
Abstract
Aging imposes a barrier to somatic cell reprogramming through poorly understood mechanisms. Here, we report that fibroblasts from old mice express higher levels of Zeb2, a transcription factor that activates epithelial-to-mesenchymal transition. Synthesis of Zeb2 protein is controlled by a natural antisense transcript named Zeb2-NAT. We show that transfection of adult fibroblasts with specific LNA Gapmers induces a robust downregulation of Zeb2-NAT transcripts and Zeb2 protein and enhances the reprogramming of old fibroblasts into pluripotent cells. We further demonstrate that Zeb2-NAT expression is precociously activated by differentiation stimuli in embryonic stem (ES) cells. By knocking down Zeb2-NAT, we were able to maintain ES cells challenged with commitment signals in the ground state of pluripotency. In conclusion, our study identifies a long noncoding RNA that is overlapping and antisense to the Zeb2 locus as a target for rejuvenation strategies.
Similar content being viewed by others


Introduction
Somatic cell reprogramming resets aged cells back into an embryonic-like state, offering unprecedented opportunities for studying cellular rejuvenation. Cells from centenarian individuals have been successfully reprogrammed1, 2 demonstrating the potential for aging reversibility at the cellular level. However, reprogramming efficiency declines with aging3,4,5,6 possibly because old tissues accumulate senescent and genetically unstable cells that do not reprogram (reviewed by Mahmoudi and Brunet7). Indeed, it is well established that cell senescence, which can be triggered by multiple stresses such as DNA damage and replicative exhaustion associated with telomere shortening, causes a p53-dependent block to reprogramming4, 8,9,10. Improved reprogramming efficiency has been achieved by the knocking-down expression of senescence-associated proteins p53, p21CIP1, p16INK4A and p14ARF 3, 10, 11, and microRNA-19512. However, these manipulations raise the question of whether it is safe to generate iPS cells from old donors by blocking natural defenses against cancer.
Here, we set out to investigate novel molecular pathways that constitute age-associated barriers for somatic cell reprogramming. The reprogramming of mouse fibroblasts is initiated by a mesenchymal-to-epithelial (MET) transition that is activated inside the nucleus13. In particular, Sox2/Oct4 suppresses the transcription of the epithelial-to-mesenchymal (EMT) transition mediator Snail, c-Myc downregulates TGF-β receptors, and Klf4 induces epithelial genes such as E-cadherin13. An additional transcription factor involved in this process is Zeb2, which activates EMT by repressing the expression of E-cadherin and other genes required for epithelial intercellular junctions14, 15. We found that fibroblasts from old mice express higher levels of Zeb2 compared to fibroblasts from young animals. As reprogramming requires suppression of pro-EMT signals, we hypothesized that Zeb2 overexpression contributes to the inefficient reprogramming of old fibroblasts. In human cells, Zeb2 expression is controlled by a natural antisense long noncoding RNA (lncRNA) named Zeb2-NAT, by a mechanism that involves retention of the first intron of Zeb2 pre-mRNA16. This intron contains an IRES sequence required for translation of Zeb2 protein, and a strong correlation was observed between expression of Zeb2-NAT, Zeb2 intron 1 retention, and Zeb2 protein synthesis16. Because the human and murine Zeb2 locus shares a conserved structure17, we asked whether a similar correlation exists in mouse fibroblasts. Indeed, we observed that transfection of adult fibroblasts with specific LNA Gapmers18 induced a robust downregulation of Zeb2-NAT transcripts and Zeb2 protein, and we demonstrate that silencing Zeb2-NAT suffices to enhance reprogramming of old fibroblasts. We also found that Zeb2-NAT expression is activated by differentiation stimuli in embryonic stem (ES) cells and we show that knocking down Zeb2-NAT maintained ES cells challenged with commitment signals in the ground state of self-renewal and pluripotency. Thus, our study unravels the potential of targeting a long noncoding antisense RNA for rejuvenation strategies.
Results
Zeb2 and Zeb2-NAT levels increase with aging
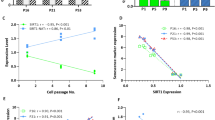
We used a reprogrammable mouse line (i4F, Fig. 1a) that carries the transcriptional activator (rtTA) within the ubiquitously expressed Rosa26 locus and a doxycycline-inducible polycistronic cassette encoding the murine transcription factors Oct4, Sox2, Klf4, and c-Myc19. Upon addition of doxycycline, embryonic fibroblasts (MEFs) from i4F mice were efficiently reprogrammed in vitro based on the appearance of sharply defined cell colonies composed of small, round, and tightly packed cells that stained positive for alkaline phosphatase (AP) activity (Supplementary Fig. 1a, b). After 2 weeks of doxycycline induction, we observed an average of 936 ± 236 AP+ clones in culture wells plated with 1 × 106 MEFs (Fig. 1b). In contrast, only 462 ± 245 AP+ clones were observed in wells plated with the same number of early-passage (P1) fibroblasts from adult (10–30-week old) mice, and 28 ± 21 AP+ clones were detected in wells plated with the same number of P1 fibroblasts from old (70–100-week old) animals (Fig. 1b). Quantification of reprogramming efficiency of fibroblasts isolated from adult and old mice relative to embryonic fibroblasts (MEFs) reveals a clear age-dependent decay (Fig. 1c). Accordingly, we observed that unlike fibroblasts from old mice, embryonic fibroblasts start to undergo epithelial-like morphological changes (i.e., cells became rounded, aggregated, and formed well-defined epithelial-like intercellular junctions) after 3–5 days in culture in the presence of doxycycline (Supplementary Fig. 1c). As expected, reprogramming fibroblasts showed downregulation of mRNA coding for Zeb2 (Fig. 1d) and Snail1 (Fig. 1e). However, these mRNAs were less reduced in doxycycline-treated adult and old fibroblasts compared to embryonic fibroblasts (Fig. 1d, e). The difference was particularly striking for Zeb2 mRNA, which undergoes a sixfold decrease during MEF reprogramming but is not significantly altered in adult and old fibroblasts induced with doxycycline (Fig. 1d). We further noticed that fibroblasts from aged animals express increased levels of Zeb2 protein compared to MEFs (Fig. 1f, g and Supplementary Fig. 1d). Because expression of the Zeb2 protein in human cells is controlled by the natural antisense RNA Zeb2-NAT16, we measured the levels of Zeb2-NAT using primers that target the 3′ end of the transcript at a region without correspondence to the sequence of Zeb2 pre-mRNA (Fig. 2a), and we found that Zeb2-NAT was significantly upregulated in adult and old fibroblasts compared to embryonic fibroblasts (Fig. 2b). Moreover, reprogramming i4F fibroblasts into iPSCs resulted in significantly reduced Zeb2-NAT levels (Fig. 2c). Next, we designed primers to distinguish spliced and unspliced Zeb2 transcripts, and observed much higher levels of transcripts containing intron 1 than transcripts containing intron 2 (Fig. 2d), suggesting that splicing of the first intron of murine Zeb2 pre-mRNA is specifically regulated. Compared to MEFs, old fibroblasts have significantly higher levels of Zeb2 transcripts containing the first intron (Fig. 2d), consistent with the previously described correlation between increased expression of Zeb2-NAT and retention of the first intron in Zeb2 transcripts16. However, the level of Zeb2 transcripts containing either spliced intron 1 or spliced intron 2 is also higher in old fibroblasts (Fig. 2d), raising the possibility of a concomitant age-dependent upregulation of Zeb2 transcription.

