
Abstract
The two p190RhoGAP proteins, p190RhoGAP-A and -B, are key regulators of Rho GTPase signaling and are essential for actin cytoskeletal structure and contractility. Here we report the discovery of two evolutionarily conserved GTPase-like domains located in the ‘middle domain’, previously thought to be unstructured. Deletion of these domains reduces RhoGAP activity. Crystal structures, MANT-GTPγS binding, thermal denaturation, biochemical assays and sequence homology analysis all strongly support defects in nucleotide-binding activity. Analysis of p190RhoGAP proteins therefore indicates the presence of two previously unidentified domains which represent an emerging group of pseudoenzymes, the pseudoGTPases.
Similar content being viewed by others


Introduction
In recent years, ‘pseudoenzymes’ have shown key roles for enzymatic folds in regulation and control of signal transduction pathways using noncatalytic methods. For example, the kinase family has an extensive ‘pseudo-’ group with members that adopt the kinase fold but are degraded in one or more of the consensus motifs required for adenosine triphosphate (ATP) binding and catalysis1. Instead, the pseudokinases are implicated in scaffolding/adaptor roles in signal transduction. The Ras superfamily of small GTPases consists of over 150 proteins that act as molecular switches in broad and diverse cellular pathways and processes2, but relatively few pseudoGTPases have been discovered. GTPases bind to guanosine triphosphate (GTP), hydrolyze γ-phosphate, release guanosine diphosphate (GDP) and then re-bind GTP, a process termed ‘GTPase cycling’. This cycling, and consequent signal transduction, is regulated by GTPase activating proteins (GAP) (for GTP hydrolysis) and guanine nucleotide exchange factors (GEF) (for GDP release). The five Ras superfamily subgroups (Ras, Rho, Rab, Ran and Arf) each contain five highly conserved sequence motifs, termed ‘G-motifs’, required for nucleotide-binding and catalytic activity2. Like pseudokinases, pseudoGTPases by definition would consist of a GTPase fold lacking one or more of these G motifs.
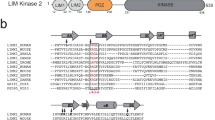
Small GTPases in the Rho subgroup (including RhoA, Cdc42 and Rac1) mediate signaling from the cell membrane to the actin cytoskeleton and play key roles in cellular functions such as adhesion, migration and cytokinesis, and in disease-associated processes such as cell growth and metastasis in cancer3. The p190RhoGAP proteins, p190RhoGAP-A (ARHGAP35) and p190RhoGAP-B (ARHGAP5)4,5,6, are key regulators of Rho GTP hydrolysis and are highly important for maintenance of proper Rho signaling. They share over 50% sequence identity and a domain organization containing a GTP-binding GTPase domain, four FF domains and a C-terminal GAP domain. An ~700 amino-acid stretch between the FF and GAP domains is termed the ‘middle domain’7 (Fig. 1a), and is thought to be unstructured.

p190RhoGAP proteins contain pseudoGTPase domains. a Current domain assignment for p190RhoGAP proteins (top). Secondary structure predictions of p190RhoGAP-A from humans (below) shown as probability of secondary structure (y axis) against residue number (x axis). Predicted helices in red and strands in blue. b Updated domain assignment for p190RhoGAP proteins
In the current study, we set out to gain a more comprehensive description of the domain architecture of the p190RhoGAP proteins in order to better understand their functions. We identify two GTPase-like folds in the middle domain of both p190RhoGAP-A and -B which we term pG1 and pG2, and show that they are important for GAP activity toward RhoA. Crystal structures of pG1 reveal similarities to the Ras-like GTPase superfamily. However, conserved GTPase motifs are notably absent from both domains, and binding assays support a lack of nucleotide-binding activity. Thus, we classify p190RhoGAP pG1 and pG2 as psuedoGTPases.
Results
Two GTPase-like folds are predicted in p190RhoGAP
Although our initial analyses indicate that the middle domainsof p190RhoGAP proteins indeed contain unstructured regions, secondary structure prediction algorithms unexpectedly indicate extensive order for the amino-terminal half of the middle domain, corresponding to residues 550–960 of p190RhoGAP-A (human) (Fig. 1a). Homology detection within this region strongly predicts two distinct domains encompassing residues 592–767 and 779–950 of human p190RhoGAP-A. Despite sequence identities below 20%, both domains have very high predicted homology (probability scores over 99% and E-values of 6 × 10−19) and matched secondary structure with members of the Ras superfamily of small GTPases (Supplementary Fig. 1 and Supplementary Table 1). Consistently, these tandem GTPase-like domains are predicted by homology detection in human p190RhoGAP-B, in p190RhoGAP-A and -B from diverse vertebrate species, and in the single p190RhoGAP protein from lower species including Drosophila melanogaster and the sponge Amphimedon queenslandica (Supplementary Table 1). These domains, however, show low sequence identity with the known N-terminal GTPase in p190RhoGAP (12 and 21% in p190RhoGAP-A). Overall, these putative GTPase-like domains appear to be conserved across evolution in p190RhoGAP proteins. We term these domains ‘pG1’ and ‘pG2’ (Fig. 1b).
