
Abstract
Preterm birth (PTB), defined as the birth of a baby at <37 weeks of gestation, is known to be the main cause of neonatal morbidity and mortality. Here, we report genetic associations between preterm birth and gestational age in a Japanese population. We conducted a genome-wide association study (GWAS) of 384 cases who delivered prematurely and 644 controls and considered gestational age as a quantitative trait in 1028 Japanese women. Unfortunately, we were unable to identify any significant variants associated with PTB or gestational age using the current sample. We also examined genetic associations previously reported in European populations and identified no associations, even with the genome-wide subthreshold (p value < 10–6). This data report aims to provide summary statistics of current GWASs on PTB in a Japanese population for future meta-analyses of genetics and PTB with larger sample sizes.
Similar content being viewed by others

Introduction
Preterm birth (PTB) is defined as delivery at <37 weeks of gestation. The estimated global PTB rate was 9.8% in 2000 and increased to 10.6% in 2014, ranging from 13.4% in North Africa to 8.7% in Europe1. The percentage of PTB in Japan was 3.71% between 1979 and 1983 but increased to 4.77% between 2009 and 20142. Although the rate of PTB in Japan is lower than that worldwide, it is clear that PTB is closely associated with morbidity and mortality in immature infants, and elucidating the cause of PTB is of interest. Many maternal and fetal characteristics have been associated with PTB, including maternal demographic characteristics, nutritional status, pregnancy history, infection, and biological and genetic markers3. However, 31–50% of PTB are due to spontaneous onset of labor, making it difficult to identify the cause4. The association between PTB and genetic variants has been reported previously. It was reported that women whose mothers or sisters had a history of PTB were 55% more likely to deliver prematurely than those with no family history of PTB5, and that genetic factors accounted for 34% of delivery timing in twin analysis6. Moreover, it was reported that maternal and fetal genetic factors influenced gestational age7. Recently, several genome-wide association studies (GWASs) on PTB have been conducted. The largest study, in which the EBF1, EEFSEC, AGTR2, WNT4, ADCY5, and RAP2G loci were associated with gestational week, and the EBF1, EEFSEC, and AGTR2 loci were associated with PTB, was reported in 2017 and used a discovery dataset from 23andMe comprising over 40 thousand women of European ancestry8. In addition, a genome-wide association study (GWAS) of the Finnish population in 2019 reported a novel association between the SLIT2 locus and PTB9. Although a few GWASs on PTB and gestational age have been reported, no GWASs have been conducted in an Asian population, including the Japanese population. Therefore, in this study, we conducted a GWAS of PTB and gestational age to identify related variants in a Japanese population.
Materials and methods
We collected 1158 samples of postpartum maternal blood from Keio University Hospital between 2011 and 2020. This study was approved by the Keio University School of Medicine Ethics Committee (20100154) and the Institutional Review Board of the National Research Institute for Child Health and Development (406) and was conducted in accordance with the ethical standards outlined in the 1964 Helsinki Declaration and later amendments. All women provided written informed consent.
Of the 1158 samples, 80 medically-indicated PTB samples, such as those from women with multiple pregnancies and placenta previa, were excluded. After excluding these 80 samples, analysis was performed, as shown in the flowchart (Supplementary Fig. 1). Genome-wide SNP genotyping was performed using the Infinium Omni2.5-8 v1.0 Kit and v1.1 Kit (Illumina, San Diego, CA, USA) by using the standard cluster file provided by Illumina. The ped file generated using GenomeStudio v2.0.5 (Illumina, San Diego, CA, USA) was converted to the Variant Call Format (VCF) file using PLINK 1.90 beta10. The VCF file was converted from Illumina strands to reference sequence strands (GRCh37) using bcftools 1.911. The VCF files were converted from GRCh37 to GRCh38 using CrossMap 0.6.0 (default: no option)12.
By using the converted VCF files, we filtered out low-quality samples by applying the following conditions. For sample call rates, the PLINK command with the “--missing” option was used, and no samples were removed because all samples had call rates above 0.98. For heterozygosity rates, the PLINK command with the “--het” option was used, and nine samples were removed because their heterozygosity rates were outliers ±3 SD from the 1078 samples. For sex inconsistency, we calculated the heterozygosity rate of each sample on chromosome X, and four samples were removed because their heterozygosity rates were less than 0.01.
