-hydrosilylation of allenes
Allenes can undergo hydrosilylation with hydrosilanes in the presence of transition metal catalysts to produce allylsilanes or vinylsilanes (Fig. 1a)15,16,17,18,19,20,21. The major difficulty in allene hydrosilylation is to control the regio- and stereoselectivity because multiple vinylsilane and allylsilane products may be generated (Fig. 1a). The majority of metal-catalyzed hydrosilylation of terminal allenes show high selectivity for β,γ-hydrosilylation, affording allylsilanes or vinylsilanes with a terminal alkene group without an issue of the control over Z/E-selectivity (Fig. 1b)15,16,17,18,19,20. However, α,β-hydrosilylation of terminal allenes is more challenging because it can potentially form four isomeric Z/E-allylsilane and Z/E-vinylsilane products (Fig. 1a). Selective formation of one organosilane product out of four possible isomers is of high synthetic importance. Except for a recent example of molybdenum catalyst for α,β-hydrosilylation of allenes with modest Z/E selectivity at 110 °C or under UV irradiation (Fig. 1c)21, catalysts for selective allene α,β-hydrosilylation that can combine high catalyst activity, high Z-stereoselectivity, broad substrate scope and mild reaction conditions are conspicuously unknown.
Platinum complexes are the most frequently encountered catalysts for hydrosilylation reactions in industry22,23,24,25,26. However, there is a growing interest in replacing platinum catalysts with earth-abundant base-metal catalysts for hydrosilylation27,28. Accordingly, a tremendous development has been made in cobalt-catalyzed hydrosilylation of alkenes and alkynes29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. The hydrosilylation of allenes has been studied with stoichiometric amounts of the cobalt complex Co2(CO)8, but this ligandless cobalt catalyst shows low selectivity for α,β-hydrosilylation44. A Co-catalyzed allene α,β-hydrosilylation that can selectively produce (Z)-allylsilanes still remains unknown. Driven by our continuous interest in developing base-metal-catalyzed hydrofunctionalization of unsaturated organic molecules, we are interested in identifying a highly Z-selective cobalt catalyst for allene α,β-hydrosilylation. An improved Z/E-selectivity is anticipated for ligated cobalt catalysts due to a greater stereochemical communication between ligand and allene substrate from a relatively smaller cobalt center, comparing with the molybdenum catalyst Mo(CO)6 21. Herein we report a cobalt-catalyzed stereoselective α,β-hydrosilylation of terminal allenes to prepare (Z)-allylsilanes. Furthermore, we have developed a practical one-pot procedure to access synthetically challenging trisubstituted (Z)-allylic alcohols by combining this cobalt-catalyzed allene hydrosilylation and subsequent oxidation of the resulting (Z)-allylsilanes. In addition, we show that Co(acac)2 can be reduced by PhSiH3 in the presence of bisphosphine ligands to generate well-defined, but catalytically active, Co(I) hydride complexes.
Results
Evaluation of reaction conditions
We initiated our studies of Co-catalyzed hydrosilylation of allenes by evaluating reaction conditions for the reaction of cyclohexylallene with phenylsilane. This reaction can potentially produce six organosilanes from either 1,2-hydrosilylation ((E)-1a, (Z)-1a, (E)-1a′, and (Z)-1a′) or 2,3-hydrosilylation (1a″ and 1a‴), as depicted in Table 1. We tested this reaction with various cobalt catalysts that were generated in situ from bench-stable Co(acac)2 and phosphine ligands. In general, these experiments were conducted with 2 mol% cobalt catalysts in THF at room temperature for 18 h. The results of these experiments are summarized in Table 1.