Inefficient reprograming of old fibroblasts. a Schematics of the reprogrammable mouse line i4F in which the OSKM reprogramming factors (Oct4, Sox2, Klf4, and c-Myc) are expressed from a single genomic locus using a doxycycline-inducible transgene. Mouse embryonic fibroblasts (MEFs) and fibroblasts isolated from adult (10–30-week old) and old (70–100-week old) mice were induced with doxycycline for 2 weeks. b Quantification of AP+ clones. c Reprogramming efficiency relative to MEFs. d, e qRT-PCR analysis of Zeb2 (d) and Snail (e) mRNA in MEFs and fibroblasts from adult and old mice that were either not exposed to doxycycline or induced for 2 weeks. For all graphics depicted, Student’s t test (two-tailed) statistics, *p < 0.05, **p < 0.01, and ***p < 0.001; error bars represent standard deviation; n=experiments with independent cell cultures. f Immunofluorescence for Zeb2 in MEFs and old fibroblasts. The corresponding images stained with DAPI are shown (scale bars correspond to 10 μm). g Immunoblot for Zeb2 and β-actin in total cell lysates from two independent early-passage cultures of MEFs and old fibroblasts

. Zeb2-NAT expression is upregulated in old fibroblasts and correlates with retention of Zeb2 intron 1. a Schematics of the murine Zeb2 and Zeb2-NAT locus. An internal ribosome entry site (IRES) is located in the first intron of the Zeb2 transcript. Double arrows indicate the position of primers used to detect Zeb2-NAT RNA. b qRT-PCR analysis of Zeb2-NAT RNA in MEFs and fibroblasts from adult and old mice. Transcript levels were normalized to GAPDH mRNA and depicted as fold change relative to MEFs. c qRT-PCR analysis of Zeb2-NAT RNA in MEFs and in a iPS cell line derived from i4F fibroblasts. d qRT-PCR analysis of spliced and unspliced Zeb2 RNA in MEFs and old fibroblasts. Double arrows indicate the position of primers. For all graphics depicted, Student’s t test (two-tailed) statistics, *p < 0.05, **p < 0.01; error bars represent standard deviation; n=experiments with independent cell cultures
Downregulation of Zeb2-NAT enhances reprogramming of aged cells
Having shown that Zeb2-NAT expression in fibroblasts increases with age and is reduced during reprogramming to iPSCs, we next asked whether higher levels of Zeb2-NAT transcripts contribute to the low reprogramming efficiency of aged fibroblasts. To determine the effect of reducing the levels of Zeb2-NAT on reprogramming old fibroblasts, we designed LNA Gapmers that specifically target Zeb2-NAT and Zeb2 transcripts (Supplementary Fig. 2). These are chimeric antisense oligonucleotides with a central block of DNA designed to hybridize with the target RNA. Upon hybridization, the endogenous and ubiquitous ribonuclease (RNase) H hydrolyzes the RNA strand of the resulting RNA–DNA heteroduplexes. Locked nucleic acid (LNA)-modified nucleotides flanking the DNA oligonucleotide improve the stability and binding affinity for the target RNA18. Transfection of adult fibroblasts with antisense oligonucleotides targeting Zeb2-NAT resulted in a robust decrease of Zeb2-NAT RNA (Fig. 3a), total Zeb2 RNA (Fig. 3b), and Zeb2 protein (Fig. 3c). Transfection with antisense oligonucleotides targeting Zeb2 reduced the levels of Zeb2 transcripts (Fig. 3b) but not Zeb2-NAT (Fig. 3a). Further analysis of spliced and unspliced Zeb2 transcripts revealed that oligonucleotides targeted to Zeb2 RNA efficiently reduced the level of Zeb2 mRNA devoid of intron 1 and 2 (Fig. 3d), but had a marginal effect on the level of Zeb2 transcripts with intron 1 retention (Fig. 3d). In contrast, oligonucleotides targeted to Zeb2-NAT caused a significant reduction of both spliced Zeb2 mRNA and Zeb2 transcripts with intron 1 retention (Fig. 3d). Similar results were observed using digital droplet PCR (Supplementary Fig. 3a), which reduces PCR bias and provides a higher degree of sensitivity and precision20, 21. It is also evident from the results obtained by digital PCR that Zeb2-NAT RNA molecules are much less abundant than Zeb2 transcripts. To determine whether knocking down Zeb2-NAT affects Zeb2 transcription, we used metabolic labeling with modified uridine (4-thiouridine, 4sU), which allows to distinguish recently transcribed RNA from steady-state RNA levels22,23,24. Analysis of RNA-4sU shows that targeting Zeb2-NAT leads to increased levels of newly transcribed total Zeb2 RNA and lower levels of newly transcribed Zeb2 carrying the first intron (Supplementary Fig. 3b). Taken together, these experiments suggest that knockdown of Zeb2-NAT enhances both the transcriptional rate of the Zeb2 gene and retention of intron 1 in Zeb2 transcripts. We next transfected fibroblasts from aged i4F mice with LNA Gapmers targeting either Zeb2 or Zeb2-NAT, and 24 h later, doxycycline was added to the culture to induce reprogramming. Upon 3 weeks of culture in a medium supplemented with doxycycline, we assessed reprogramming efficiency based on the number of AP+ colonies. As shown in Fig. 4 (Supplementary Fig. 4a, b), significantly more colonies were observed in cultures of fibroblasts from adult and old mice transfected with anti-Zeb2-NAT and anti-Zeb2 oligonucleotides compared to cultures treated with nonspecific oligonucleotides. To determine whether these AP+ colonies correspond to bona fide iPS cell lines, individual colonies with a morphology similar to those formed by ES cells were picked (Supplementary Fig. 4c), and cells were transferred to a chemically defined medium containing the cytokine leukemia inhibitory factor LIF25, 26 and the 2i small-molecule inhibitors CHIR99021 and PD032590127, without doxycycline and in the absence of feeder cells. Of all colonies derived from old animals, only those that had been transfected with anti-Zeb2-NAT oligonucleotides survived (Fig. 4c). The surviving colonies expressed the markers of pluripotency Nanog and SSEA1, as shown by immunofluorescence (Fig. 4d). Moreover, when cells from these colonies were subcutaneously injected into immune-deficient mice, they induced the formation of tumoral masses that are reminiscent of teratomas28 with disorganized differentiation into endodermal, mesodermal, and ectodermal derivatives (Fig. 4e), thus confirming their in vivo differentiation potential. We therefore conclude that downregulation of Zeb2-NAT is sufficient to liberate aged fibroblasts from endogenous constraints to reprogramming without compromise to developmental potency.

Knocking down Zeb2-NAT affects Zeb2 expression. Fibroblasts from adult mice were transfected with either control LNA Gapmers or oligonucleotides targeting Zeb2-NAT and Zeb2. a qRT-PCR analysis of Zeb2-NAT RNA. b qRT-PCR analysis of total Zeb2 transcripts (primer Zeb2 #1, see Supplementary Table 2). c Immunoblot for Zeb2 and β-actin in total cell lysates after transfection of anti-Zeb2-NAT Gapmers #1 and #2 (see Supplementary Fig. 2b). d qRT-PCR analysis of spliced and unspliced Zeb2 RNA. Double arrows indicate the position of primers. For all graphics depicted, Student’s t test (two-tailed) statistics, *p < 0.05, **p < 0.01, ***p < 0.001; error bars represent standard deviation; n=experiments with independent cell cultures

Knocking down Zeb2-NAT is sufficient to enhance reprogramming of old fibroblasts. Fibroblasts were transfected with the indicated LNA Gapmers and induced to reprogram for 3 weeks. a, b Reprogramming efficiency in adult and old fibroblasts based on quantification of AP+ colonies. Student’s t test (two-tailed) statistics, *p < 0.05; error bars represent standard deviation; n=experiments with independent cell cultures. c Proportion of colonies that survived re-plating in medium containing LIF and 2i, without doxycycline; colonies were counted at day 8 after replating. At least ten independent colonies where picked from each condition. Fischer’s exact test was used for statistical analysis. d Immunofluorescence for Nanog and SSEA1 in reprogrammed old fibroblasts transfected with anti-Zeb2-NAT oligonucleotides; the corresponding images stained with DAPI are shown (scale bars, 20 μm). e Histological section of a subcutaneous teratoma containing mesoderm (mes), endoderm (end), and ectoderm (ect) 15 weeks after injection of cells into immune-deficient mice. Magnifications as indicated
Although AP+ colonies derived from control old fibroblasts did not survive in feeder-free culture conditions in the absence of doxycycline (Fig. 4c), a few clones were able to grow when cultured in the presence of feeder cells. These clones expressed the markers of pluripotency Nanog and SSEA1 (Supplementary Fig. 4d) and induced the formation of teratomas, confirming that they correspond to iPSCs. These results show that knocking down Zeb2 expression enhances the reprogramming potential of old fibroblasts but is not essential for this process, which is in agreement with previous studies reporting the successful reprograming of cells from centenarian individuals1, 2. As an alternative to antisense Gapmer oligonucleotides, we tested the ability of short hairpin RNAs (shRNAs) to downregulate Zeb2 and Zeb2-NAT RNA by RNA interference (RNAi). Infection of fibroblasts from old mice with lentiviral particles containing a DNA sequence that codes for a shRNA targeting Zeb2 had no significant effect on Zeb2-NAT RNA (Supplementary Fig. 5a), but resulted in a robust decrease of total Zeb2 RNA (Supplementary Fig. 5b), and Zeb2 protein (Supplementary Fig. 5c). Although expression of a shRNA targeting Zeb2-NAT also decreased the levels of Zeb2 protein (Supplementary Fig. 5c), reduction of Zeb2-NAT and total Zeb2 transcripts (Supplementary Fig. 5a, b) was much less striking than the effect observed with anti-Zeb2-NAT Gapmers (Fig. 3a, b). Possibly, the low abundant Zeb2-NAT transcripts remain mostly restricted to the nucleus and therefore are not efficiently degraded by cytoplasmic RNAi, while RNase-H-mediated Gapmer knockdown efficiently suppresses RNA targets localized both in the nucleus and in the cytoplasm29. As expected from the lower levels of Zeb2 protein induced by shRNAs, RNAi-mediated knockdown of Zeb2 and Zeb2-NAT expression resulted in increased reprogramming efficiency of old fibroblasts (Supplementary Fig. 5d–f). To determine whether the knockdown of Zeb2 and Zeb2-NAT transcripts affects the expression of previously known modulators of the reprogramming process3, 8, 30, we analyzed the tumor suppressors p16, p21, and p53. Fibroblasts from old mice were transfected with control LNA Gapmers and oligonucleotides targeting Zeb2-NAT and Zeb2. The results show no significant change in either p16, p21, and p53 RNA levels (Supplementary Fig. 6a) or proliferation capacity (Supplementary Fig. 6b), indicating that Zeb2 and Zeb2-NAT are implicated in the reprogramming process through a distinct mechanism.