After removing 13 samples by sample QC, we also filtered out low-quality variants by applying the following conditions. For variant call rates, the PLINK command with the “--missing” option was used, and 42,469 variants in the VCF files of ver1.0 and 65,843 variants in the VCF files of ver1.1 were removed because these variants had call rates less than 0.98. For Hardy–Weinberg equilibrium (HWE), the PLINK command with the “--hardy” option was used, and 408 variants in the VCF files of ver1.0 and 624 variants in those of ver1.1 were removed (p value of HWE <1e−6).
After removing low-quality variants by variant QC, the resultant VCF files of ver1.0 and ver1.1 were transformed into a form that conforms to Beagle 5.4 (default: no option)13 and integrated with shared SNPs before imputation. By using this conformed file, whole genome imputation was performed with the 1000 Genomes Project reference haplotype data (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/). Beagle software was used to estimate the genotype dosages for the GWASs, and the variants with DR2 > 0.7 were retained. We also filtered out low-quality imputed variants by applying the Hardy–Weinberg equilibrium (HWE) test with a p > 0.001 and minor allele frequency (MAF) > 0.001.
After removing low-quality variants by variant QC, we checked the genetic relatedness. For the detection of relatedness, the PLINK command with the “--genome --min 0.1875” option was used, and 37 pairs were indicated to show relatedness. Of these 37 pairs, 7 pairs (14 samples) were from the same person who delivered twice: one delivery was a term birth and the other was a preterm birth. Of these pairs, 7 samples from the preterm birth were used for the subsequent analysis. Twenty-one pairs (42 samples) were from the same person who delivered twice, and both were term births. In these pairs, 21 samples with higher sample call rates were used for the subsequent analysis. The remaining 9 pairs (18 samples) were not from the same person. In those pairs, 9 samples with higher sample call rates than the other samples were used for the subsequent analysis. In summary, by analyzing relatedness, 37 samples were unrelated and used for the subsequent analysis. After filtering based on genetic relatedness, a total of 1028 samples and 11,183,581 variants were analyzed.
By using the resultant file, detection of population stratification was conducted by using principal component analysis (PCA) (Supplementary Fig. 2). Thirteen samples were identified as Han Chinese samples, but they were not removed because the top 10 genotype principal components (PCs) were used as covariates in the subsequent analysis.
GWASs were independently performed for both preterm birth as a binary trait and gestational age as a continuous trait by using PLINK 1.90 beta. In the analysis of preterm birth, the phenotype data were divided into two groups: the term-birth group (control group; N = 644) and the preterm-birth group (case group; N = 384) (Supplementary Fig. 3a). Of 1028 samples, 578 were derived from the VCF files of ver1.0, and 450 samples were derived from the VCF files of ver1.1 (Supplementary Fig. 3b). In the analysis of gestational age, because of the variability of the gestational age in the sample, quantile normalization was performed; the values were considered quantitative traits in the GWASs (Supplementary Fig. 3c). In both analyses, the version of Illumina cluster files (1.0 and 1.1) and the first 10 principal components (generated as a result of PCA using samples from the Japanese population in Tokyo and the Han Chinese population extracted from the 1000 Genome Project data) were used as covariates. We set the genome-wide significance level at p < 5.0 × 10–8.
Results
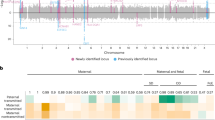
A GWAS of PTB was conducted, and no SNPs were identified as significant at p < 5.0 × 10–8 (Fig. 1a). Then, a GWAS of gestational age (as a quantitative trait) was conducted, and no SNPs were identified as significant at p < 5.0 × 10–8 (Fig. 1b). Thus, in this study, no significant SNPs were found to be associated with PTB or gestational age with the current sample (Supplementary Fig. 4).

Each dot represents a variant. The x-axis represents genomic coordinates, and the y-axis represents −log10 (p value). Two colors, blue and gray, are used alternately for dots on a chromosome for easy viewing. The blue line represents the suggestive significant threshold (α) of p = 1.0 × 10−5. a Manhattan plot of genome-wide associations between variants and PTB among the Japanese population (384 cases and 644 controls). b Manhattan plot of genome-wide associations between variants and gestational age among the Japanese population.