Crystal structure of pG1 domains from p190RhoGAP-A and -B
We obtained crystals of the p190RhoGAP-A and p190RhoGAP-B pG1 domains that diffract to 1.9 and 2.6 Å resolution, respectively (Table 1 and Supplementary Fig. 2a). The resulting crystal structures clearly reveal a small GTPase fold in both cases, with a central 6-stranded β-sheet surrounded by four α-helices8 (Fig. 2a andSupplementary Fig. 2b, c). The root mean square deviation (r.m.s.d.) between the two pG1 structures is 1.8 Å over 128 Cαs (Fig. 2b), and 2.7 Å (135 Cαs) between the prototypical small GTPase H-Ras and p190RhoGAP-A pG1 (Fig. 2c and Supplementary Fig. 2b, c). Importantly, neither pG1 domain crystal structure contains any electron density that could be interpreted as bound nucleotide.

Structural analysis of p190RhoGAP pseudoGTPase domain pG1. a Crystal structures of pG1 for p190RhoGAP-A (left, cyan) and p190RhoGAP-B (right, blue). Secondary structure and N and C termini are labeled. Unmodeled loops are indicated by a dashed line. b Superposition of p190RhoGAP-A and -B pG1 domains. c Superposition of p190RhoGAP-A pG1 domain with GTP-analog bound H-Ras in gray; PDB ID: 5P2156
Nucleotide-binding and catalytic activity of small GTPases requires correct positioning of the G motifs termed G1 (phosphate-binding P-loop), G2 (Switch I), G3 (Switch II), G4 and G52. All five G motifs are degraded in p190RhoGAP pG1 (Fig. 3a). In canonical GTPases, the P-loop (G1) forms a cavity to bind phosphates of GDP/GTP. In p190RhoGAP, helix α1 (which follows the P-loop) is extended and sterically occludes the phosphate-binding cavity (Fig. 3a, b). Switch I (G2) is normally associated with regulated effector binding to small GTPases, undergoes conformational changes during nucleotide cycling and contains a conserved threonine that contacts the GTP γ-phosphate and Mg2+ (Fig. 3a). Switch I (G2) is completely absent in p190RhoGAP pG1 due to an 11-residue deletion (Fig. 3a and Supplementary Fig. 2b, c). Switch II (G2) is also degraded in p190RhoGAP pG1, and places a glutamate side chain (Glu636) where the γ-phosphate of GTP binds to canonical small GTPases (Fig. 3b). The G4 and G5 motifs are disordered in our structures, but their sequence divergence (Fig. 3a) suggests degraded ability to bind guanine base, and in our structures the guanosine-binding site appears to be sterically hindered by the side chains of Glu606 and Arg739, which form a predicted salt bridge with one another (Fig. 3b). Together, the substantially degraded G motifs in the p190RhoGAP pG1 domain indicate an inability to bind nucleotide.

Analysis of the degraded nucleotide binding pocket of p190RhoGAP pG1. a Alignment (top) and structural comparison (bottom) of conserved G motifs for H-Ras (PDB ID: 5P2156) and p190RhoGAP-A pG1 (p190A). Conserved threonine T35 for H-Ras is shown. Dashed box indicates region shown in b. b Close-up of the disrupted nucleotide-binding site of p190RhoGAP-A pG1 domain (left) and additional space filling model indicating the location of GTP analog when H-Ras is superposed onto the structure (right)
pG1 is not a nucleotide-binding domain
To test nucleotide-binding directly, we performed binding assays using the fluorescent nonhyhdrolyzable GTP analog MANT-GTPγS (2′/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-(γ-thio)-triphosphate). We tested binding in the presence of magnesium, and in both the presence and absence of EDTA, since our positive control protein Rac1 requires EDTA for strong MANT-GTPγS binding9. Consistent with our hypothesis that pG1 does not bind nucleotide, we find no change in fluorescence of MANT-GTPγS upon addition of either p190RhoGAP-A or -B pG1 in these conditions9 (Fig. 4a). We find that this lack of nucleotide binding for pG1 domains is evolutionarily conserved across diverse species, including pG1 from the single p190RhoGAP of D. melanogaster (Fig. 4b). We further tested MANT-GTPγS binding in the presence of a panel of atypical divalent cations including Mn2+, Ca2+ and Zn2+ and find that pG1 does not bind MANT-GTPγS in the presence of these atypical divalent cations (Fig. 4c).