It was recently reported that ectopic expression of Zeb2-NAT in human cells prevents splicing of intron 1 in Zeb2 mRNA and increases Zeb2 protein synthesis16, arguing for a direct regulatory role of the antisense RNA product rather than the act of antisense transcription. It was further proposed that the antisense RNA interferes with spliceosome assembly through base-pairing with sequences in Zeb2 pre-mRNA that are required for intron 1 splicing16. To investigate whether such a mechanism would explain the phenotype observed in murine fibroblasts, we designed two Zeb2-NAT mimics composed of 2′-O-methyl (2′OMe) RNA oligonucleotides31 (Fig. 5a). The 2′OMe RNA oligonucleotides are complementary to Zeb2 pre-mRNA sequences predicted to be involved in splicing32. Upon simultaneous transfection of old fibroblasts with the LNA Gapmer anti-Zeb2-NAT and either 2′OMe RNA #1 or 2′OMe RNA #2, the levels of total Zeb2 RNA and Zeb2 transcripts with intron 1 retention were similar to those observed in untreated cells, and significantly higher than in cells transfected with LNA Gapmer anti-Zeb2-NAT (Fig. 5b, c). We further observed that after transfection with anti-Zeb2-NAT Gapmer and 2′OMe RNAs, the level of Zeb2 protein was similar to control and higher than in cells transfected with Gapmer only (Fig. 5d). Transfection of old fibroblasts with 2′OMe-RNAs in the absence of anti-Zeb2-NAT Gapmer did not cause statistically significant changes, although the levels of Zeb2 transcripts tended to be increased relative to control, untreated cells (Fig. 5b, c). Taken together, these results indicate that 2′OMe RNAs can rescue the knockdown of endogenous Zeb2-NAT transcripts. We therefore reasoned that if 2′OMe RNAs act as functional mimics of Zeb2-NAT, they should prevent the increase in reprogramming efficiency caused by Zeb2-NAT downregulation. Indeed, the reprogramming efficiency was similar in control, untreated old fibroblasts and in cells simultaneously transfected with the LNA Gapmer anti-Zeb2-NAT and either 2′OMe-RNA #1 or 2′OMe RNA #2, in contrast with the higher efficiency observed in old fibroblasts transfected with the LNA Gapmer only (Fig. 5e, f).

Expression of 2′OMe RNA oligonucleotides mimicking Zeb2-NAT. a Position of 2′OMe RNA oligonucleotides relative to Zeb2 RNA. b, c qRT-PCR analysis of Zeb2 transcripts (primers Zeb2 #1 and Zeb2 intron, see Supplementary Table 2) in fibroblasts from old mice transfected as indicated. d Immunoblot for Zeb2 and β-actin in total cell lysates of old fibroblasts transfected as indicated. e Representative reprogramming experiment stained for Alkaline Phosphatase after 3 weeks in culture with doxycycline. f Reprogramming efficiency of old fibroblasts transfected as indicated. For all graphics depicted, Student’s t test (two-tailed) statistics, *p < 0.05. At least three independent experiments were carried per condition
Zeb2-NAT expression correlates with stem cells commitment
Having shown that downregulation of Zeb2-NAT enhances reprogramming of old fibroblasts into iPSCs, we next asked whether this antisense transcript also plays a role in stem cell pluripotency. First, we analyzed the expression of Zeb2-NAT in the murine ES cell line E14Tg2a.4 (E14)33 and found that Zeb2-NAT transcripts are expressed at very low levels (Fig. 6a), similarly to that observed in iPSCs derived from i4F fibroblasts (Fig. 2c). Compared to MEFs, the level of Zeb2-NAT is 41-fold lower in ES cells (Fig. 6a). The level of total Zeb2 RNA and Zeb2 transcripts with retention of the first intron is also significantly lower in ES cells (Fig. 6a), and Zeb2 protein is not detected by immunoblot (Fig. 6b). The observation that Zeb2-NAT levels are much lower in pluripotent cells compared to fibroblasts suggests that increased expression of this antisense RNA may be required during the differentiation process. To determine whether Zeb2-NAT expression increases when stem cells shift from self-renewal to lineage commitment, E14 cells were challenged by removing LIF and 2i from the culture medium. The combined presence of these inhibitors, which act by shielding cells from inductive differentiation stimuli, is required to maintain ES cells in the ground state of self-renewal and pluripotency27. We found that at 20 h after LIF/2i removal, Zeb2-NAT was already significantly upregulated (Fig. 6c). At the same time point, the levels of Zeb2, Nanog, Oct4, and Sox2 transcripts remained unchanged. Downregulation of the pluripotency genes Nanog, Oct4, and Sox2 was detected later, after 72 h of culture in a medium without LIF/2i (Fig. 6c). At the 72-h time point, the level of total Zeb2 transcripts increased (Fig. 6c), as expected since Zeb2 expression is higher in differentiated cells than in pluripotent cells. Additionally, we took advantage of the fact that ES cells grown to high confluence tend to spontaneously differentiate. Consistent with the view that loss of pluripotency correlates with activation of Zeb2-NAT expression, cells grown to high confluence expressed higher levels of Zeb2-NAT transcripts compared to cells grown at low density (Supplementary Fig. 7a, b). In contrast, the level of total Zeb2 RNA remained similar in cells grown at low and high confluence (Supplementary Fig. 7c), providing further evidence that activation of Zeb2-NAT expression precedes upregulation of Zeb2 expression. These results argue that Zeb2-NAT transcription in ES cells is tightly controlled by differentiation stimuli and that activation of Zeb2-NAT expression represents an early marker for loss of pluripotency.

