
Abstract
GJA1 is the causative gene for oculodentodigital dysplasia (ODDD). A novel de novo GJA1 variant, NM 000165:c263C > T [p.P88L], was identified in a mosaic state in a patient with short stature, seizures, delayed myelination, mild hearing loss, and tooth enamel hypoplasia. Although the patient exhibited severe neurodevelopmental delay, other clinical features of ODDD, including limb anomalies, were mild. This may be due to differences in the mosaic ratios in different organs.
Similar content being viewed by others


Oculodentodigital dysplasia (ODDD [MIM: 164200]) is characterized by multiple congenital anomalies1. The major clinical features include (1) distinctive facial findings, including a thin nose with hypoplastic ala nasi, small anteverted nares, prominent columella, and microcephaly; (2) eye findings, such as microphthalmia and glaucoma; (3) syndactyly, typically digital malformation, including bilateral complete syndactyly of digits 4 and 5 (type III syndactyly), camptodactyly, and permanent joint flexion of the digits; (4) teeth anomalies and enamel hypoplasia; and (5) cardiac dysfunctions2,3,4. Some patients exhibit dysplastic ears and conductive hearing loss5,6. Various neurological symptoms, including dysarthria, spastic paraparesis, ataxia, and seizures, occur in ~30% of patients with ODDD7,8,9,10. ODDD is a genetic disorder inherited as an autosomal dominant trait. In 2003, connexin-43 (gap junction protein alpha 1; GJA1) [MIM*121014] on chromosome 6q22 was identified as the gene responsible for this condition11. Recently, we identified a novel GJA1 variant in an undiagnosed patient with a neurodevelopmental disorder; however, the variant was identified as mosaic. The details of this study are as follows.
The patient was a 5-year-old Japanese girl who was the third child of healthy parents. She was born at 37 weeks and 6 days of gestation by vaginal delivery in the cephalic position without any abnormalities. Her birth weight was 2738 g (25–50th centile). Her parents noticed that she showed her eye movements pursuing moving objects but showed no social smiles at 2 months of age. At 12 months of age, she was referred to our hospital because of developmental delay with no acquisition of a sitting position.
On admission, her vital signs were normal, and no cardiac arrhythmias were detected. She displayed a flat nasal bridge and bilateral epicanthus. Very mild webbing was observed between her fingers and toes, except between the first and second fingers and toes. Camptodactyly was also noted in both the fingers and toes. No abnormalities were observed in the skull bone. Neurological examination revealed deep tendon hyperreflexia but no evidence of extrapyramidal signs. Hypotonia or muscle weakness was not observed. An auditory brainstem response revealed mild hearing loss in the left ear. Cranial computed tomography showed bilateral basal ganglia calcifications (Fig. 1a). Brain magnetic resonance imaging revealed mild dilatation of the extracerebral space and lateral ventricles. Delayed myelination of the subcortical white matter and a thin corpus callosum were also observed (Fig. 1b–d).

a Cranial computed tomography image. Calcification is shown in the bilateral basal ganglia (arrows). b Sagittal image of brain magnetic resonance imaging, indicating thinning of the corpus callosum (arrow). c, d T1- and T2-weighted brain magnetic resonance axial images indicating abnormalities in several regions, including the cavus septum pellucidum (arrow).
At the age of 2 years, the patient experienced her first epileptic seizure. Electroencephalography revealed focal epileptic discharges in the right frontal lobe, and zonisamide was prescribed. She can currently hold a sitting position but has difficulty walking. She does not speak meaningful words but can communicate through babbling and eye contact, indicating severe neurodevelopmental delay. She can eat orally but needs assistance. The erupted lower anterior teeth are fused and have weak enamel. No visual loss or visual field defects were observed. The intelligence scale test showed that her developmental quotient was <20. At present (5 years of age), her height is 97.5 cm (<3rd centile), her weight is 14.1 kg (3rd~10th centile), and her occipitofrontal circumference is 49.5 cm (25–50th centile), indicating short stature.
For precise diagnosis, this patient was enrolled in the research project “Initiative on Rare and Undiagnosed Disorders”12, which was performed in accordance with the Declaration of Helsinki and approved by the ethics committee of our institution. After informed consent was obtained from the family, blood samples were collected from the patient and her parents. Genomic DNA was extracted from peripheral blood samples following a standard protocol, and exome sequencing was performed using trio samples, including parental samples, as previously described13. GATK HaplotypeCaller was used for variant calling (https://www.broadinstitute.org/). The results revealed a de novo variant of GJA1 (NM 000165:c263C > T [p.P88L]). This variant is not included in the gnomAD or ClinVar databases. CADD_phred was 28.2, and the MutationTaster_score was 1, suggesting that the variant is damaging. According to the ACMG/AMP guidelines14, four scores (PS2, PM2, and PP3) were adaptive. Thus, this variant was classified as “likely pathogenic”.