Assessment of nucleotide binding by p190RhoGAP pG1. a–c Fluorescence of MANT-GTPγS monitored over time upon addition of proteins as indicated. a pG1 from 190RhoGAP-A or -B, or Rac1 as a control, in the presence of 20 mM EDTA plus 10 mM MgCl2. b p190RhoGAP-A pG1 of Xenopus laevis (frog), Gallus gallus (chicken) and Danio rerio (zebrafish) or the single p190RhoGAP of Drosophila melanogaster. c p190RhoGAP-A pG1 in the presence of different divalent cations. Rac1 in Mg2+ is included as a positive control. Uncorrected for photobleaching. d–i Thermal shift assay. d–f Thermal denaturation curves (from a representative experiment) of pG1 domains or Rac1 in the presence of divalent cation, nucleotide or both as listed in f. Buffer alone curves (black) are labeled with arrows. g–i Histograms of melting temperature changes (ΔT m) compared to buffer alone, determined by fitting the thermal denaturation curves (d–f) to a sigmoidal model. Error bars indicate s.e.m. (n=3). Positive shifts indicate a stabilization in the presence of ligand
We next employed a thermal shift assay10 to investigate potential nucleotide and divalent cation binding by pG1. We find that the melting temperatures (T m) of pG1 from p190RhoGAP-A and -B are unchanged in the presence of divalent cation, nucleotide or both. This contrasts with Rac1 which exhibits a positive shift in T m in the presence of ligands, reflective of direct binding (Fig. 4d–i). Consistent with our biochemical studies, co-crystallography of p190RhoGAP-A pG1 with GTPγS and Mg2+ yields no conformational differences and no electron density corresponding to bound nucleotide. Taken together, these studies establish that the isolated pG1 domain of p190RhoGAP is not a nucleotide-binding domain.
p190RhoGAP pG2 is a predicted pseudoGTPase
Due to a current lack of structural information for the p190RhoGAP pG2 domain, we employed sequence alignment and homology prediction. Similar to pG1, the five G motifs of pG2 are highly degraded (Supplementary Fig. 3 and Supplementary Table 1). From these alignments it is evident that the predicted G motifs in pG2 are degraded from the consensus sequences of active small GTPases like Rab8, Ras3 and Rad.
Rhotekin-binding assay to assess active RhoA
To assess a potential role for pG1–pG2 in p190RhoGAP activity toward RhoA, we performed Rhotekin pulldown assays comparing levels of active RhoA at varying expression levels of p190RhoGAP-A wild-type or a deletion mutant lacking pG1–pG2 (ΔpG1–pG2) (Fig. 5). We observe that p190RhoGAP-A ΔpG1–pG2 leads to higher levels of active RhoA compared to wild-type p190RhoGAP-A (Fig. 5a, b). When data are grouped into bins of similar expression levels of p190RhoGAP (1–10%, 10–25%, 25–50% and 50–100% of maximum expression in each experiment), we find that a significantly higher level of active RhoA is found in cells expressing mutant p190RhoGAP-A compared to wild-type p190RhoGAP-A (Fig. 5b, c). Thus, removal of pG1–pG2 from p190RhoGAP-A appears to decrease its activity toward RhoA in this system. This raises the exciting possibility that pG1–pG2 is important for full p190RhoGAP activity. This observation may be due to biological effects such as differences in p190RhoGAP localization, as it has recently been suggested that regions near pG1–pG2 drive p190RhoGAP localization to membrane protrusions11. Additionally, reported binding partners of p190RhoGAP such as Rnd3 may bind the pG1–pG2 region and affect p190RhoGAP activity12. Interestingly, a conserved surface on p190RhoGAP-A pG1 is observed (Supplementary Fig. 4) potentially suggesting a protein interaction surface.

Rho activation assays. a Active endogenous RhoA levels assessed by pulldown with a GST-rhotekin Rho binding domain (GST-RBD) from HEK293T cells transfected with a gradient of Flag-p190RhoGAP-A, pG1-pG2 deletion mutant (ΔpG1–pG2), or empty Flag vector (−). A representative set of immunoblots is shown. b Scatter plot of active RhoA as a function of Flag-p190RhoGAP expression. Active RhoA is normalized to maximum levels in each experiment (empty vector control) and Flag-p190RhoGAP expression as a percentage of maximum expression within each experiment. Shown is the compilation of data from 8 independent experiments. c Scatter/bar plot of data from (b), with data points binned according to Flag-p190RhoGAP expression as indicated (1–10%, 10–25%, 25–50% and 50–100%). The mean and s.e.m. (error bars) of each group is shown. Unpaired t-tests (two-tailed) were performed in GraphPad Prism, with statistical significance indicated above pairs and p-values as follows: 1–10% bin p=0.0053; 10–25% bin p=0.0019; 25–50% bin p=0.0006; and 50–100% bin p=0.0133
Discussion
The pseudokinases represent a paradigm for analysis of other pseudoenzyme folds1. Similar to the G motifs of small GTPases, canonical kinases contain consensus motifs responsible for catalysis and ATP binding13. Pseudokinases are degraded in one or more of these motifs, and can be further classified based on whether they are: (i) unable to bind nucleotide, (ii) able to bind nucleotide but have no catalytic activity or whether they (iii) retain catalytic activity (albeit low in some cases)14. There are numerous examples of pseudokinases that fall into this classification system (Table 2)15, 16. These classifications have important implications for function of the pseudokinases, with the non-enzymatic groups thought to assume scaffolding/adaptor roles in signal transduction17.