Zeb2-NAT expression in ES cells increases in response to differentiation stimuli. a qRT-PCR analysis of Zeb2-NAT and Zeb2 transcripts in MEFs and in the embryonic stem cell line E14. b Immunoblot for Zeb2 and tubulin in total cell lysates from MSFs (mouse skin fibroblasts), E14 cells and iPSCs. c qRT-PCR analysis of the indicated transcripts in E14 cells cultured in medium with LIF/2i or incubated for 20 and 72 h after removal of LIF and 2i. d qRT-PCR analysis of the indicated transcripts in old fibroblasts and TNG-A cells grown in the presence or absence of 2i. For all graphics depicted, Student’s t test (two-tailed) statistics was used, *p < 0.05, **p < 0.01, ***p < 0.001; error bars represent standard deviation; n=experiments with independent cell cultures
Blocking Zeb2-NAT safeguards ES cells pluripotency
We additionally investigated how activation of Zeb2-NAT expression relates to loss of pluripotency in ES cells challenged by removal of 2i from the culture medium. We analyzed E14 cells and the transgenic ES strain TNG-A that carries a green fluorescence protein (GFP) reporter inserted into one Nanog allele (Nanog +/GFP)34. It was previously reported that TNG-A cells are more prone to exit the ground state of pluripotency35. Accordingly, and in contrast to E14 cells, TNG-A cells maintained in a medium without 2i rapidly changed their morphology (Supplementary Fig. 8a, b). We observed that 20 h after removing 2i from the culture medium of TNG-A cells, Zeb2-NAT expression was already significantly upregulated (Fig. 6d). The level of Zeb2 transcripts retaining intron 1 was also increased at 20 h in culture without 2i (Fig. 6d), consistent with the proposed role of Zeb2-NAT in preventing splicing of the first Zeb2 intron. The total level of Zeb2 RNA was similar to control at 20 h, but increased at 48 h (Fig. 6d), in further agreement with the view that upregulation of Zeb2-NAT expression precedes activation of Zeb2 transcription. Transfection with LNA Gapmers targeting Zeb2-NAT blocked upregulation of Zeb2 expression, in agreement with the proposed model that ZEB2-NAT expression activates Zeb2 transcription. Both E14 and TNG-A cells grown in a medium supplemented with 2i presented a robust expression of Nanog RNA (Fig. 7a) and protein (Fig. 7b and Supplementary Fig. 8c). At 48 h after removal of 2i from the culture medium, the levels of Nanog decreased, particularly in TNG-A cells (Fig. 7a, b and Supplementary Fig. 8c). The levels of E-cadherin were also significantly decreased in TNG-A cells cultured without 2i (Fig. 7c). Transfection with anti-Zeb2-NAT or anti-Zeb2 oligonucleotides was sufficient to counteract this effect (Fig. 7a–c). Namely, in TNG-A cells maintained for 48 h without 2i but transfected with anti-Zeb2-NAT or anti-Zeb2 oligonucleotides, the levels of E-cadherin mRNA were similar to those detected in cells cultured in a medium with 2i (Fig. 7c). Compared to E14 cells (Supplementary Fig. 8d), TNG-A cells grown in a medium without 2i additionally showed reduced capacity to form teratomas upon injection into immune-deficient mice (Fig. 7d–f and Supplementary Fig. 9). Although tumors were detected in animals injected with cells pretreated with both anti-Zeb2-NAT and anti-Zeb2 oligonucleotides, larger and more differentiated teratomas were observed when Zeb2-NAT was targeted (Fig. 7d–f and Supplementary Fig. 9). This could be because LNA Gapmers were more efficient in knocking down Zeb2-NAT transcripts than Zeb2 mRNA. Altogether, these results suggest that blocking Zeb2-NAT upregulation in stem cells was challenged with differentiation stimuli safeguards pluripotency. E14 cells are probably better shielded against differentiation stimuli than TNG-A cells, and thus maintain the ground state of self-renewal and pluripotency when cultured for 48 h in the absence of 2i. However, a decrease in expression of the pluripotency marker Oct4 was observed in E14 cells maintained in a medium without 2i at a later time point (96 h) (Supplementary Fig. 10). When E14 cells were first transfected with anti-Zeb2-NAT Gapmer (Supplementary Fig. 10a, b) and then cultured for 96 h in a medium without 2i, the level of Oct4 was significantly higher (Supplementary Fig. 10c). We also observed that transfection of E14 cells with anti-Zeb2-NAT and anti-Zeb2 Gapmers before culture for 96 h in the absence of 2i yielded more AP+ cells (Supplementary Fig. 10d). This suggests that, as observed in TNG-A cells, blocking Zeb2-NAT expression in E14 cells challenged by removal of 2i from the culture medium maintains self-renewal and pluripotency.