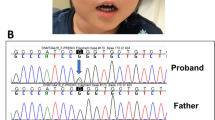
The identified variant was analyzed using Sanger sequencing. Because there are many homologous genes with similar nucleotide sequences to GJA1, PCR primer sets (3′-TTGTCTCTTTGTTTCTTTCAG-5′ and 3′-GTACCACTGGATCAGCAAGAA-5′) were designed at unique sequence sites. For PCR amplification, we used GoTaq® (Promega, Madison, WI). As shown in Fig. 2a, neither of the parents showed p.P88L, and de novo occurrence was confirmed; however, the peak of the minor variant in the patient was lower than that in the wild-type, suggesting the possibility of mosaicism. The read depth of the variant was retrospectively checked, and the read ratio of the minor allele was 8/59 (no contradiction for mosaic status). In this study, GATK HaplotypeCaller was used for variant calling. GATK HaplotypeCaller uses probability calculations to determine whether a variant is heterozygous, homozygous, or wild-type homozygous, even if the proportion of variant reads at depth is less than half. This explains why even a mosaic variant at a low depth could be detected. To confirm the mosaicism ratio, the PCR products were subcloned and inserted into a pGEM-T Vector® (Promega) and transformed into E. coli. After transformation, colonies were selected on ABPC-supplemented agar culture medium. The plasmids were extracted from each colony and sequenced. Finally, a minor variant was found in 7 out of 34 colonies (Fig. 2b), indicating that the estimated mosaicism rate in lymphocytes was 41%.

a Electropherogram of PCR-direct sequencing showing a very low peak of “T”. b After subcloning the PCR products, two types of clones, namely, the wild-type allele and altered minor variant allele, were detected.
Previously, 77 GJA1 variants have been reported in patients with ODDD or erythrokeratodermia4,6,15,16,17,18,19,20,21,22,23. These findings have been reported in a wide range of medical fields, including pediatrics, dermatology, dentistry, ophthalmology, and orthopedics. This is due to the wide variety of clinical symptoms and severity of this disease, as connexin-43 is pleiotropically expressed in many tissues and regulates intercellular signaling. As shown in Table 1, the present patient exhibited most of the possible clinical findings in ODDD, such as camptodactyly, enamel hypoplasia, hearing loss, and neurological findings. In particular, abnormal brain imaging, including brain calcification, is not rare in patients with ODDD7,17. On the other hand, a thin nose and hypoplastic alae, the typical facial findings of ODDD, were not observed. Syndactyly, one of the major findings of ODDD, was also not observed; however, ODDD cases without syndactyly are not rare, and the webbed fingers identified in the present patient were considered to be the consequence of incomplete syndactyly. Therefore, we diagnosed the present patient as having ODDD. Although arrythmia and ocular involvement were not observed, the development of these findings is dependent on the clinical course and may appear in the future. Therefore, careful clinical follow-up is important for this patient.
GJA1 consists of four transmembrane domains, two extracellular loops, and exposed amino- and carboxyl-termini in the cytoplasm15. The p.P88L variant identified in this study is located in one of the transmembrane regions. As shown in Supplementary Fig. 1, most variants in the transmembrane regions are related to ODDD. There were some variants in the region neighboring p.P88L. p.S86Y was identified as a de novo variant in a patient with ODDD19 who was first diagnosed with syndactyly and later developed distinctive facial findings and ophthalmological involvement. Psychomotor development was mildly delayed. p.T89I was identified in a familial case of syndactyly with no neurological features21. In contrast, the present patient showed severe neurodevelopmental delay but very mild signs of syndactyly (mild webbing of fingers and toes). This may be due to the mosaicism in the present patient. Mosaicism is known to produce atypical or attenuated clinical symptoms due to masking by normal cells24. Therefore, we hypothesized that different mosaic ratios in different organs may contribute to the differences in severity of the clinical features in the present patient.
Patients with ODDD who exhibit neurological features as the main finding, but mild syndactyly may not be rare. By applying comprehensive genetic analysis, the existence of such atypical cases has become apparent, and the disease concept is being expanded.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3354.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Gorlin, R. J., Miskin, L. H. & St Geme, J. W. Oculodentodigital dysplasia. J. Pediatr. 63, 69–75 (1963).
Delmar, M. & Makita, N. Cardiac connexins, mutations and arrhythmias. Curr. Opin. Cardiol. 27, 236–241 (2012).
Wittlieb-Weber, C. A., Haude, K. M., Fong, C. T. & Vinocur, J. M. A novel GJA1 mutation causing familial oculodentodigital dysplasia with dilated cardiomyopathy and arrhythmia. HeartRhythm Case Rep. 2, 32–35 (2016).