The reactions catalyzed by the combination of Co(acac)2 and monophosphine ligands, such as PPh3 and PCy3, proceeded to low conversions of cyclohexylallene and produced a mixture of six products with low selectivities for (Z)-1a (Table 1, entries 1 and 2). Improved conversions and selectivities to (Z)-1a (83−91%) were achieved for reactions that were conducted with the combination of Co(acac)2 and bisphosphine ligands, such as dppe, dppp, dppb, dcpe or dppf (Table 1, entries 3−7). In particular, the reaction with the catalyst generated from Co(acac)2 and rac-binap occurred to full conversion with excellent selectivity (98%) for (Z)-1a (Table 1, entry 8). Similarly high selectivity for (Z)-1a was obtained for the reaction catalyzed by Co(acac)2 and xantphos, but this reaction produced a significant amount of bis((Z)−3-cyclohexylallyl)(phenyl)silane 2 (Fig. 2a), which was generated by the hydrosilylation of cyclohexylallene with (Z)-1a as a hydrosilylating reagent (Table 1, entry 9). The result of entry 9 indicates that the hydrosilylation of cyclohexylallene with secondary silanes is likely feasible. Indeed, cyclohexylallene reacted with secondary silanes Ph2SiH2 and MePhSiH2, in the presence of 1 mol% Co(acac)2 and 1 mol % xantphos, affording the corresponding (Z)-allylsilanes 1b and 1c in high yields with excellent stereoselectivities (Fig. 2b, c)11. In addition, we tested a nitrogen-based ligand mesPDI for this transformation, and the reaction afforded a mixture of (Z)-1a, (E)-1a and (E)-1a with low regio- and stereoselectivity (Table 1, entry 10). Furthermore, we also tested various solvents for this hydrosilylation catalyzed by Co(acac)2/binap (Table 1, entries 11–14). The reactions conducted in toluene, diethylether and tert-butylmethylether proceeded to full conversions of cyclohexylallene with excellent selectivity for (Z)-1a (Table 1, entries 11, 13 and 14), but the reaction did not occur in hexane, likely due to the poor solubility of the cobalt catalyst in hexane (Table 1, entry 12). In addition, this reaction occurred smoothly in the absence of any solvent and afforded the desired product (Z)-1a in 85% isolated yield with Z/E of 99:1 (Table 1, entry 15).

Hydrosilylation of cyclohexylallene with secondary silanes. a The reaction with phenylsilane PhSiH3. b The reaction with diphenylsilane Ph2SiH2. c The reaction with methylphenylsilane Me(Ph)SiH2; Z/E ratios were determined with gas chromatography (GC) analysis on crude reaction mixtures
Substrate scope of allenes
Under the identified conditions (Table 1, entry 8), we studied the scope of monosubstituted allenes for this reaction. These results are summarized in Fig. 3. In general, a variety of monosubstituted allenes reacted to produce the desired disubstituted (Z)-allylsilanes (1a−1q) in high yields (74−92%) with excellent stereoselectivities (Z:E = 99:1). This reaction shows good functional group tolerance and a range of reactive groups, such as chloro (1g), bromo (1h and 1p), iodo (1i), siloxy (1j), ester (1k), pinacol boronic ester (1l), and imide (1m), are compatible with the reaction conditions. Under the identified conditions, the bromine- and iodine-containing allenes (1h, 1p and 1i) were not fully consumed and this accounts for the relatively lower yields of 1h, 1p and 1i compared with other entries.

Scope of monosubstituted allenes. Conditions: allene (0.500 mmol), PhSiH3 (0.500 mmol), Co(acac)2 (10.0 μmol), binap (10.0 μmol), THF (1 mL), room temperature, 18 h; yield of isolated product; Z/E ratios were determined with gas chromatography (GC) analysis on crude reaction mixtures. #3 mol % catalyst, 50 °C
Similarly, disubstituted terminal allenes also reacted with PhSiH3 under the conditions identified for hydrosilylation of monosubstituted allenes (Fig. 3). For example, the reaction between 1-methyl-1-phenylallene and PhSiH3 proceeded to full conversion in 24 h in the presence of 2 mol% Co(acac)2 and rac-binap. However, the same reaction conducted with 1 mol% Co(acac)2/xantphos proceeded to full conversion in only 3 h, affording (Z)-allylsilane 3a in 94% isolated yield with excellent stereoselectivity. Therefore, we chose Co(acac)2/xantphos to study the scope of disubstituted terminal allenes and the results are listed in Fig. 4. A series of disubstituted terminal allenes containing aromatic or aliphatic groups readily reacted with PhSiH3 in the presence of 1 mol% Co(acac)2/xantphos at room temperature for 3 h, affording the desired (Z)-allylsilanes (3b−3s) in high yields (74−95%) with excellent stereoselectivities (Z:E = 99:1). Decreased Z:E ratio (88:12) was obtained for allene with decreased steric difference between R and Rʹ (3q). A variety of reactive groups, such as siloxy (3f), chloro (3g and 3n), bromo (3h), ester (3i), and acetal (3j), are tolerated under the reaction conditions. In addition, this allene hydrosilylation was tested with secondary hydrosilanes (Ph2SiH2, Ph(Me)SiH2, and Et2SiH2), and these reactions produced the corresponding (Z)-allylsilanes (3s−3v) in high yields with high stereoselectivities. However, this hydrosilylation of allene did not occur when tertiary hydrosilanes, such as (EtO)3SiH and (Me3SiO)2MeSiH, were used.