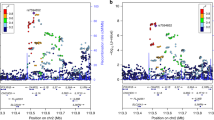
The regions centered on SNPs known to be associated with PTB and gestational age were examined (Fig. 2 and Supplementary Table 1). None of the known SNPs were associated with PTB (Fig. 2a) or gestational age (Fig. 2b), even with a genome-wide subthreshold of p < 10–6. Odds ratios of known SNPs8 associated with PTB and effect sizes of SNPs8 associated with gestational age were compared to those from our study (Supplementary Fig. 5).

The x-axis represents the chromosomal position. The y-axis represents −log10 (p value). Each dot represents a variant, and its color shows the LD index (r2) with the known associated SNP in each locus. Gene names and nucleotide coordinates are shown in each panel. a Regional plots for associations between variants and PTB among the Japanese population (384 cases and 644 controls). b Regional plots for associations between variants and gestational age among the Japanese population.
Discussion
We conducted a GWAS in a Japanese population and were unable to identify any significant variants associated with PTB and gestational age. We also examined variants around the variants known to be associated with PTB and gestational age reported previously, but no significant variants were confirmed. There are a couple of reasons that could explain these negative findings in this study. Although we excluded samples from medically-indicated PTBs in this study, we did not completely focus on spontaneous PTB because the sample size was expected to be extremely small. The sample size of this study was much smaller than that of other studies. Further studies with larger sample sizes and detailed clinical information are indispensable to detect significant genetic variants associated with PTB and gestational age in the Japanese population.
Data availability
The summary statistics of GWASs on PTB and gestational weeks will be deposited in the GWAS Catalog (Accession Numbers: GCST90268004 and GCST90268005).
References
Blencowe, H. et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet 379, 2162–2172 (2012).
Sakata, S., Konishi, S., Ng, C. F. S. & Watanabe, C. Preterm birth rates in Japan from 1979 to 2014: analysis of national vital statistics. J. Obstet. Gynaecol. Res. 44, 390–396 (2018).
Goldenberg, R. L., Culhane, J. F., Iams, J. D. & Romero, R. Epidemiology and causes of preterm birth. Lancet 371, 75–84 (2008).
Slattery, M. M. & Morrison, J. J. Preterm delivery. Lancet 360, 1489–1497 (2002).
Boyd, H. A. et al. Maternal contributions to preterm delivery. Am. J. Epidemiol. 170, 1358–1364 (2009).
Kistka, Z. A. et al. Heritability of parturition timing: an extended twin design analysis. Am. J. Obstet. Gynecol. 199, 43.e1–5 (2008).
York, T. P., Strauss, J. F. 3rd, Neale, M. C. & Eaves, L. J. Estimating fetal and maternal genetic contributions to premature birth from multiparous pregnancy histories of twins using MCMC and maximum-likelihood approaches. Twin Res. Hum. Genet. 12, 333–342 (2009).
Zhang, G. et al. Genetic associations with gestational duration and spontaneous preterm birth. N. Engl. J. Med. 377, 1156–1167 (2017).
Tiensuu, H. et al. Risk of spontaneous preterm birth and fetal growth associates with fetal SLIT2. PLoS Genet. 15, e1008107 (2019).
https://www.cog-genomics.org/plink/. Accessed 7 December 2022.
https://github.com/samtools/bcftools. Accessed 7 December 2022.
https://crossmap.sourceforge.net/. Accessed 7 December 2022.
https://faculty.washington.edu/browning/beagle/beagle.html. Accessed 7 December 2022.
Acknowledgements
We would like to thank Dr. Kei Miyakoshi (Seibo Hospital) and Prof. Daigo Ochiai (Kitasato University) for sample collection. We are grateful to all medical staff at Keio University Hospital for providing excellent perinatal care. This study was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (21H02887), the National Center for Child Health and Development (NCCHD 2022A-3) and Gunma University for the promotion of scientific research.
Author information
Authors and Affiliations
Contributions
M.T. and K. Hata conceived the project. K. Hasegawa drafted the initial manuscript with critical input from N.K. H.K. performed the experiments and obtained data. K. Hasegawa, N.K. and K.N. performed data analysis. N.K., K.N., K.M., Y.K., K. Hata and M.T. reviewed and N.K. finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hasegawa, K., Kumasaka, N., Nakabayashi, K. et al. Genome-wide association study of preterm birth and gestational age in a Japanese population. Hum Genome Var 10, 19 (2023). https://doi.org/10.1038/s41439-023-00246-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00246-9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}