
Abstract
Nail–patella syndrome (NPS) is a hereditary disease caused by pathogenic variants in LMX1B and characterized by nail, limb, and renal symptoms. This study revealed a likely pathogenic LMX1B variant, NM_002316.4: c.723_726delinsC (p.Ser242del), in Japanese twins with clubfoot. The patients’ mother, who shared this variant, developed proteinuria after delivery. p.Ser242del is located in the homeodomain of the protein, in which variants that cause renal disease tend to cluster. Our findings highlight p.Ser242del as a likely pathogenic variant, expanding our knowledge of NPS.
Similar content being viewed by others

Nail–patella syndrome (NPS; OMIM: 161200) is an autosomal dominant (AD) disease characterized by four major symptoms: nail dysplasia, patellar abnormalities, elbow dysplasia, and iliac horns. Other presentations of NPS patients include other skeletal abnormalities; nephropathy; hearing loss; and neurological, ophthalmological, and dental problems1,2. NPS exhibits full penetrance in heterozygous individuals, but its severity varies markedly. To date, more than 130 LMX1B pathogenic/likely pathogenic variants causative of NPS have been registered in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed 5 August, 2023). LMX1B is a member of the LIM homeodomain protein family; all proteins in this family have a homeodomain and a pair of LIM domains. The homeodomain binds to DNA to perform transcription factor functions by interacting with specific DNA elements in target genes3. A previous report showed an association between this homeodomain and nephropathy2,3. LMX1B has been reported to be essential for dorsoventral patterning during development, so the skeletal phenotype of NPS patients is considered to result from a deficiency in dorsoventral patterning regulated by LMX1B3.
The transcription factor LMX1B first gained prominence because of its involvement in dorsoventral axis formation during limb development3. One of the major determinants of the prognosis of NPS is kidney involvement, which is thought to be caused by arrested development of podocytes and glomeruli due to a pathogenic variant of LMX1B. NPS with kidney involvement has a broad spectrum of severity, ranging from asymptomatic proteinuria and/or hematuria to symptomatic kidney failure4.
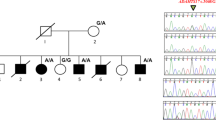
In this paper, we describe a likely pathogenic variant of LMX1B identified by clinical genetic analysis of Japanese twins born to an NPS-affected mother. The pedigree of this family is shown in Fig. 1(i). III-3 was diagnosed with NPS in infancy and had typical symptoms, such as triangular deformities of the nail, underdeveloped patellae, hearing loss in the right ear, and asymptomatic hypercalciuria. Her proteinuria became apparent after delivery of the twins (described below). III-1 was diagnosed with NPS and presented with symptoms of malformed finger and toenails and elbow anomalies with difficulty extending. II-2 was also diagnosed with NPS, which presented as malformed nails.

(i) Pedigree diagram showing segregation of the LMX1B variant in an autosomal dominant manner. I-2, II-1, and II-2 had previously been diagnosed with nail–patella syndrome (NPS), although the clinical phenotypes were unclear. III-1 (older) had malformed finger and toenails and elbow anomalies with difficulty in extension. III-3 (younger, profoundly affected) was diagnosed with NPS in infancy and exhibited typical symptoms such as triangular appearance of the lunulae, underdeveloped patellae, loss of hearing in the right ear, and proteinuria developing after delivery. (ii) LMX1B pathogenic and likely pathogenic variants in NPS patients from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). LMX1B is composed of 402 amino acids, described by line segments of 10 amino acids each. The gray squares indicate three domains in LMX1B: the LIM1 domain, LIM2 domain, and homeodomain (residues 56–106, 115–168, and 219–278, respectively). Some pathogenic (or likely pathogenic) missense variants (gray circles) and an in-frame deletion (gray diamond) were clustered within the homeodomain. p.Ser242del (black diamond and dashed line) is located in the homeodomain.