The pseudoGTPases can be classified in a similar manner. The existence of a pseudoGTPase subgroup unable to bind nucleotide (class i) is supported by our findings for p190RhoGAP, and by previous studies of the human centromere protein M (CENP-M)18 and a fungal dynein motor protein light intermediate chain (LIC)19 (Supplementary Table 2 and Table 2). Likewise, a pseudoGTPase subgroup able to bind nucleotide but with no catalytic activity (class ii) contains members of the Rnd family that have degraded sequences near the Switch II motif (Supplementary Table 2) and permanently bind GTP but do not catalyze hydrolysis20 (Table 2). Lastly, a pseudoGTPase subgroup that retains catalytic activity (class iii) includes the RGK family, which can catalyze hydrolysis but diverges at several G-motif sequences21 (Table 2 and Supplementary Table 2). We note that the G protein-like domain of the AGAP proteins (subgroup of the ArfGAP family) also lacks several conserved G motifs; however, reports of nucleotide binding are controversial22. Similar to pseudokinases, pseudoGTPases also exist across phylogenetic groups; for example, EhRabX3, a protein in the parasitic protozoan Entamoeba hystolytica contains a GTPase domain lacking multiple G motifs23, and the bacterial CheY response regulator receiver superfamily comprises a small GTPase fold with degraded G motifs and no nucleotide binding24, 25. Therefore, our discovery of evolutionarily conserved pseudoGTPase domains in p190RhoGAP illustrates that, similar to pseudokinases, pseudoGTPases exist in mammalian cytosolic signaling cascades, and represent a growing class of pseudoenzymes with diverse nucleotide-binding, enzymatic activity and physiological functions. As with recently discovered pseudophosphatases, pseudoproteases and pseudodeubiquitinases26, 27, understanding the function of pseudoGTPases will be an important future challenge with new mechanistic lessons in signal transduction.
Methods
Bioinformatics
Domain assignment searches on full-length p190RhoGAP proteins (p190RhoGAP-A from Homo sapiens, UniProtQ9NRY4; and p190RhoGAP-B from Homo sapiens, UniprotQ13017) were conducted using the National Center for Biotechnology Information (NCBI) Blast/CDD28 and InterPro29. Secondary structure predictions were also conducted on full-length protein sequences using NetSurfP30, PSIPRED31 and JPred32. Primary sequence alignments were carried out in CLUSTALO33. Residues 550–960 of Homo sapiens p190RhoGAP-A, 590–950 of Homo sapiens p190RhoGAP-B, 593–927 of D. melanogaster p190RhoGAP (UniProt ID: Q9VX32) and 610–950 of sponge p190RhoGAP (A. queenslandica, NCBI Reference sequence XP_003385690.2) were submitted for homology detection used the HHpred server34, 35. Structural similarity searches were conducted using the Dali server36. All structural figures generated using CCP4MG37.
Expression constructs
The complementary DNA (cDNA) encoding full-length Rattus norvegicus (rat) p190RhoGAP-A (ARHGAP35) protein (NCBI Reference Sequence: NP_001258061.1, UniP rotA0A0G2KB46) was inserted into a modified pCDNA-3.1 (Invitrogen) vector containing an N-terminal Flag tag. The full-length cDNA was used as a PCR template to amplify the predicted pG1 (residues 592–767) and pG2 (residues 766–958) regions. Similarly, regions encoding pG1 (590–763) or pG2 (764–954) of Homo sapiens (human) p190RhoGAP-B (ARHGAP5) (UniProt ID: Q13017) were generated by PCR using full-length p190RhoGAP-B cDNA as template. Codon-optimized synthetic cDNAs (Supplementary Table 3) encoding the pG1 region from Gallus gallus (chicken) p190RhoGAP-A (UniProt ID: A0A1D5P6Q7, residues 592–764, 90% identical to rat p190RhoGAP-A), Xenopus laevis (frog) p190RhoGAP-A (UniProt ID: Q6NU25, residues 587–762, 89% identical to rat p190RhoGAP-A), Danio rerio (zebrafish) p190RhoGAP-A (UniProt ID: F1R0X6, residues 592–772, 69% identical to rat p190RhoGAP-A) and D. melanogaster p190RhoGAP, which contains a single gene for p190 (UniProt ID: Q9VX32, residues 593–753, 22% identical to rat p190RhoGAP-A), were purchased from GenScript. All cDNA fragments encoding the pG1 and/or pG2 domains of p190RhoGAP-A and -B were inserted into a modified pET vector containing an N-terminal hexahistidine (His6) tag followed by a recognition sequence for tobacco etch virus (TEV) protease for expression in Escherichia coli. Human Rac1 (UniProt ID: P63000) residues 2–177 was inserted into the pET28a plasmid for expression in E. coli as a His-tagged protein9. GST-RBD (Rhotekin) was a gift from Martin Schwartz (Addgene plasmid no. 15247)38. Mutant constructs were generated with QuikChange Lightning Site-Directed mutagenesis kit (Agilent).The sequences of primers used in this study are listed in Supplementary Table 4.
