
Abstract
Congenital tooth agenesis is caused by the impairment of crucial genes related to tooth development, such as Wnt signaling pathway genes. Here, we investigated the genetic causes of sporadic congenital tooth agenesis. Exome sequencing, followed by Sanger sequencing, identified a novel single-nucleotide deletion in WNT10A (NC_000002.12(NM_025216.3):c.802del), which was not found in the healthy parents of the patient. Thus, we concluded that the variant was the genetic cause of the patient’s agenesis.
Similar content being viewed by others
The Wingless-related integration site (WNT) ligand family comprises 19 highly conserved genes across species, ranging from invertebrates to mammals1. These genes encode secreted glycoproteins linked to the canonical intracellular beta-catenin signaling pathway, which plays a critical role in multiple tissues and organ development. One of the family members, WNT10A, is known to have specific relevance to the skin, skin appendages, and teeth2. Pathogenic variants in the WNT10A gene are the most frequent cause of nonsyndromic tooth agenesis in humans, including the Japanese population3.
Congenital tooth agenesis is a condition characterized by missing teeth, which can vary in number and type. It ranges from selective tooth agenesis to syndromic conditions such as ectodermal dysplasia4. Nonsyndromic hypodontia, a mild selective tooth agenesis defined as missing fewer than six teeth, is a common congenital disorder in humans. A more severe phenotype, oligodontia, involves the loss of six or more permanent teeth. In the Japanese population, the frequency is 6.8% for hypodontia and 0.1% for oligodontia3. The etiology of congenital tooth agenesis has been investigated, and several causative genes have been identified5,6,7,8,9,10,11,12. Many genes have been reported as etiologic agents of tooth agenesis, including MSX1, PAX9, LRP6, WNT10A, and WNT10B13. Recent studies have highlighted WNT10A as a significant causative gene with diverse effects on gene/protein function14,15,16,17,18. According to genetic studies on families with tooth agenesis, the WNT10A pathogenic variant is the most frequent cause of human tooth agenesis (STHAG4, OMIM:150400)17.
Here, we report the clinical genetic analysis of a patient with a sporadic form of nonsyndromic hypodontia with three congenitally missing teeth.
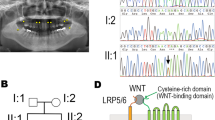
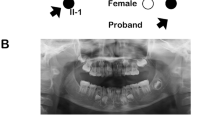
Saliva samples were obtained from the proband (II-1), her unaffected siblings (II-2), and both parents (I-1 and I-2; Fig. 1a). The patient’s parents provided written informed consent. The Institute for Developmental Research and the Aichi-Gakuin University Committee approved this clinical and molecular genetic study, which was conducted in accordance with the Declaration of Helsinki. The patient (II-1; Fig. 1a) was a 7-year-old girl who presented with missing teeth 17, 24, and 27 (Fédération Dentaire International tooth numbering system). Orthopantomography confirmed a missing tooth in the mandible (Fig. 1b). The patient had no systemic abnormalities except for tooth number, including the crown morphology of the other teeth or the jawbone. Furthermore, no abnormalities in tooth number were found in the other family members (Fig. 1a).

a Male and female members are shown by squares and circles, respectively. The filled circle denotes the affected participant. b Phenotypic characteristics of the patient. X-ray imaging of the proband. c X indicates the missing teeth of the patient.
According to the manufacturer’s protocol, the Oragene DISCOVER kit was used to extract genomic DNA from 2 ml of saliva. Whole-exome sequencing was performed using a SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA), and the captured libraries were sequenced using an Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) with 150 base pair paired-end reads. Whole-exome sequencing (WES) of the patient’s genomic DNA identified a WNT10A variant, NC_000002.12(NM_025216.3):c.802del, in the proband (II:1). Trio-based Sanger sequencing with a specific primer set (5’-CTCAGCGTTTGCCTCTGTA-3,’ 5’-ACGAAACAGCACCAGTGGAA-3’) confirmed that this was a de novo variant (Fig. 2). This variant is not found in the online gnomAD database (https://gnomad.broadinstitute.org/). According to the ACMG-AMP Guidelines (PVS1 and PS2), the variant was classified as pathogenic.

The Sanger sequencing chromatogram demonstrated heterozygosity for the WNT10A variant (c.802delT) in the proband (II-1).
The C-terminal region of the WNT ligand plays a pivotal role in binding to FRIZZLED (FZD), a WNT receptor with seven transmembrane domains. The WNT10A variant in the current case, p.Ser268fs, would result in a loss of FZD binding activity, similar to other variants included in our previous report19. WNT ligands, including WNT10A, are cysteine-rich morphogens that can interact with the FZD receptor and LDL receptor-related protein 5/6 (LRP5/6). The nucleotide substitution identified in the current case resulted in a frameshift at nucleotide 802. Hence, the WNT10A gene variant product had an unrelated peptide consisting of 11 amino acid residues, QAAASSRRAGRX, after the 268th Lys at the C-terminus: NM_025216.3(NP_079492.2):p.(Ser268Glnfs*12). Thus, the variant product lacks a functional domain that interacts with the WNT receptors of FZD.