The affected mother (III-3) delivered monochorionic diamniotic (MD) monozygotic twins (IV-1 and IV-2) at 36 weeks and 4 days of gestation by cesarean section. Both IV-1 and IV-2 are female and weighed 2090 g and 1902 g at birth, respectively. The twins had ankle varus deformities (Supplementary Fig. A, B) with no other dysmorphic features. Approximately 18 h after birth, they were found to have asymptomatic hypoglycemia (blood glucose of ~30 mg/dL). The patients were admitted to the neonatal intensive care unit and intravenously infused with glucose via enteral feeding. After admission, their blood glucose levels were stable, and their oral intake gradually increased. Hence, continuous intravenous glucose supplementation was stopped for IV-1 and IV-2 on the 3rd and 4th days after birth, respectively. Subsequently, no recurrence of hypoglycemia occurred, and the twins were discharged on the 15th day after birth. At the 1-month follow-up, no notable changes in their general condition were noted. However, the triangular deformities of the nails had become more pronounced (Supplementary Fig. C, D).
Although ankle varus deformities were not observed in other affected family members, the family history strongly suggested that the twins suffered from NPS. After obtaining informed consent, peripheral whole-blood samples were obtained from the twins for genetic analysis. Written informed consent was also obtained from the parents for the anonymized publication of the patients’ details.
The sequences of the protein-coding region of LMX1B along with the intronic boundary regions were analyzed by targeted next-generation sequencing using the hybrid capture method at Kazusa DNA Research Institute (Kisarazu, Chiba, Japan). The obtained nucleotide sequences were compared with published human genome reference sequences, and the presence or absence of low-frequency nucleotide substitutions and deletions/insertions of short nucleotide sequences was analyzed.
DNA sequencing revealed that the affected siblings (IV-1 and IV-2) harbored a heterozygous deletion-insertion [NM_002316.4: c.723_726delinsC (p.Ser242del)] in exon 4 of LMX1B, which is located on chromosome 9q33.3 (Fig. 1(ii)). c.723_726delinsC was not registered in either the Human Genome Mutation Database (HGMD) or ClinVar, but it was also absent from the control databases [the Genome Aggregation Database (gnomAD) and Tohoku Medical Megabank Organization (ToMMo)]. The identification of this variant was consistent with the analysis previously performed for their mother (III-1), and the combined annotation-dependent depletion score was 325. In accordance with the American College of Medical Genetics and Genomics (ACMG) guidelines, c.723_726delinsC was classified as a likely pathogenic variant5.
In the present case, considering that the affected twins were MD monozygotic, we were concerned about similar development of kidney involvement. To follow up for their kidney-related issues, they were scheduled for regular outpatient consultation at our pediatric nephrology department in infancy. NPS with kidney involvement has various characteristic patterns, such as proteinuria, hematuria, and end-stage kidney failure4. In a previous study, kidney failure was found in 3% (3/123) of NPS patients6. This study also revealed the exacerbation of kidney involvement in the perinatal period6, with the mother developing proteinuria after delivery of the twins. Another study showed that NPS patients with variants in the homeodomain are more likely than those with LIM domain site variants to suffer from proteinuria2 and that variants in patients with severe nephropathy tend to cluster in specific regions of the homeodomain. In addition, pathogenic variants identified in LMX1B-related nephropathy patients were also clustered in the homeodomain3.
NPS patients require regular follow-up consultations at a nephrology department to track renal function in infancy or early childhood. It has been suggested that screening for nephropathy should be performed annually, including measurements of spot urine protein/creatinine or the albumin/creatinine ratio4. Since some NPS patients have an earlier onset of glaucoma than does the general population, consultation with an ophthalmologist to screen for signs and symptoms of glaucoma is recommended for these patients6.
Pathogenic variants in LMX1B result in skeletal dysplasia affecting dorsal tissues, especially the nail and patella. These variants are also significantly expressed in the dorsal mesenchyme of developing joints7, consistent with clubfoot. A report from the early 1990s on patients with clubfoot8 suggested that the presence of clubfoot in the affected twins in the present study was a symptom of NPS, although there was no family history of this condition.