Blocking expression of Zeb2-NAT in ES cells enhances pluripotency and self-renewal. a qRT-PCR analysis of Nanog RNA in TNG-A cells transfected with the indicated oligonucleotides and grown in the presence (+) or absence (−) of 2i for 48 h. Transcript levels were normalized to GAPDH mRNA and depicted as fold change. Student’s t test (two-tailed) statistics, *p < 0.05, ***p < 0.001; error bars represent standard deviation. At least three independent experiments were carried per condition. b Immunoblot for Nanog and β-actin in total cell lysates from TNG-A cells transfected as indicated and grown in the presence (+) or absence (-) of 2i for 48 h. Relative quantification was carried out using Scion Image Software. c qRT-PCR analysis of E-cadherin RNA in TNG-A cells transfected as indicated and grown in the presence (+) or absence (−) of 2i for 48 h. Transcript levels were normalized to GAPDH mRNA and depicted as fold change. Student’s t test (two-tailed) statistics, *p < 0.05; error bars represent standard deviation. At least three independent experiments were carried per condition. d Histological section of a subcutaneous teratoma containing mesoderm (mes), endoderm (end), and ectoderm (ect), at 7 weeks after injection of cells treated with anti-Zeb2-NAT oligonucleotides. e Quantification of tumor volume (mm3). Student’s t test (two-tailed) statistics, *p < 0.05; error bars represent standard deviation; n=teratomas from independent cell cultures. f Representative images of tumors observed at 7 weeks after cell transplantation. Magnifications as indicated
Discussion
Here, we describe antisense transcription as a novel regulatory layer with implications in reprogramming efficiency of aged cells and conservation of pluripotent features. Sense and antisense transcripts of the same locus have been shown to be common occurrences36. Yet, few antisense lncRNAs have been shown to be functional37, 38. Our results show that a lncRNA that is overlapping and antisense to the Zeb2 locus regulates expression of the Zeb2 protein in mouse fibroblasts and ES cells. Reducing the levels of Zeb2-NAT RNA facilitates OSKM-induced iPSC generation of aged cells by liberating fibroblasts from endogenous constraints without compromising their developmental potency. Additionally, we found that Zeb2-NAT expression is rapidly upregulated when ES cells receive commitment signals, and blocking the expression of this antisense transcript is sufficient to maintain challenged ES cells in a state of self-renewal and pluripotency. In summary, our results underscore the physiological role of a long noncoding antisense RNA in fine-tuning the expression of a protein-coding gene implicated in pluripotency, differentiation, and reprogramming.
Methods
Fibroblast reprogramming
Fibroblasts were derived from the reprogrammable transgenic mouse line i4F19. MEFs were prepared from total mouse embryos as previously described39. Adult (10–30-weeks old) and old (70–100-weeks old) fibroblasts were obtained from the mouse ears as described40. For reprogramming, passage 1 fibroblasts were plated at a density of 5 × 105 cells per well in six-well gelatin-coated plates, or as stated in the figure legends, and cultured in iPS cell medium with doxycycline (1 µg ml−1). The medium was changed every 48 h until iPS-like cell colonies appeared (after ~7 days of treatment). Reprogramming plates were stained for AP activity (AP detection kit, Chemicon International).
Culture conditions
Cells were maintained in an incubator at 37 °C and 5% CO2. Primary fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; Promocell), 2 mM GlutaMAX (Gibco), and 100 μg/ml penicillin–streptomycin (Sigma). E14, TNG-A, and iPS cells were maintained in feeder-free conditions using gelatin-coated plates (0.1% gelatin) and iPS cell medium that contains high-glucose DMEM supplemented with KSR (15%, Invitrogen), LIF (1000 U/ ml), nonessential amino acids, penicillin–streptomycin, glutamax, and β-mercaptoethanol. The 2i chemical inhibitors CHIR99021 and PD0325901 were purchased from Axon Medchem, dissolved in DMSO, and used at 3 μM and 250 μM, respectively.
LNA Gapmers
RNase H-activating Gapmers were designed and purchased from Exiqon. Antisense oligonucleotides with a phosphorothioate backbone and sequences complementary to Mus musculus Zeb2-NAT (Genbank accession no. NR_110572.1) and Zeb2 (Genbank accession no. NM_001289521.1) were flanked by LNA-modified bases. As negative control, we used the antisense LNA Gapmer provided by Exiqon (172392—seq: AACACGTCTATACGC), which is designed to be similar to specific Gapmers but has no homology to any known microRNA, lncRNA, or mRNA sequence in the mouse. The sequences are detailed in Supplementary Table 1. LNA Gapmers were transfected twice with a 24-h interval using Lipofectamine RNAiMAX (Invitrogen) and used at a final concentration of 10 mM per transfection. Unless otherwise stated in figure legends, cells were analyzed one day after the last transfection.
shRNAs
From Sigma Mission, we purchased sequence-verified viral vector against mZeb2 (TRCN0000070887 and TRCN0000070883) and was custom made against mZeb2-NAT (the sequences are detailed in Supplementary Table 1) in the pLK0.1-puro-CMV-tGFP vector. Lentiviral particles against the same transcript were mixed equimolarly. For cell transduction, we used lentiviral particles at a multiplicity of infection of 5, overnight, as previously described41. Briefly, cells were plated at a final confluency of 50–60% in a six-well plate the day before infection. The virus was mixed in culture media with 8 μg/ml of polybrene and added directly to the cells. After incubation for 24 h, the media was changed and cells were harvested 48–72 h post infection.
2′OMe-RNA oligonucleotides
2′-OMe RNA oligonucleotides mimicking different regions of Zeb2-NAT were synthesized by Sigma. The sequences of the oligonucleotides are shown in Supplementary Table 1. 2′-OMe RNA oligonucleotides were transfected twice with a 24-h interval using Lipofectamine RNAiMAX (Invitrogen), at a final concentration of 10 mM per transfection.