Protein expression and purification
His6-tagged p190RhoGAP-A and -B pG1 proteins, and His-Rac1, were expressed in BL21 (DE3) cells (Millipore Sigma) or Rosetta (DE3) cells (Millipore Sigma) by induction with 0.2–0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) overnight at 16 °C. Cells were harvested by centrifugation at 2,000×g and lysed in nickel binding buffer (50 mM HEPES pH 7.3, 500 mM NaCl, 5 mM imidazole) by addition of lysozyme followed by freeze/thaw cycles and sonication. Lysates were clarified by centrifugation at 5000×g for 1 hand filtration, and applied to nickel beads for affinity purification (Ni Sepharose 6 Fast Flow, GE Healthcare). Following elution of bound proteins by increasing concentrations of imidazole in nickel-binding buffer, the His6 tag was removed from p190RhoGAP proteins by incubation with TEV protease overnight during dialysis against buffer containing 20 mM Tris pH 7.5 and 150 mM NaCl. The cleavage reaction was then flowed over a nickel affinity column (HisTrap Fast Flow, GE Healthcare) to remove the His6 tags, uncleaved His6-tagged protein and the His6-tagged TEV protease. The flow-through containing untagged p190RhoGAP proteins was concentrated in a centrifugal filter (Amicon Ultra, Millipore Sigma) and applied to size exclusion chromatography (Superdex 75 prep grade, GE Healthcare) in 20 mM Tris pH 7.5 and 150 mM NaCl. We were unable to express soluble protein for pG2 or tandem pG1–pG2 for either p190RhoGAP-A or -B using either E. coli, insect cell or mammalian expression systems. His6-tagged wild-type human Rac1 was eluted from nickel beads with increasing concentrations of imidazole in nickel-binding buffer, loaded onto a size exclusion chromatography column (Superdex 75 prep grade, GE Healthcare) in 20 mM Tris pH 7.5 and 150 mM NaCl and 1 mM dithiothreitol (DTT) and fractions pooled and concentrated in a centrifugal filter (Amicon Ultra, Millipore Sigma). GST-RBD (Rhotekin) was expressed in BL21 (DE3) cells (Millipore Sigma) by induction with 0.5 mM IPTG overnight at 16 °C. Cells were harvested by centrifugation at 2.000×g and lysed in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT, 5 mM MgCl2, 0.1 M phenylmethylsulfonyl fluoride (PMSF) and Roche cOmplete EDTA-Free protease inhibitor tablet) by addition of lysozyme followed by freeze/thaw cycles and sonication. Lysates were clarified by centrifugation at 5.000×g for 1 h, filtered and applied to Glutathione Sepharose 4B beads (GE Healthcare) for 1 h rocking at 4 °C. Beads were washed with RBD buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT, 1% Triton X-100, 5 mM MgCl2 and 0.05 M PMSF). After the final wash, beads were resuspended in RBD buffer supplemented with 10% glycerol, flash frozen in liquid nitrogen and stored at −80 °C in single-use aliquots. The GST-Rhotekin RBD purification protocol is adapted from ref. 38.
Crystallization/data collection and structure determination
Initial small needle clusters of X. laevis p190RhoGAP-A pG1 crystals were obtained by sparse matrix screening using a TTP Labtech Mosquito by vapor diffusion in sitting drops at room temperature with a 1:1 (v/v) ratio of purified protein to reservoir solution containing 1.7 M lithium sulfate, 0.1 M sodium acetate pH 4.5. The pH/buffer screening was then carried out using the Slice Screen (Hampton Research), which yielded improvement in crystal singularity and size with substitution of N-(2-Acetamido) iminodiacetic acid (ADA) at pH 6.3 in 1.7–1.8 M lithium sulfate. Crystals were harvested from the drop, quickly incubated in 3.4 M sodium malonate pH 6.3 as a cryoprotectant and flash-cooled in liquid nitrogen. Three sets of diffraction data were collected from a single crystal at Northeastern Collaborative Access Team (NE-CAT) Beamline 24-ID-E at Argonne National Laboratory Advanced Photon Source, processed separately in HKL200039, and scaled together in SCALEPACK39 to improve completeness and resolution to 1.9 Å. The data were initially processed in P6, and PhenixXtriage indicated a point group of P622. Matthews probability calculation indicated a single copy of pG1 in the asymmetric unit for P622. For molecular replacement, initial search models were selected according to HHpred34, 35 homology detection, which predicted a Ras-like GTPase fold for the p190RhoGAP-A pG1 sequence. Many molecular replacement runs in Phenix Phaser40, 41 using multiple small GTPase structures as search models failed to discover a structure solution. Next, a more robust search model preparation was performed in mr_rosetta in Phenix42, which generates search models based on HHpred sequence alignments with homologous structures. A top scoring HHpred homology alignment was that of the small GTPase-like domain of human Arf-GAP with GTPase, Ankyrin repeat and PH domain-containing protein 3 (AGAP3; PDB accession code 3IHW, unpublished, Structural Biology Consortium, probability 99.8%, E-score 2E−17, 13% identical, 129 residues aligned), which was modified by mr_rosetta to generate an improved search model with 1.9 Å r.m.s.d. over 134 aligned residues compared to the original search model. This mr_rosetta model was further modified manually to remove loops connecting predicted secondary structure elements. Finally, the modified models were used as Phaser search models40, with the best model yielding a final translation function Z-score (TFZ) of 6.7 for a single copy in space group P6522. Initial building was performed in Phenix AutoBuild43, which built 121 Cα positions, 85 of which were built in correct location (compared to the final refined model) with 18 of these correctly docked in the sequence (residues 657–674) with R free value of 44.9%. A subsequent round of autobuilding in the ARP/wARP module44 of CCP4i45, 46 successfully built 142 residues with 134 correctly docked into sequence for a R free of 34.5%. Manual model building was then carried out in Coot47, and refinement in Phenix48 including TLS parameters determined in Phenix. The final model contains 157 residues. PISA analysis of crystallographic interfaces supports that the isolated pG1 domains behave as monomers, consistent with gel filtration chromatography. Two malonate ions and three sodium ions are built in p190RhoGAP-A pG1.