Scope of disubstituted allenes. Reaction conditions: allene (0.500 mmol), PhSiH3 (0.500 mmol), Co(acac)2 (5.0 μmol), xantphos (5.0 μmol), THF (1 mL), rt, 3 h; yield of isolated product; Z/E ratios were determined with gas chromatography (GC) analysis on crude reaction mixtures. *2 mol % catalyst, RT, 12 h. #2 mol% catalyst, 70 °C, 24 h
Synthetic potential
As both Co(acac)2 and the xantphos ligand employed in this transformation are bench-stable, we tested the reaction between 1-methyl-1-phenylallene and PhSiH3 or Ph2SiH2 on a 10 mmol scale in the presence of 0.5 mol% of Co(acac)2 and xantphos that were weighed on the benchtop without using a drybox. These reactions afforded (Z)-allylsilanes 3a and 3s in high isolated yield with Z:E of 99:1 (Fig. 5a). In addition, we demonstrated that (Z)-allylsilane 3v underwent Ir-catalyzed intramolecular dehydrogenative silylation to afford a six-membered silacyclic compound 4 in 51% isolated yield (Fig. 5b); [dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl, nbe = norbornene]45,46.

Synthetic potential. a A concentration of 10 mmol scale reactions. b Ir-catalyzed dehydrogenative silylation of (Z)-allylsilane 3v. c One-pot synthesis of (Z)-allylic alcohols; The Z/E ratios were determined with gas chromatography (GC) analysis on crude reaction mixtures
Allylic alcohols are synthetically valuable intermediates in various organic transformations. The synthesis of stereodefined allylic alcohols, particularly ones containing a trisubstituted Z-alkene unit47,48,49,50,51, is a persisting challenge in synthetic chemistry. Here we developed a one-pot procedure to synthesize (Z)-allylic alcohols containing disubstituted or trisubstituted alkenes by combining the cobalt-catalyzed allene hydrosilylation and the subsequent oxidation of the resulting (Z)-allylsilanes with H2O2 under basic conditions. A series of (Z)-allylic alcohols (5a–5h) can be prepared in good isolated yields using this one-pot procedure (Fig. 5c, see Supplementary Methods for the detailed procedure).
Discussion
(Z)-allylsilanes are thermodynamically less stable than (E)-allylsilanes and are susceptible to Z/E-isomerization to form the thermodynamically more stable (E)-allylsilanes in the presence of a Co-H species40,52. The high Z-selectivities obtained for reactions listed in Fig. 2 suggest that the cobalt catalyst generated from Co(acac)2 and binap does not catalyze the Z/E-isomerization of these Z-allylsilanes. Indeed, the isolated (Z)-allylsilane (Z)-1l does not undergo Z/E-isomerization in the presence of 2 mol % Co(acac)2/binap and 1 equivalent of PhSiH3 (Fig. 6a). However, (Z)-allylsilane (Z)-1l was isomerized to (E)-1l at room temperature in 24 h in the presence of 1 mol% Co(acac)2/xantphos and 1 equivalent of PhSiH3 (Fig. 6a). We also tested these isomerization reactions in the absence of 1 equivalent of PhSiH3 and comparable results were obtained (Fig. 6b). Due to this Z/E-isomerization, the reaction in Fig. 2b afforded a mixture of (Z)-1b and (E)-1b with a ratio of 63:37 when the reaction time was extended to 48 h (Fig. 6e). Interestingly, the Z/E-isomerization of the isolated (Z)-3a did not occur in the presence of Co(acac)2/xantphos catalyst (Fig. 6c, d). We attributed the lack of Z/E-isomerization for the trisubstituted allylsilane (Z)-3a to the increased steric hindrance around the C=C bond, compared to the disubstituted allylsilane (Z)-1l.