4sU pulse
4sU was added to the cell culture medium for 30 min to a final concentration of 500 μM. Afterward, the medium was removed and the cells were immediately lysed with Trizol. Newly transcribed tagged RNA fractions were isolated as previously described23, 42. Briefly, RNA was labeled with Biotin-HPDP (2 µl of Biotin-HPDP (Pierce) (1 mg/ml dimethylformamide) per 1 µg of RNA) for 1.5 h at room temperature and further precipitated with chloroform/isoamyl alcohol (24:1). To separate labeled and unlabeled RNA, we used streptavidin-coated magnetic beads. Biotinylated RNA samples were heated to 65 °C for 10 min and immediately placed on ice for 5 min. Biotinylated RNA was mixed with streptavidin beads (1:1 v/v) and incubated at RT for 10 min with rotation. RNA was washed (wash buffer 1×: 100 mM Tris, pH 7.5; 10 mM EDTA; 1 M NaCl; and 0.1% Tween20) into µMacs columns in a magnetic strand and eluted (100 µl of elution buffer (100 mM DTT)) directly into RLT buffer. RNA was further recovered with a RNAeasy MinElute Spin Column before analysis.
RNA analysis
Total RNA was extracted from cells with Trizol (Life Technologies), DNase I treated, and retrotranscribed into cDNA using random hexamers as primers following the manufacturer’s protocol (Roche First-Strand cDNA Synthesis Kit). Quantitative real-time PCR was performed using Syber Green Power PCR Master Mix (Applied Biosystems) in an Applied Biosystems 7500 fast or ViiA 7 equipment. For input normalization, we used the housekeeping genes Actb (β-actin) and Gapdh. The primers used are listed in Supplementary Table 2. The relative expression of each RNA of interest was determined by calculating 2−ΔΔct values, which express the difference between the cycle threshold for the primer pair targeting the RNA of interest and the primer pair targeting a housekeeping gene. Quantitative PCR data were obtained from independent biological replicates (n values indicated in the corresponding figures) and were tested for normal distribution using the Shapiro–Wilk test and for equal variance using the F test. Normal distribution and equal variance were confirmed in the large majority of data and, therefore, we assumed normality and equal variance for all samples. Based on this, we used the Student’s t test (two-tailed, unpaired) to estimate statistical significance.
Droplet digital PCR
Droplet digital PCR (ddPCR) was carried out according to manufacturer recommendations and as described43, 44. Briefly, the ddPCR mixture contained 7.5 μl of 2× ddPCR Evagreen Supermix (Bio-Rad, Hercules, USA), 10 nM of both the Zeb2-NAT, Zeb2 intron 1, or GAPDH forward and reverse primers, and 5 ng of cDNA (RNA equivalent) in each 15-μl reaction. The 15-μl reaction was assembled into a droplet cartridge (Bio-Rad, Hercules, USA), according to the Bio-Rad protocol and the cartridge was placed in the droplet generator (Bio-Rad #186-3002) giving rise to around 20,000 individual droplets in an emulsion43. Droplets were transferred to a 96-well plate (Eppendorf, Hamburg, Germany) and thermal sealed with a foil lid (Eppendorf, Hamburg, Germany). Sealed plates were cycled using a Veriti DX thermal cycler (ThermoFisher Scientific) under the following conditions: 2 min at 30 °C; 10-min hold at 95 °C; 48 cycles of 95 °C for 50 s and then 59 °C for 120 s; 5 min at 4 °C; and 5 min at 90 °C. After amplification, the plate was transferred to a Bio-Rad droplet reader (QX200) from which data were extracted and analyzed with the Quantasoft software following manufacturer recommendations.
Protein analysis
Whole-cell extracts were prepared in lysis buffer (150 mM NaCl, 50 mM Tris, pH 8, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, and protein inhibitors cocktail). A total protein amount of 30 µg was loaded per lane in a PAGE 10% Bis–Tris gel of 1.0 mm and electrophoresed in MES SDS Running Buffer (Invitrogen). The antibodies used for immunoblotting are listed in Supplementary Table 3 and the full blots are depicted in Supplementary Fig. 11.
Immunofluorescence
Cells previously seeded on coverslips were fixed with 2% paraformaldehyde for 15 min at room temperature, washed with PBS, and permeabilized (0.02% Triton X-100 in PBS) for 20 min. Samples were then blocked for 1.5 h in PBS containing 5% BSA, incubated for 2 h with primary antibodies diluted in PBS containing 4% BSA, washed with PBS containing 0.01% Tween20, and incubated for 2 h with secondary antibodies conjugated with Cy3 or Alexa488 (diluted in PBS with 4% BSA). The primary antibodies used for immunofluorescence are listed in Supplementary Table 3. Cells were imaged using Zeiss confocal microscope (Zeiss LSM 880).
Animal procedures
Animal experimentation at Instituto de Medicina Molecular was conducted strictly within the rules of the Portuguese official veterinary directorate, which complies with the European Guideline 86/609/EC concerning laboratory animal welfare, according to a protocol approved by the institute’s Animal Ethics Committee. To assess the capacity for teratoma formation, cells were trypsinized and 2 × 106 cells were subcutaneously injected into the flanks of 8-week-old immunocompromised mice (NOD-SCID mice from Charles River). Animals were killed with anesthetic overdose and necropsy was performed. Subcutaneous tumor and ipsilateral inguinal lymph node were harvested, fixed in 10% neutral-buffered formalin, embedded in paraffin, and 3-μm sections were stained with hematoxylin and eosin. Tissue sections were examined by a pathologist blinded to experimental groups in a Leica DM2500 microscope coupled to a Leica MC170 HD microscope camera.
Statistical analysis
Comparisons of data between two groups were analyzed with the Student’s t test (two-tailed, unpaired) or as detailed in the corresponding figure legend.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Lapasset, L. et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253 (2011).
Yagi, T. et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS ONE 7, e41572 (2012).
Li, H. et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136–1139 (2009).
Banito, A. et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 23, 2134–2139 (2009).
Wang, B. et al. Reprogramming efficiency and quality of induced pluripotentstem cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS Curr. 3, RRN1274 (2011).
Soria-Valles, C. et al. NF-kappaB activation impairs somatic cell reprogramming in ageing. Nat. Cell Biol. 17, 1004–1013 (2015).
Mahmoudi, S. & Brunet, A. Aging and reprogramming: a two-way street. Curr. Opin. Cell Biol. 24, 744–756 (2012).