Although functional null variants of the WNT10A gene cause autosomal recessive ectodermal dysplasia, patients with a heterozygous null variant of the WNT10A gene are often diagnosed with nonsyndromic tooth agenesis but rarely with mild ectodermal dysplasia. This is because slight anomalies in other ectodermal tissues are more difficult to detect than those related to the number of teeth.
The number of missing teeth varies among patients, even among family members carrying the same variant14,20. Therefore, while the WNT10A variant caused hypodontia in the current patient, the same variant can also cause oligodontia. The molecular mechanisms underlying the phenotypic variation in tooth number among patients carrying identical gene variants should be elucidated in the future.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3345.
References
Cadigan, K. M. & Nusse, R. Wnt signaling: a common theme in animal development. Genes Dev. 11, 3286–3305 (1997).
Doolan, B. J., Onoufriadis, A., Kantaputra, P. & McGrath, J. A. WNT10A, dermatology and dentistry. Br. J. Dermatol. 185, 1105–1111 (2021).
Machida, J. et al. Genetic epidemiology of tooth agenesis in Japan: a population- and family-based study. Clin. Genet. 88, 167–171 (2015).
Visinoni, A. F., Lisboa-Costa, T., Pagnan, N. A. & Chautard-Freire-Maia, E. A. Ectodermal dysplasias: clinical and molecular review. Am. J. Med. Genet. A 149A, 1980–2002 (2009).
Zhou, M. et al. Analyses of oligodontia phenotypes and genetic etiologies. Int. J. Oral Sci. 13, 32 (2021).
Kamamoto, M. et al. Clinical and functional data implicate the Arg(151)Ser variant of MSX1 in familial hypodontia. Eur. J. Hum. Genet. 19, 844–850 (2011).
Goto, H. et al. A novel LRP6 variant in a Japanese family with oligodontia. Hum. Genome Var. 8, 30 (2021).
Adachi, J. et al. Novel MSX1 frameshift mutation in a Japanese family with nonsyndromic oligodontia. Hum. Genome Var. 8, 29 (2021).
Tatematsu, T. et al. An aberrant splice acceptor site due to a novel intronic nucleotide substitution in MSX1 gene is the cause of congenital tooth agenesis in a Japanese family. PLoS ONE 10, e0128227 (2015).
Song, S. et al. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 88, 126–131 (2009).
Dinckan, N. et al. Whole-exome sequencing identifies novel variants for tooth agenesis. J. Dent. Res. 97, 49–59 (2018).
Kantaputra, P. N. et al. Mutations in LRP5 and BMP4 are associated with mesiodens, tooth agenesis, root malformation, and oral exostoses. Clin. Genet. 102, 333–338 (2022).
Meade, M. J. & Dreyer, C. W. Tooth agenesis: an overview of diagnosis, aetiology and management. Jpn Dent. Sci. Rev. 59, 209–218 (2023).
Machida, J. et al. WNT10A variants isolated from Japanese patients with congenital tooth agenesis. Hum. Genome Var. 4, 17047 (2017).
Mostowska, A. et al. Nucleotide variants of genes encoding components of the Wnt signalling pathway and the risk of non-syndromic tooth agenesis. Clin. Genet. 84, 429–440 (2013).
Sun, R., Li, S., Xia, B. & Zhu, J. Detection of novel variant and functional study in a Chinese family with nonsyndromic oligodontia. Oral Dis. 29, 2177–2187 (2023).
Grejtakova, D. et al. WNT10A variants in relation to nonsyndromic hypodontia in eastern Slovak population. J. Genet. 97, 1169–1177 (2018).
Arzoo, P. S., Klar, J., Bergendal, B., Norderyd, J. & Dahl, N. WNT10A mutations account for ¼ of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 164A, 353–359 (2014).
Adachi, J. et al. Novel WNT10A variant in a Japanese case of nonsyndromic oligodontia. Hum. Genome Var. 10, 3 (2023).
Vastardis, H., Karimbux, N., Guthua, S. W., Seidman, J. G. & Seidman, C. E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 13, 417–421 (1996).
Acknowledgements
We thank the participants for their involvement in this study. This work was supported in part by AMED under the grant numbers JP17nk0101334 and JP20ek0109397 (to Y.T.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ando, M., Aoki, Y., Sano, Y. et al. Novel frameshift variant of WNT10A in a Japanese patient with hypodontia. Hum Genome Var 11, 5 (2024). https://doi.org/10.1038/s41439-023-00259-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00259-4