Initial crystals of human p190RhoGAP-B were obtained by sparse matrix screening and grew as needle clusters in 2 M ammonium sulfate (AmSO4), 5% isopropanol. Crystal size could be improved by optimizing reservoir buffer to 2.2 M AmSO4 and 1% isopropanol, but diffraction studies indicated that freezing was poor. AmSO4 was substituted with sodium malonate pH 4.7 and crystals grew larger and in the absence of isopropanol. Final crystals were grown in 2.15 M sodium malonate pH 4.7 and cryoprotected in 3.4 M sodium malonate pH 4.7 before freezing in liquid nitrogen for diffraction studies. A 2.6 Å data set was collected from a single crystal at NECAT beamline 24-ID-E and processed in HKL200039. Matthews probability calculator indicated a single copy of p190RhoGAP-B pG1 in the asymmetric unit. A molecular replacement search model was generated using the nearly completed model of X. laevis p190RhoGAP-A pG1, which was modified by sculptor49 to replace nonidentical residues with the p190RhoGAP-B sequence. A single molecular replacement solution was found with Phaser40 with a TFZ score of 7.2 in space group P43. PhaserAutoBuild was carried out43, which resulted in a model containing 113 residues, of which 87 were correctly docked into p190RhoGAP-B sequence, with R free value of 31.9%. Manual model building was carried out in Coot47, and refinement in Phenix48 and Refmac546 including TLS parameters determined by TLSMD50. The final model contains 136 residues. For both crystal structures, model quality was assessed in Molprobity51. Crystallographic software is compiled by SBGrid52.
Nucleotide-binding assays
MANT-GTPγS was purchased from Jena Bioscience. Binding of 0.5 μM MANT-GTPγS to 1–2 μM purified protein was measured at room temperature in buffer containing 20 mM Tris pH 7.5, 50 mM NaCl, 10 mM divalent salt (MgCl2, MnCl2, CaCl2, ZnCl2, NiCl2, CoCl2) plus or minus 20 mM EDTA in 100 μl reaction volumes in a black-bottomed microplate. Fluorescence data were collected at excitation and emission wavelengths of 360 and 440 nm, respectively, on a TECAN Infinite M1000 plate reader, or on a BioTek Synergy 2 multi-mode reader with excitation/emission filters of 360/40 and 450/50, respectively. A time course of binding was carried out with fluorescence measurements taken every 30 s for 30 min, with protein added to nucleotide after 3 baseline measurements (which were averaged as signal at time zero). In each experiment, the fluorescence signal was normalized to time zero. Purified wild-type Rac1 was used as a positive control9.
Thermal shift assays were performed as described previously for pseudokinases10. Protein at 2–5 μM in 20 mM Tris pH 7.4 and 150 mM NaCl was mixed with SYPRO Orange (ThermoFisher; stock solution at 5000×) at a final concentration of 2×–5×, in the absence or presence of 1 mM MgCl2 or MnCl2, and in the absence or presence of 200 μM nucleotide: GTP, GTPγS, GDP, GMP or ATP in a total reaction volume of 25 μl. A Bio-Rad CFX Connect Real-time PCR machine with FAM filters was used to collect data. The mixture was pre-equilibrated to 4 °C for 5 min, followed by thermal ramping of 1 °C per min from 4 to 95 °C, with fluorescene measurements taken after each 1 °C increment. Fluorescence signal was normalized and plotted as a function of temperature, and data were fit to a sigmoidal curve in Prism 7 (Graphpad) with R 2 values of >0.99. The midway inflection point of the curve represents the melting temperature (T m). Points after the fluorescence maximum were excluded from fitting. Changes in the melting temperature (ΔT m) compared to the buffer-only control curve were calculated for each ligand pair and reported as the difference in T m. The mean and s.e.m. of three separate experiments was determined. Positive ΔT m of ≥3 °C is generally accepted as a significant stabilization of the protein in the presence of ligand10.