Isomerization of (Z)-allylsilanes. a, b Isomerization of a disubstituted (Z)-allylsilane (Z)-1l. c, d Isomerization of a trisubstituted (Z)-allylsilane (Z)-3a. e Hydrosilylation of cyclohexylallene with Ph2SiH2 conducted for 48 h
To understand the mechanism of this cobalt-catalyzed allene hydrosilylation, we conducted a deuterium-labeling reaction between buta-2,3-dien-2-ylbenzene and PhSiD3. This reaction afforded the corresponding (Z)-allylsilane with a deuterium atom located trans to the phenyl group (Fig. 7a). In addition, a mercury test suggests the homogeneous nature of this catalytic hydrosilylation reaction (Fig. 7b). To provide insight into the cobalt intermediate for this allene hydrosilylation, we tested the reaction of Co(acac)2 and PhSiH3 in the presence of various bisphosphine ligands and found that the reaction using 2 equivalents of dppbz ligand generated a well-defined CoI-H complex (dppbz)2CoH (6) in 70% isolated yield (Fig. 7c)53. Complex 6 was active for allene hydrosilylation with regio- and stereoselectivity matching those of the corresponding reaction catalyzed by Co(acac)2 and dppbz ligand (see Supplementary Table 1 for the detailed evaluation of hydrosilylation with complex 6).

Reaction mechanism. a A deuterium-labeling reaction. b Mercury test. c A cobalt hydride complex 6 prepared by reduction of Co(acac)2 with PhSiH3. d proposed catalytic cycle for cobalt-catalyzed stereoselective 1,2-hydrosilylation of allenes. e hydrosilylation of 1-cyclopropyl-1-phenylallene, an allene containing a radical clock
On the basis of the result of the deuterium-labeling experiment (Fig. 7a), the generation of a catalytically active Co(I)–H intermediate (Fig. 7c), and the precedent of the cobalt-catalyzed hydrosilylation of alkenes27,28, we propose a hydrometalation pathway with a Co(I) hydride intermediate for this Co-catalyzed stereoselective hydrosilylation of allenes (Fig. 7d). In such a mechanism, migratory insertion of the allene substrate into a Co(I)-H species forms an η 1-bound allylcobalt intermediate (I). The steric repulsion between the RL of the allyl group and the ligand of the cobalt catalyst makes the formation of η 3-bound allylcobalt intermediate (II) unfavorable. Subsequent σ-bond metathesis54 between the η 1-bound allylcobalt intermediate (I) and hydrosilane produces the (Z)-allylsilane product and regenerates the Co(I)-H species. Minimizing the steric interaction between the Co(I)-H species and the substituents on the allene substrate accounts for the observed (Z)-selectivity, which suggests that reducing the steric difference between two substituents of 1,1-disubstituted allenes will decrease the Z/E-selectivity. Indeed, the hydrosilylation of 1-cyclopropyl-1-phenylallene produces a mixture of (Z/E)-allylsilanes with a Z/E-ratio of 63:37 (Fig. 7e). Such steric interaction has been proposed in a Rh-catalyzed stereoselective hydroformylation of terminal allenes55.
In summary, we have developed a highly regio- and stereoselective allene hydrosilylation catalyzed by cobalt complexes generated from bench-stable Co(acac)2 and binap or xantphos ligand. A wide range of monosubstituted and disubstituted terminal allenes reacted to afford the corresponding linear (Z)-allylsilanes in high yields with excellent stereoselectivities. This cobalt-catalyzed allene hydrosilylation coupled with sequential oxidation of the resulting (Z)-allylsilane provided a practical one-pot approach to prepare synthetically challenging (Z)-allylic alcohols. Further studies to develop cobalt-catalyzed selective hydrofunctionalization of other types of multiply unsaturated molecules are on going.
Methods
General procedure for cobalt-catalyzed allene hydrosilylation
In an argon-filled dry box, Co(acac)2, phosphine ligand, THF(1 mL) and a magnetic stirring bar were added to a 4-mL screw-capped vial and the mixture was stirred for 5 min. Then terminal allenes (0.500 mmol) and silane (1.1 eq, 0.550 mmol) were added. The vial was sealed with a cap containing a PTFE septum and removed from the dry box. The reaction mixture was stirred at room temperature for 18 h (Fig. 3) or 3 h (Fig. 4) and the resulting solution was concentrated in vacuum. The crude product was purified by column chromatography on silica gel with a mixture of ethyl acetate and hexane as eluent.
Data availability
The authors declare that all the data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request.
References
Brook, M. A. Silicon in Organic, Organometallic, and Polymer Chemistry. (J. Wiley, New York, 2000).