Pace, N. P. et al. Two novel GJA1 variants in oculodentodigital dysplasia. Mol. Genet. Genom. Med. 7, e882 (2019).
Kelly, S. C. et al. A novel GJA 1 mutation in oculo-dento-digital dysplasia with curly hair and hyperkeratosis. Eur. J. Dermatol. 16, 241–245 (2006).
Dwarakanathan, A., Bhat, M., Gn, S. & Shetty, S. Missense and deletion mutations in GJA1 causing oculodentodigital dysplasia in two Indian families. Clin. Dysmorphol. 24, 159–162 (2015).
Loddenkemper, T., Grote, K., Evers, S., Oelerich, M. & Stögbauer, F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J. Neurol. 249, 584–595 (2002).
Harting, I. et al. Oculodentodigital dysplasia: a hypomyelinating leukodystrophy with a characteristic MRI pattern of brain stem involvement. AJNR Am. J. Neuroradiol. 40, 903–907 (2019).
De Bock, M., Kerrebrouck, M., Wang, N. & Leybaert, L. Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front. Pharmacol. 4, 120 (2013).
Barzegar, M., Sayadnasiri, M. & Tabrizi, A. Epilepsy as a rare neurologic manifestation of oculodentodigitalis dysplasia. Iran. J. Child Neurol. 6, 39–43 (2012).
Paznekas, W. A. et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 72, 408–418 (2003).
Takahashi, Y. et al. Six years’ accomplishment of the Initiative on Rare and Undiagnosed Diseases: nationwide project in Japan to discover causes, mechanisms, and cures. J. Hum. Genet. 67, 505–513 (2022).
Narita, K. et al. Whole-exome analysis of 177 pediatric patients with undiagnosed diseases. Sci. Rep. 12, 14589 (2022).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Paznekas, W. A. et al. GJA1 mutations, variants, and connexin 43 dysfunctions as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 30, 724–733 (2009).
Jamsheer, A. et al. A novel GJA1 missense mutation in a Polish child with oculodentodigital dysplasia. J. Appl. Genet. 50, 297–299 (2009).
Tumminelli, G. et al. Oculodentodigital dysplasia with massive brain calcification and a new mutation of GJA1 gene. J. Alzheimers Dis. 49, 27–30 (2016).
Boyden, L. M. et al. Dominant de novo mutations in GJA1 cause erythrokeratodermia variabilis et progressiva without features of oculodentodigital dysplasia. J. Invest. Dermatol. 135, 1540–1547 (2015).
Jamsheer, A. et al. Three novel GJA1 missense substitutions resulting in oculo-dento-digital dysplasia (ODDD) - further extension of the mutational spectrum. Gene 539, 157–161 (2014).
Choi, J. et al. Oculodentodigital dysplasia with a novel mutation in GJA1 diagnosed by targeted gene panel sequencing: a case report and literature review. Ann. Clin. Lab Sci. 48, 776–781 (2018).
Bilal, M. et al. Sequence variants in MEGF8 and GJA1 underlying syndactyly. Mol. Syndromol. 14, 201–207 (2023).
Huang, X., Wang, N., Xiao, X., Li, S. & Zhang, Q. A novel truncation mutation in GJA1 associated with open-angle glaucoma and microcornea in a large Chinese family. Eye 29, 972–977 (2015).
Li, C. et al. Two de novo GJA1 mutation in two sporadic patients with erythrokeratodermia variabilis et progressiva. Mol. Genet. Genom. Med. 7, e670 (2019).
Mohiuddin, M., Kooy, R. F. & Pearson, C. E. De novo mutations, genetic mosaicism and human disease. Front. Genet. 13, 983668 (2022).
Funding
This work was supported by the Initiative on Rare and Undiagnosed Diseases (Grant Number 22ek0109549) from the Japan Agency for Medical Research and Development (AMED); Grants-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare, Japan (22FC1005); and Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (JSPS) (Grant Number 21K07873).
Author information
Authors and Affiliations
Contributions
T.Y. designed this study. R.S. was involved in organizing this study and drafting the manuscript. T.Y., K.I., M.S., T.S., K.I., and S.N. contributed to the acquisition of clinical data. K.S.Y., M.N., Y.I., Y.M., and Y.A. contributed to the acquisition of genomic data. All the authors contributed to the analysis and interpretation of the data. All the authors agree to be accountable for all the aspects of the work and ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shimomura, R., Yanagishita, T., Ishiguro, K. et al. Rare mosaic variant of GJA1 in a patient with a neurodevelopmental disorder. Hum Genome Var 11, 2 (2024). https://doi.org/10.1038/s41439-023-00262-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00262-9