The affected twins had the same symptoms, such as triangular deformities of the nails and clubfoot, that led to a diagnosis of NPS despite the abundant variation generally reported among NPS patients. In previous case reports of MD twins with AD diseases, namely, Ehlers–Danlos syndrome9 and maturity-onset diabetes of the young10, similar symptoms in the twins helped achieve a diagnosis via genetic analysis. In addition, a comparison between siblings and twins affected by autosomal dominant polycystic kidney disease showed that there was a significantly smaller difference in age at end-stage renal disease in twins than in siblings11. The presentation of similar clinical symptoms in MD twins, who share much of their genetic information, suggests that the phenotypic variation observed in individuals with the same genotype is influenced by other modifying factors.
In the patients reported here, we identified a likely pathogenic variant, c.723_726delinsC (p.Ser242del). Hamlington et al. reported 22 pathogenic variants, including p.Ser242del, in NPS patients12; however, they did not report the exact clinical manifestations of the NPS patients. Here, we have described the clinical manifestations of limb and kidney involvement, which are considered to be associated with p.Ser242del.
The affected mother was shown to have proteinuria as a result of the progression of kidney disease, with which p.Ser242del can be associated, considering its location within the variant hotspot in the LMX1B homeodomain. For the appropriate follow-up of NPS-affected children, consultations with nephrologists, orthopedists, and other specialists are needed. Continuous follow-up of patient growth, development, and symptoms, as well as clarification of the relationships between genetic variants and symptoms, are also needed in these cases. The expansion of a database containing this information is highly important for the future diagnosis and follow-up of genetic diseases.
HGV DATABASE
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3368.
References
Witzgall, R. Nail-patella syndrome. Pflug. Arch. 469, 927–936 (2017).
Bongers, E. M. et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur. J. Hum. Genet. 13, 935–946 (2005).
Harita, Y., Kitanaka, S., Isojima, T., Ashida, A. & Hattori, M. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy. Pediatr. Nephrol. 32, 1845–1850 (2017).
Lemley, K. V. Kidney disease in nail-patella syndrome. Pediatr. Nephrol. 24, 2345–2354 (2009).
Richards, S. et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Sweeney, E., Fryer, A., Mountford, R., Green, A. & McIntosh, I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J. Med. Genet. 40, 153–162 (2003).
Dreyer, S. D. et al. LMX1B transactivation and expression in nail-patella syndrome. Hum. Mol. Genet. 9, 1067–1074 (2000).
Guidera, K. J., Satterwhite, Y., Ogden, J. A., Pugh, L. & Ganey, T. Nail Patella Syndrome: A Review of 44 Orthopaedic Patients. J. Pediatr. Orthop. 11, 737–742 (1991).
Ruggeri, P., Calcaterra, S. & Girbino, G. Bullous emphysema as first presentation of Ehlers-Danlos syndrome in monozygotic twins. Respir. Med. Case Rep. 14, 40–42 (2014).
Faguer, S. et al. Massively enlarged polycystic kidneys in monozygotic twins with TCF2/HNF-1beta (hepatocyte nuclear factor-1beta) heterozygous whole-gene deletion. Am. J. Kidney Dis. 6, 1023–1027 (2007).
Persu, A. et al. Comparison between siblings and twins supports a role for modifier genes in ADPKD. Kidney Int. 6, 2132–2136 (2004).
Hamlington, J. D., Jones, C. & McIntosh, I. Twenty-two novel LMX1B mutations identified in nail patella syndrome (NPS) patients. Hum. Mutat. 5, 458 (2001).
Acknowledgements
We thank the family members for their participation in this study. We also thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. This study was supported in part by the medical staff at Yokohama City University Medical Center.
Author information
Authors and Affiliations
Contributions
N.K. and K.I. designed the case study, collected the data, and wrote the manuscript. S.N., R.S., M.K., and M.T. performed medical examinations. M.Y., N.N., A.T., M.H., and K.T. provided advice. S.I. supervised the study. All of the authors have read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kishio, N., Iwama, K., Nakanishi, S. et al. A deletion variant in LMX1B causing nail–patella syndrome in Japanese twins. Hum Genome Var 11, 10 (2024). https://doi.org/10.1038/s41439-024-00266-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00266-z