Fleming, I., Barbero, A. & Walter, D. Stereochemical control in organic synthesis using silicon-containing compounds. Chem. Rev. 97, 2063–2192 (1997).
Gaspar, P. & West, R. in The Chemistry of Organic Silicon Compounds Vol 2 (eds Rappoport, Z. and Apeloig, Y.) (John Wiley and Sons, Chester, UK, 1998).
Angle, S. R. & El-Said, N. A. Stereoselective synthesis of tetrahydrofurans via formal [3+2]-cycloaddition of aldehydes and allylsilanes. formal total synthesis of the muscarine alkaloids (−)-allomuscarine and (+)-epimuscarine. J. Am. Chem. Soc. 124, 3608–3613 (2002).
Judd, W. R., Ban, S. & Aubé, J. Remote control of diastereoselectivity in intramolecular reactions of chiral allylsilanes. J. Am. Chem. Soc. 128, 13736–13741 (2006).
Masse, C. E. & Panek, J. S. Diastereoselective reactions of chiral allyl and allenyl silanes with activated C: X. pi.-Bonds. Chem. Rev. 95, 1293–1316 (1995).
Suginome, M., Matsumoto, A. & Ito, Y. New Synthesis of (E)-allylsilanes with high enantiopurity via diastereoselective intramolecular bis-silylation of chiral allylic alcohols. J. Am. Chem. Soc. 118, 3061–3062 (1996).
Kamachi, T., Kuno, A., Matsuno, C. & Okamoto, S. Cobalt-catalyzed mono-coupling of R3SiCH2MgCl with 1,2-dihalogenoethylene: a general route to γ-substituted (E)-allylsilanes. Tetrahedron Lett. 45, 4677–4679 (2004).
Suginome, M., Iwanami, T., Ohmori, Y., Matsumoto, A. & Ito, Y. Stereoselective synthesis of highly enantioenriched (E)‐allylsilanes by palladium‐catalyzed intramolecular bis‐silylation: 1,3‐chirality transfer and enantioenrichment via dimer formation. Chem. Eur. J. 11, 2954–2965 (2005).
Wrighton, M. S. & Schroeder, M. A. Chromium carbonyl photocatalyzed 1, 4-hydrosilation of 1, 3-dienes. Synthesis of allylsilanes. J. Am. Chem. Soc. 96, 6235–6237 (1974).
Guptill, D. M., Cohen, C. M. & Davies, H. M. L. Rhodium(II)-catalyzed stereoselective synthesis of allylsilanes. Org. Lett. 15, 6120–6123 (2013).
Parasram, M., Iaroshenko, V. O. & Gevorgyan, V. Endo-selective pd-catalyzed silyl methyl heck reaction. J. Am. Chem. Soc. 136, 17926–17929 (2014).
Koh, M. J., Khan, R. K. M., Torker, S. & Hoveyda, A. H. Broadly applicable Z‐and diastereoselective ring‐opening/cross‐metathesis catalyzed by a dithiolate Ru complex. Angew. Chem. Int. Ed. 53, 1968–1972 (2014).
Sawaki, R., Sato, Y. & Mori, M. Ligand-controlled highly stereoselective syntheses of E- and Z-allylsilanes from dienes and aldehydes using nickel complex. Org. Lett. 6, 1131–1133 (2004).
Sudo, T., Asao, N., Gevorgyan, V. & Yamamoto, Y. Lewis acid catalyzed highly regio-and stereocontrolled trans-hydrosilylation of alkynes and allenes. J. Org. Chem. 64, 2494–2499 (1999).
Miller, Z. D., Li, W., Belderrain, T. R. & Montgomery, J. Regioselective allene hydrosilylation catalyzed by n-heterocyclic carbene complexes of nickel and palladium. J. Am. Chem. Soc. 135, 15282–15285 (2013).
Miller, Z. D. & Montgomery, J. Regioselective allene hydroarylation via one-pot allene hydrosilylation/Pd-catalyzed cross-coupling. Org. Lett. 16, 5486–5489 (2014).
Kidonakis, M. & Stratakis, M. Ligandless regioselective hydrosilylation of allenes catalyzed by gold nanoparticles. Org. Lett. 17, 4538–4541 (2015).
Miller, Z. D., Dorel, R. & Montgomery, J. Regiodivergent and stereoselective hydrosilylation of 1, 3‐disubstituted allenes. Angew. Chem. Int. Ed. 54, 9088–9091 (2015).