Marion, R. M. et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460, 1149–1153 (2009).
Marion, R. M. et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141–154 (2009).
Kawamura, T. et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460, 1140–1144 (2009).
Hong, H. et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460, 1132–1135 (2009).
Kondo, H. et al. Blockade of senescence-associated microRNA-195 in aged skeletal muscle cells facilitates reprogramming to produce induced pluripotent stem cells. Aging Cell 15, 56–66 (2016).
Li, R. et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 (2010).
Cano, A. et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 (2000).
Vandewalle, C. et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 33, 6566–6578 (2005).
Beltran, M. et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 22, 756–769 (2008).
Nelles, L., Van de Putte, T., van Grunsven, L., Huylebroeck, D. & Verschueren, K. Organization of the mouse Zfhx1b gene encoding the two-handed zinc finger repressor Smad-interacting protein-1. Genomics 82, 460–469 (2003).
Bennett, C. F. & Swayze, E. E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259–293 (2010).
Abad, M. et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 502, 340–345 (2013).
Sanders, R. et al. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 83, 6474–6484 (2011).
Hindson, B. J. et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610 (2011).
Amorim, M. J., Cotobal, C., Duncan, C. & Mata, J. Global coordination of transcriptional control and mRNA decay during cellular differentiation. Mol. Syst. Biol. 6, 380 (2010).
Dolken, L. et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972 (2008).
Rabani, M. et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436–442 (2011).
Smith, A. G. et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336, 688–690 (1988).
Williams, R. L. et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336, 684–687 (1988).
Ying, Q. L. et al. The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008).
Hentze, H. et al. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2, 198–210 (2009).
Lennox, K. A. & Behlke, M. A. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 44, 863–877 (2016).
Hanna, J. et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462, 595–601 (2009).
Goyenvalle, A. & Davies, K. E. Challenges to oligonucleotides-based therapeutics for Duchenne muscular dystrophy. Skelet. Muscle 1, 8 (2011).
Desmet, F. O. et al. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Stryke, D. et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 31, 278–281 (2003).
Chambers, I. et al. Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–1234 (2007).
Munoz Descalzo, S., Rue, P., Garcia-Ojalvo, J. & Martinez Arias, A. Correlations between the levels of Oct4 and Nanog as a signature for naive pluripotency in mouse embryonic stem cells. Stem Cells 30, 2683–2691 (2012).
Luo, S. et al. Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 18, 637–652 (2016).
Pelechano, V. & Steinmetz, L. M. Gene regulation by antisense transcription. Nat. Rev. Genet. 14, 880–893 (2013).
Wang, Y. et al. Identification, stability and expression of Sirt1 antisense long non-coding RNA. Gene 539, 117–124 (2014).
Palmero, I. & Serrano, M. Induction of senescence by oncogenic Ras. Methods Enzymol. 333, 247–256 (2001).
Li, H., Vogel, H., Holcomb, V. B., Gu, Y. & Hasty, P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 27, 8205–8214 (2007).
Feng, Y. et al. Methods for the study of long noncoding RNA in cancer cell signaling. Methods Mol. Biol. 1165, 115–143 (2014).
Friedel, C. C. & Dolken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271–1278 (2009).
Strain, M. C. et al. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS ONE 8, e55943 (2013).
Jones, M. et al. Low copy target detection by droplet digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J. Virol. Methods 202, 46–53 (2014).
Acknowledgements
We thank members of the Carmo-Fonseca laboratory, in particular Sandra Martins and Teresa Carvalho for their insightful discussions and advice. We are also grateful to José Rino for help with the confocal microscope, and to Joana Marques and colleagues for animal husbandry. We further acknowledge Manuel Serrano, Sandrina Nóbrega, and Dafni Chondronasiou (CNIO, Spain) for their valuable comments and for providing the reprogrammable i4F mouse, i4F tissues, E14, and JCW9545 cell lines and Austin Smith (University of Edinburgh, UK) for sharing the TNG-A cell line. This work was supported by an AXA research fund, Fundação para a Ciência e Tecnologia (FCT), and FEDER (PTDC/BIM-MED/0032/2014; PTDC/BEX-BCM/5899/2014; and LISBOA-01-0145-FEDER-007391, a project cofunded by FEDER, through POR Lisboa 2020—Programa Operacional Regional de Lisboa, PORTUGAL 2020, and Fundação para a Ciência e a Tecnologia). B.B.d.J. is an FCT investigator (IF/00166/2014). C.A.-V. was a Gulbenkian Foundation Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
B.B.d.J. and M.C.-F. conceived the study and wrote the paper. B.B.d.J. designed the experimental strategy and performed most of the experiments. S.P.M., A.S.-F., and C.A.-V. contributed to molecular biological analysis. S.B. contributed to cell culture analysis. T.C. performed the histopathological analysis of the subcutaneous tumors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commonslicense, unless indicated otherwise in a credit line to the material. If material is not included in the article’sCreative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bernardes de Jesus, B., Marinho, S.P., Barros, S. et al. Silencing of the lncRNA Zeb2-NAT facilitates reprogramming of aged fibroblasts and safeguards stem cell pluripotency. Nat Commun 9, 94 (2018). https://doi.org/10.1038/s41467-017-01921-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-01921-6
This article is cited by
-
Imprinting fidelity in mouse iPSCs depends on sex of donor cell and medium formulation
Nature Communications (2022)
-
The Mechanism of Stem Cell Aging
Stem Cell Reviews and Reports (2022)
-
Divergent regulation of lncRNA expression by ischemia in adult and aging mice
GeroScience (2022)
-
LncRNA MEG8 Attenuates Cerebral Ischemia After Ischemic Stroke Through Targeting miR-130a-5p/VEGFA Signaling
Cellular and Molecular Neurobiology (2021)
-
The ageing epigenome and its rejuvenation
Nature Reviews Molecular Cell Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
