
Abstract
A heterozygous mutation in GRID2 that causes SCAR18 was first reported in an Algerian family with autosomal dominant cerebellar ataxia (ADCA). We identified the second ADCA family with a heterozygous GRID2 mutation. The Algerian family had cognitive impairment and hearing loss associated with cerebellar ataxia. However, the Japanese family presented here showed pure cerebellar ataxia. Therefore, we should also screen for the GRID2 mutation in ADCA families with pure cerebellar ataxia.
Similar content being viewed by others

Spinocerebellar ataxias are heterogeneous disorders that include chronic progressive cerebellar ataxia. Spinocerebellar ataxias with autosomal dominant inheritance are termed SCAs, and those with autosomal recessive inheritance are termed SCARs1,2. Molecular genetic research has revealed many causative genes and variants for the hereditary forms of spinocerebellar ataxia. Interestingly, SCAs and SCARs have overlapping causative genes, including SPTBN2 (SCA5/SCAR14), GRM1 (SCA44/SCAR13), and STUB1 (SCA48/SCAR16)3,4,5,6,7,8. A homozygous GRID2 mutation was first reported to be a cause of SCAR18 in 20139. In 2015, a heterozygous GRID2 mutation was also reported as a cause of autosomal dominant cerebellar ataxia (ADCA)10. Here, we report the clinical and genetic features of the second ADCA family with a heterozygous GRID2 mutation.
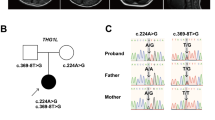
The family tree is shown in Fig. 1A. The affected individuals in this family were the proband (II-1), his mother (I-2), and his sisters (II-2 and II-3). The proband’s father (I-1) did not show any neurological symptoms. We collected their clinical information from medical records and from our examination.

A Family tree. Squares and circles indicate males and females, respectively. Filled symbols indicate affected individuals. The arrow indicates the proband. B Brain MRI. Axial and sagittal slices of a T1-weighted image obtained from brain MRI in Patient II-1 showing cerebellar atrophy. C SPECT. 123I-IMP SPECT shows reduced cerebellar blood flow. D Sanger sequencing. Green, red, black, and blue curves represent adenine (A), thymine (T), guanine (G), and cytosine (C), respectively. The arrows indicate the position of the heterozygous GRID2 mutation in the affected individuals and the unaffected one.
We also collected genomic DNA from all family members (I-1, I-2, II-1, II-2, and II-3). We first excluded pathogenic repeat expansions in SCA1 and 2, MJD/SCA3, 6, 7, 8, 17, 31, and 36, and DRPLA. Then, we conducted whole-exome sequence analysis for all participants. We screened for the known genes in SCAs (SCA5, 11, 13, 14, 15, 17, 19, 21, 22, 23, 26, 27, 28, 34, 35, 37, 38, 40, 41, 42, 43, 44, 45, 46, 47, and 48) and SCARs (SCAR1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 13, 14 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, and 31). We performed Sanger sequencing to confirm the variant in all participants. Functional prediction was conducted by in silico analysis, including PolyPhen211, SIFT12, and CADD13. Furthermore, we classified the variants according to ACMG guidelines14. This study was approved by our institutional review board, and written informed consent was obtained from all participants.
The present family included four patients with cerebellar ataxia in two generations. Their clinical information is shown in Table 1. The ages of onset in the four patients ranged from their 30s to their 50s. Symptoms at onset were gait disturbance and/or dysarthria. Their grandmother had gait instability, although we could not collect neurological information or genomic DNA for her. All patients exhibited truncal and limb ataxia without eye movement disorders, pyramidal tract signs, or sensory disturbance. Cognitive impairment and hearing loss were absent in all patients. The Scale for the Assessment and Rating of Ataxia (SARA) scores at the first examination for I-2, II-1 and II-2 (disease duration: approximately 15–20 years) were all approximately 10. Moreover, the SARA score for II-3 was 7 (disease duration: two years). The SARA scores slightly worsened to 12, 11, and 17 for I-2, II-1, and II-2 after five, seven, and seven years of disease duration, respectively. Brain MRI showed cerebellar atrophy in all patients who underwent this procedure (representative MRI in II-1, Fig. 1B). 123I-IMP SPECT in II-1 showed markedly reduced cerebellar blood flow (Fig. 1C).
There were no pathogenic repeat expansions in SCA1 or 2 or MJD/SCA3, 6, 7, 8, 17, or 31. Through gene screening by whole-exome sequencing, we found a heterozygous GRID2 variant (NM_001510.4: c.1966C>G, p.Leu656Val) in all patients in this family. The GRID2 variant allowed segregation of the affected individuals from a normal family member (I-1), as confirmed by Sanger sequencing (representative data for II-1 and I-1, Fig. 1D). The results of in silico analysis indicated this variant was probably damaging with Polyphen2, damaging with SIFT, and 27.7 with CADD. The amino acid at this position was conserved across species. According to the ACMG guidelines, the variant (NM_001510.4: c.1966C>G, p.Leu656Val) was classified as “pathogenic”.