Tafazolian, H. & Schmidt, J. A. Highly efficient regioselective hydrosilylation of allenes using a [(3IP)Pd(allyl)]OTf catalyst; first example of allene hydrosilylation with phenyl-and diphenylsilane. Chem. Commun. 51, 5943–5946 (2015).
Asako, S., Ishikawa, S. & Takai, K. Synthesis of linear allylsilanes via molybdenum-catalyzed regioselective hydrosilylation of allenes. ACS Catal. 6, 3387–3395 (2016).
Lewis, L. N., Stein, J., Gao, Y., Colborn, R. E. & Hutchins, G. Platinum catalysts used in the silicones industry. Platin. Met. Rev. 41, 66–75 (1997).
Markó, I. E. et al. Selective and efficient platinum(0)-carbene complexes as hydrosilylation catalysts. Science 298, 204–206 (2002).
Markó, I. E. et al. Highly active and selective platinum(0)‐carbene complexes. efficient, catalytic hydrosilylation of functionalised olefins. Adv. Synth. Catal. 346, 1429–1434 (2004).
Marciniec, B., Posała, K., Kownacki, I., Kubicki, M. & Taylor, R. New Bis (dialkynyldisiloxane) triplatinum(0) cluster: synthesis, structure, and catalytic activity in olefin‐hydrosilylation reactions. ChemCatChem 4, 1935–1937 (2012).
Bernhammer, J. C. & Huynh, H. V. Platinum(II) complexes with thioether-functionalized benzimidazolin-2-ylidene ligands: Synthesis, structural characterization, and application in hydroelementation reactions. Organometallics 33, 172–180 (2013).
Sun, J. & Deng, L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 6, 290–300 (2016).
Du, X. & Huang, Z. Advances in base-metal-catalyzed alkene hydrosilylation. ACS Catal. 7, 1227–1243 (2017).
Mo, Z., Liu, Y. & Deng, L. Anchoring of silyl donors on a N‐heterocyclic carbene through the cobalt‐mediated silylation of benzylic C-H bonds. Angew. Chem. Int. Ed. 52, 10845–10849 (2013).
Mo, Z., Xiao, J., Gao, Y. & Deng, L. Regio-and stereoselective hydrosilylation of alkynes catalyzed by three-coordinate cobalt(I) alkyl and silyl complexes. J. Am. Chem. Soc. 136, 17414–17417 (2014).
Huang, K.-H. & Isobe, M. Highly regioselective hydrosilylation of unsymmetric alkynes using a phenylthio directing group. Eur. J. Org. Chem. 2014, 4733–4740 (2014).
Chen, C. et al. Rapid, regioconvergent, solvent-free alkene hydrosilylation with a cobalt catalyst. J. Am. Chem. Soc. 137, 13244–13247 (2015).
Noda, D., Tahara, A., Sunada, Y. & Nagashima, H. Non-precious-metal catalytic systems involving iron or cobalt carboxylates and alkyl isocyanides for hydrosilylation of alkenes with hydrosiloxanes. J. Am. Chem. Soc. 138, 2480–2483 (2016).
Schuster, C. H., Diao, T., Pappas, I. & Chirik, P. J. Bench-stable, substrate-activated cobalt carboxylate pre-catalysts for alkene hydrosilylation with tertiary silanes. ACS Catal. 6, 2632–2636 (2016).
Ibrahim, A. D., Entsminger, S. W., Zhu, L. & Fout, A. R. A highly chemoselective cobalt catalyst for the hydrosilylation of alkenes using tertiary silanes and hydrosiloxanes. ACS Catal. 6, 3589–3593 (2016).
Rivera-Hernández, A. et al. Regio-and stereoselective hydrosilylation of unsymmetrical alkynes catalyzed by a well-defined, low-valent cobalt catalyst. Org. Lett. 18, 4242–4245 (2016).
Guo, J. & Lu, Z. Highly chemo‐, regio‐, and stereoselective cobalt‐catalyzed markovnikov hydrosilylation of alkynes. Angew. Chem. Int. Ed. 55, 10835–10838 (2016).
Zuo, Z., Yang, J. & Huang, Z. Cobalt‐catalyzed alkyne hydrosilylation and sequential vinylsilane hydroboration with markovnikov selectivity. Angew. Chem. Int. Ed. 55, 10839–10843 (2016).