Rho activity assays
To measure levels of active Rho53, HEK293T cells (ATCC) were plated in 6-well culture dishes with Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and penicillin/streptomycin. Cells were transfected with a gradient of pCMV-derived plasmid DNAs from 0.1–5 μg encoding Flag-p190RhoGAP-A wild-type and mutant or empty Flag vector using Lipofectamine 2000 and OPTIMEM (Thermo Fisher). At 68–72 h after transfection, the cells were harvested. Rhotekin pulldown assays were used to assay active Rho in transfected HEK293T cell lysates similarly as described previously54, 55. Cells were transferred to ice, washed with ice-cold Tris-buffered saline buffer and lysed in buffer containing 1% v/v NP-40, 25 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 10% glycerol, 1 mM PMSF, 0.2 mM Na3VO4, 10 mM NaF and complete protease inhibitor tablet EDTA-free (Roche). Lysates were clarified by centrifugation at 16,900×g at 4 °C for 5 min and transferred to tubes containing 5 μl of Rhotekin-RBD beads which were obtained commercially from Cytoskeleton, Inc. or prepared as previously described38, 53. Lysates were mixed with beads by rocking for 45 min at 4 °C. RBD beads were then washed three times with cold lysis buffer. Bound proteins were eluted with addition of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer, resolved by SDS–PAGE and transferred to polyvinylidene difluoride membranes. Bound and total endogenous RhoA was probed by anti-RhoA monoclonal antibody 26C4 at 1:500 dilution (Santa Cruz sc-418), Flag-p190RhoGAP levels in lysates were probed with anti-Flag-M2 antibody at 1:3000 dilution (Millipore Sigma F3165). Anti-mouse IRDye 800CW secondary antibody (LI-COR Biotechnology 926-32212) was used at 1:20,000 dilution. Immunoblots were imaged and quantified on an Odyssey CLx Imaging system and ImageStudio Software (LI-COR Biotechnology). Active RhoA levels, as measured by Rhotekin-bound RhoA, were standardized to total RhoA in each sample, and reported as percent normalized to the maximum level of RhoA pulled down (from empty vector control samples). P-values were calculated using the unpaired t-test, two-tailed, in GraphPad Prism, with significance indicated in the figure legend.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 5U4U and 5U4V. X-ray diffraction images are available online at SBGrid Data Bank57: doi:10.15785/SBGRID/454 (5U4U) and doi:10.1038/s41467-017-00483-x (5U4V). Other data are available from the corresponding author on reasonable request.
References
Boudeau, J., Miranda-Saavedra, D., Barton, G. J. & Alessi, D. R. Emerging roles of pseudokinases. Trends Cell. Biol. 16, 443–452 (2006).
Wennerberg, K., Rossman, K. L. & Der, C. J. The Ras superfamily at a glance. J. Cell. Sci. 118, 843–846 (2005).
Jaffe, A. B. & Hall, A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 (2005).
Burbelo, P. D. et al. p190-B, a new member of the Rho GAP family, and Rho are induced to cluster after integrin cross-linking. J. Biol. Chem. 270, 30919–30926 (1995).
LeClerc, S., Palaniswami, R., Xie, B. X. & Govindan, M. V. Molecular cloning and characterization of a factor that binds the human glucocorticoid receptor gene and represses its expression. J. Biol. Chem. 266, 17333–17340 (1991).
Settleman, J., Narasimhan, V., Foster, L. C. & Weinberg, R. A. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell 69, 539–549 (1992).
Roof, R. W. et al. Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol. Cell. Biol. 18, 7052–7063 (1998).
Bourne, H. R., Sanders, D. A. & McCormick, F. The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127 (1991).
Davis, M. J. et al. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 110, 912–917 (2013).
Murphy, J. M. et al. A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem. J. 457, 323–334 (2014).
Biname, F. et al. Cancer-associated mutations in the protrusion-targeting region of p190RhoGAP impact tumor cell migration. J. Cell. Biol. 214, 859–873 (2016).
Wennerberg, K. et al. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr. Biol. 13, 1106–1115 (2003).
Hanks, S. K., Quinn, A. M. & Hunter, T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241, 42–52 (1988).
Zeqiraj, E. & van Aalten, D. M. F. Pseudokinases-remnants of evolution or key allosteric regulators? Curr. Opin. Struct. Biol. 20, 772–781 (2010).
Murphy, J. M. et al. Insights into the evolution of divergent nucleotide-binding mechanisms among pseudokinases revealed by crystal structures of human and mouse MLKL. Biochem. J. 457, 369–377 (2014).
Scheeff, E. D., Eswaran, J., Bunkoczi, G., Knapp, S. & Manning, G. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 17, 128–138 (2009).
Hammaren, H. M., Virtanen, A. T. & Silvennoinen, O. Nucleotide-binding mechanisms in pseudokinases. Biosci. Rep. 36, e00282 (2015).
Basilico, F. et al. The pseudo GTPase CENP-M drives human kinetochore assembly. Elife 3, e02978 (2014).
Schroeder, C. M., Ostrem, J. M., Hertz, N. T. & Vale, R. D. A Ras-like domain in the light intermediate chain bridges the dynein motor to a cargo-binding region. Elife 3, e03351 (2014).
Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell. Biol. 7, 54–62 (2006).
Splingard, A. et al. Biochemical and structural characterization of the gem GTPase. J. Biol. Chem. 282, 1905–1915 (2007).
Soundararajan, M., Yang, X., Elkins, J. M., Sobott, F. & Doyle, D. A. The centaurin gamma-1 GTPase-like domain functions as an NTPase. Biochem. J. 401, 679–688 (2007).
Chandra, M. et al. Insights into the GTP/GDP cycle of RabX3, a novel GTPase from Entamoeba histolytica with tandem G-domains. Biochemistry 53, 1191–1205 (2014).
Artymiuk, P. J., Rice, D. W., Mitchell, E. M. & Willett, P. Structural resemblance between the families of bacterial signal-transduction proteins and of G proteins revealed by graph theoretical techniques. Protein Eng. 4, 39–43 (1990).
Chen, J. M., Lee, G., Murphy, R. B., Brandt-Rauf, P. W. & Pincus, M. R. Comparisons between the three-dimensional structures of the chemotactic protein CheY and the normal Gly 12-p21 protein. Int. J. Pept. Protein Res. 36, 1–6 (1990).
Eyers, P. A. & Murphy, J. M. The evolving world of pseudoenzymes: proteins, prejudice and zombies. BMC Biol. 14, 98 (2016).
Reiterer, V., Eyers, P. A. & Farhan, H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell. Biol. 24, 489–505 (2014).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226 (2015).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015).
Petersen, B., Petersen, T. N., Andersen, P., Nielsen, M. & Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51 (2009).
Buchan, D. W. A., Minneci, F., Nugent, T. C. O., Bryson, K. & Jones, D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 (2013).
Drozdetskiy, A., Cole, C., Procter, J. & Barton, G. J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 (2015).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Hildebrand, A., Remmert, M., Biegert, A. & Soding, J. Fast and accurate automatic structure prediction with HHpred. Proteins 77, 128–132 (2009).
Soding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 (2011).
Ren, X. D., Kiosses, W. B. & Schwartz, M. A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585 (1999).
Otwinowski, Z. & Minor, W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
DiMaio, F. et al. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature 473, 540–U149 (2011).
Terwilliger, T. C. et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 (2008).
Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008).
Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 (2003).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Schwarzenbacher, R., Godzik, A., Grzechnik, S. K. & Jaroszewski, L. The importance of alignment accuracy for molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 60, 1229–1236 (2004).
Painter, J. & Merritt, E. A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 39, 109–111 (2006).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Morin, A. et al. Collaboration gets the most out of software. Elife 2, e01456 (2013).
Ren, X. D. & Schwartz, M. A. Determination of GTP loading on Rho. Methods Enzymol. 325, 264–272 (2000).
Bradley, W. D., Hernandez, S. E., Settleman, J. & Koleske, A. J. Integrin signaling through Arg activates p190RhoGAP by promoting its binding to p120RasGAP and recruitment to the membrane. Mol. Biol. Cell 17, 4827–4836 (2006).
Hernandez, S. E., Settleman, J. & Koleske, A. J. Adhesion-dependent regulation of p190RhoGAP in the developing brain by the Abl-related gene tyrosine kinase. Curr. Biol. 14, 691–696 (2004).
Pai, E. F. et al. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 9, 2351–2359 (1990).
Meyer, P. A. et al. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. Nat. Commun. 7, 10882 (2016).
Acknowledgements
Anthony Koleske is thanked for helpful discussions and for providing p190RhoGAP-A cDNA. Anton Bennett is thanked for providing p190RhoGAP-B cDNA. GST-RBD was a gift from Martin Schwartz (Addgene plasmid #15247). Craig Roy and Justin McDonough are thanked for the use of the Tecan plate reader, and Anatoly Kiyatkin and Mark Lemmon for use of the BioTek Synergy 2 plate reader for the MANT assays. Bertrand Simon, Daniel Iwamoto and LeenaKuruvilla are thanked for assistance with the thermal shift assays. Mark Lemmon is also thanked for helpful comments on the manuscript. Rong Zhang, ByungHak Ha, and Alexander Scherer are thanked for technical input. Staff at beamline 24-ID-E (NE-CAT-E) at the Advanced Photon Source, Argonne National Laboratory are thanked. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by National Institutes of Health grant P41GM103403. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. National Institutes of Health grants R01NS085078, R01GM109487, R01GM114621, R01GM102262, R01GM100411, P50CA121974, S10OD018007 funded the research.
Author information
Authors and Affiliations
Contributions
A.L.S. and T.J.B. conceived the project, designed the experiments and wrote the manuscript. A.L.S. conducted the experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stiegler, A.L., Boggon, T.J. p190RhoGAP proteins contain pseudoGTPase domains. Nat Commun 8, 506 (2017). https://doi.org/10.1038/s41467-017-00483-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-00483-x
This article is cited by
-
DUSP5P1 promotes gastric cancer metastasis and platinum drug resistance
Oncogenesis (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
