
Abstract
Osteogenesis imperfecta (OI) is a rare genetic disorder characterized by brittle bones. In this case report, we describe a patient who suffered from OI type XIV with a novel splice site variant in the TMEM38B gene. Further research is needed to better understand the relationship between the phenotype of OI type XIV and this variant.
Similar content being viewed by others
Osteogenesis imperfecta (OI) is a rare genetic disorder characterized by brittle bones1. The most common molecular cause is variants in COL1A1 or COL1A22. There are several subtypes of OI caused by other molecular variants, such as IFITM5 (OI type V), SERPINF1 (OI type VI), and CRTAP (OI type VII). OI type XIV is an autosomal recessive subtype of the disorder first reported by Shaheen et al3. and caused by a homozygous truncating deletion of exon 4 of TMEM38B. In this case report, we describe a patient with OI who was found to have a splice site variant of TMEM38B. To our knowledge, this is the first report of a variant at this site in a patient with OI.
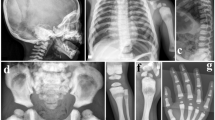
A 9-year-old Japanese boy diagnosed with OI presented to our hospital for intravenous bisphosphonate therapy. He was the first child from parents who were nonconsanguineous for at least the past three generations. No family members had a suspected diagnosis of OI. In the fetal period, long bone deformities were detected by ultrasound. He was born at 38 weeks of gestation with a birth weight of 2698 g. He was diagnosed with OI based on the radiographic findings (Fig. 1) and blue sclera. He had a history of multiple fractures of the long bones, causing severe ambulatory difficulties and short stature (-2.9 SD) despite repeated intravenous bisphosphonate therapy since he was 2 months old. Additionally, he was diagnosed with an atrial septal defect with persistent left superior vena cava, causing pulmonary overflow and pulmonary hypertension as an infant. Surgical closure of the defect was performed at 9 months of age.

The neonatal X-ray showed deformity in the long bones, consistent with in utero fractures.
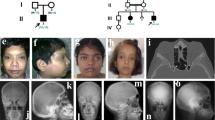
In the genetic analysis performed as part of genetic counseling, a homozygous splice donor variant of TMEM38B, NC_000009.12(NM_018112.3):c.660+1 G > A (rs144782604), was detected, which was not registered in ClinVar4. This variant was located one nucleotide downstream of exon 5 in TMEM38B. Franklin’s algorithm (https://franklin.genoox.com/; access date: 16 March 2023) classified this variant as “likely pathogenic”: PVS1_strong and PM2_moderate according to the new ClinGen recommendation5. In silico analysis by Mutation Taster6 classified this variant as deleterious. Analysis of his parents revealed a heterozygous splice site variant at the same position (Fig. 2).

Next-generation sequencing of TMEM38B in the patient revealed a homozygous splice site variant at NC_000009.12(NM_018112.3):c.660 + 1 G > A (II-1), and sequencing in the parents showed a heterozygous variant at the same site (I-1, I-2).
The truncating deletion of exon 4 of the TMEM38B gene was reported in the literature as the first known cause of OI type XIV, an autosomal recessive form of OI3,7 Subsequently, point mutations at various positions in TMEM38B have been reported8,9 The splice site variant of c.660+1 G > A described in this case is a rare variation. Its allele frequencies are known to be 0.00007 in the Japanese population10 and 0.00001 in the European population11. In this case, nonconsanguinity in the last three generations has been confirmed, but no information is available regarding earlier relationships. OI patients complicated by cardiac anomalies, including atrial septal defects, have been previously reported12,13 and atrial septal defects combined with ventricular septal defects in a patient with point mutations in TMEM38B have also been reported9. Some TMEM38B variations may have a relationship with congenital heart disease. Further research is needed to fully understand the phenotype of OI XIV and the impact of this specific variation.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3321.
References
Sillence, D. O., Senn, A. & Danks, D. M. Genetic heterogeneity in osteogenesis imperfecta. J. Med Genet. 16, 101–116 (1979).
Sykes, B., Ogilvie, D., Wordsworth, P., Anderson & Jones, N. Osteogenesis imperfecta is linked to both type I collagen structural genes. Lancet 2, 69–72 (1986).
Shaheen, R. et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J. Med Genet. 49, 630–635 (2012).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D10672018. (2018).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Volodarsky, M. et al. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum. Mutat. 34, 582–586 (2013).
Lv, F. et al. Two novel mutations in TMEM38B result in rare autosomal recessive osteogenesis imperfecta. J. Hum. Genet. 61, 539–545 (2016).
Webb, E. A. et al. Phenotypic Spectrum in Osteogenesis Imperfecta Due to Mutations in TMEM38B: Unraveling a Complex Cellular Defect. J. Clin. Endocrinol. Metab. 102, 2019–2028 (2017).
Tadaka, S. et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 6, 28 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Manoria, P. C., Misra, M. P. & Bhargava, R. K. Indian Heart J. 34, 173–174 (1982).
Vetter, U. et al. Eur. J. Pediatr. 149, 184–187 (1989).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kodama, Y., Meiri, S., Asada, T. et al. Novel splice site variant of TMEM38B in osteogenesis imperfecta type XIV. Hum Genome Var 10, 25 (2023). https://doi.org/10.1038/s41439-023-00252-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00252-x
This article is cited by
-
Update on the Genetics of Osteogenesis Imperfecta
Calcified Tissue International (2024)
-
Zebrafish Models for Skeletal and Extraskeletal Osteogenesis Imperfecta Features: Unveiling Pathophysiology and Paving the Way for Drug Discovery
Calcified Tissue International (2024)