Wang, C., Teo, W. J. & Ge, S. Cobalt-catalyzed regiodivergent hydrosilylation of vinylarenes and aliphatic alkenes: ligand-and silane-dependent regioselectivities. ACS Catal. 7, 855–863 (2016).
Liu, Y. & Deng, L. Mode of activation of cobalt(ii) amides for catalytic hydrosilylation of alkenes with tertiary silanes. J. Am. Chem. Soc. 139, 1798–1801 (2017).
Teo, W. J., Wang, C., Tan, Y. W. & Ge, S. Cobalt‐catalyzed z‐selective hydrosilylation of terminal alkynes. Angew. Chem. Int. Ed. 56, 4328–4332 (2017).
Guo, J., Shen, X. & Lu, Z. Regio‐and enantioselective cobalt‐catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Angew. Chem. Int. Ed. 56, 615–618 (2017).
Docherty, J. H., Peng, J., Dominey, A. P. & Thomas, S. P. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 9, 595–600 (2017).
Huang, G. & Isobe, M. Stereoselective synthesis of sugar allenes and their hydrosilylation catalyzed by biscobaltoctacarbonyl. Tetrahedron 57, 10241–10246 (2001).
Simmons, E. M. & Hartwig, J. F. Iridium-catalyzed arene ortho-silylation by formal hydroxyl-directed C− H activation. J. Am. Chem. Soc. 132, 17092–17095 (2010).
Kuznetsov, A. & Gevorgyan, V. General and practical one-pot synthesis of dihydrobenzosiloles from styrenes. Org. Lett. 14, 914–917 (2012).
Chen, Y. K. & Walsh, P. J. A one-pot multicomponent coupling reaction for the stereocontrolled synthesis of (Z)-trisubstituted allylic alcohols. J. Am. Chem. Soc. 126, 3702–3703 (2004).
Kerrigan, M. H. et al. One-pot multicomponent coupling methods for the synthesis of diastereo-and enantioenriched (Z)-trisubstituted allylic alcohols. J. Am. Chem. Soc. 131, 8434–8445 (2009).
Ely, R. J. & Morken, J. P. Regio-and stereoselective Ni-catalyzed 1, 4-hydroboration of 1, 3-dienes: access to stereodefined (z)-allylboron reagents and derived allylic alcohols. J. Am. Chem. Soc. 132, 2534–2535 (2010).
Koh, M. J. et al. High-value alcohols and higher-oxidation-state compounds by catalytic Z-selective cross-metathesis. Nature 517, 181–186 (2015).
Suto, T. et al. Unified total synthesis of madangamines A, C, and E. J. Am. Chem. Soc. 139, 2952–2955 (2017).
Chen, C., Dugan, T. R., Brennessel, W. W., Weix, D. J. & Holland, P. L. Z-Selective Alkene isomerization by high-spin cobalt(ii) complexes. J. Am. Chem. Soc. 136, 945–955 (2014).
Yu, S., Wu, C. & Ge, S. Cobalt-catalyzed asymmetric hydroboration/cyclization of 1, 6-enynes with pinacolborane. J. Am. Chem. Soc. 139, 6526–6529 (2017).
Waterman, R. σ-Bond metathesis: a 30-year retrospective. Organometallics 32, 7249–7263 (2013).
Köpfer, A. & Breit, B. Rhodium-catalyzed hydroformylation of 1,1-disubstituted allenes employing the self-assembling 6-DPPon system. Angew. Chem. Int. Ed. 54, 6913–6917 (2015).
Acknowledgements
This work was supported by the grant from NUS Young Investigator Award (R-143-000-630-133) and the Ministry of Education of Singapore (R-143-000-A07-112).
Author information
Authors and Affiliations
Contributions
C.W. planned and conducted most of the experiments; C.W. and W.J.T. prepared substrates for the reaction scope evaluation; S.G. directed the projects and S.G. and C.W. co-wrote the manuscript. All authors contributed to the discussion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, C., Teo, W.J. & Ge, S. Access to stereodefined (Z)-allylsilanes and (Z)-allylic alcohols via cobalt-catalyzed regioselective hydrosilylation of allenes. Nat Commun 8, 2258 (2017). https://doi.org/10.1038/s41467-017-02382-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-02382-7
This article is cited by
-
Copper-catalyzed regio- and stereo-selective hydrosilylation of terminal allenes to access (E)-allylsilanes
Nature Communications (2022)
-
Trace amount of single-atom palladium-catalyzed selective hydrosilylation of allenes
Nano Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