We described the second ADCA family with a heterozygous GRID2 mutation in the present study. To date, a heterozygous GRID2 variant has only been described in sporadic cases outside the first family10. Interestingly, our family had the same mutation of GRID2 as that in the first family10. Therefore, we could state that our family was the second family with a heterozygous p.Leu656Val GRID2 mutation. GRID2 encodes the GluRD2 protein, which generally comprises extracellular domains (S1 and S2), transmembrane segments (M1, M3, and M4), and linkers (S1M1, M3S2, and S2M4)15,16. The p.Leu656Val mutation in the two families existed in the M3S2 linker. Furthermore, according to an earlier report10, three missense mutations (p.Ala654Thr, p.Ala654Asp, and p.Leu656Val) near the M3S2 linker cause cerebellar ataxia. Additionally, seven-point variants (p.Trp18Arg, p.Thr282Met, p.Arg304*, p.Thr330Met, p.Ala654Thr, p.Ala654Asp, and p.Leu656Val) have been reported to be heterozygous GRID2 variants in five reports to date (Table 2)10,17,18,19,20. Heterozygous missense mutations could lead to a GRID2 loss of function, which would be expected due to the recessive inheritance pattern. Three missense mutations (p. Ala654Thr, p.Ala654Asp, and p.Leu656Val) and one nonsense variant (p.Arg304*) were not registered in gnomAD21. Moreover, three missense variants (p.Trp18Arg, p.Thr282Met, and p.Thr330Met) were registered in gnomAD. According to these results and the ACMG guidelines, p.Trg18Arg, p.Thr282Met, p.Arg304*, and p.Thr330Met are classified as “likely benign”, “uncertain significance”, “pathogenic”, and “likely benign”.
Thus, the GRID2 mutation of p.Leu656Val would be considered pathogenic. However, the causes of the differences in severity, including ataxia, cognitive impairment, and hearing loss, between the first family and the present remain unknown. The present family exhibited only cerebellar ataxia, which indicated a phenotype of pure cerebellar ataxia. However, some patients in the first ADCA family showed cognitive impairment and hearing loss, indicating intrafamilial clinical variability (Table 1). We only know that the disease duration and severity change in parallel.
In summary, we identified the second ADCA family with the heterozygous mutation (NM001510.4: c.1966C>G, p.Leu656Val) in the GRID2 gene; this variant was found in an Algerian family using whole-exome analysis. We should screen for GRID2 variants in the case of families with pure cerebellar ataxia in ADCA. Further studies are required to elucidate the genotype-phenotype correlation in GRID2-related ataxias.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3207.
References
Bhandari, J., Thada, P. K. & Samanta, D. Spinocerebellar Ataxia. In: StatPearls [Internet]. (Treasure Island (FL), StatPearls Publishing 2022).
Synofzik, M. & Németh, A. H. Recessive ataxias. Handb. Clin. Neurol. 155, 73–89 (2018).
Ikeda, Y., Dick, K. A., Weatherspoon, M. R., Gincel, D., Armbrust, K. R. & Dalton, J. C. et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 38, 184–190 (2006).
Elsayed, S. M., Heller, R., Thoenes, M., Zaki, M. S., Swan, D. & Elsobky, E. et al. Autosomal dominant SCA5 and autosomal recessive infantile SCA are allelic conditions resulting from SPTBN2 mutations. Eur. J. Hum. Genet. 22, 286–288 (2014).
Watson, L. M., Bamber, E., Schnekenberg, R. P., Williams, J., Bettencourt, C. & Lickiss, J. et al. Dominant mutations in GRM1 cause spinocerebellar ataxia type 44. Am. J. Hum. Genet. 101, 451–458 (2017).
Guergueltcheva, V., Azmanov, D. N., Angelicheva, D., Smith, K. R., Chamova, T. & Florez, L. et al. Autosomal-recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am. J. Hum. Genet. 91, 553–564 (2012).
Genis, D., Ortega-Cubero, S., Nicolás, H. S., Corral, J., Gardenyes, J. & Jorge, L. D. et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91, e1988–e1998 (2018).
Shi, Y., Wang, J., Li, J., Ren, H., Guan, W. & He, M. et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS ONE 8, e81884 (2013).
Utine, G. E., Haliloglu, G., Salanci, B., Cetinkaya, A., Kiper, P. O. & Alanay, Y. et al. A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J. Child Neurol. 28, 926–932 (2013).
Coutelier, M., Burglen, L., Mund Willer, E., Abada-Bendib, M., Rodriguez, D. & Chantot-Bastaraud, S. et al. GRID2 mutations span from congenital to mild adult-onset cerebellar ataxia. Neurology 84, 1751–1759 (2015).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., Kondrashov, A. S. & Sunyaev, S. R. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688 (2012).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S. & Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 5, 405–424 (2015).
Schmid, S. M. & Hollmann, M. To gate or not to gate: are the delta subunits in the glutamate receptor family functional ion channels. Mol. Neurobiol. 37, 126–141 (2008).
Talukder, I. & Wollmuth, L. P. Local constraints in either the GluN2 or GluN2 subunit equally impair NMDA receptor pore opening. J. Gen. Physiol. 138, 179–194 (2011).
Fogel, B. L., Lee, H., Deignan, J. L., Strom, S. P., Kantarci, S. & Wang, X. et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 71, 1237–1246 (2014).
Ngo, K. J., Rexach, J. E., Lee, H., Petty, L. E., Perlman, S. & Valera, J. M. et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 41, 487–501 (2020).
Shamseldin, H. E., Maddirevula, S., Faqeih, E., Ibrahim, N., Hashem, M. & Shaheen, R. et al. Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genet. Med. 19, 593–598 (2017).
Maier, A., Klopocki, E., Horn, D., Tzschach, A., Holm, T. & Meyer, R. et al. De novo partial deletion in GRID2 presenting with complicated spastic paraplegia. Muscle Nerve 49, 289–292 (2014).
Karczewski, K. J., Francioli, L. C. & Tiao, G. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Acknowledgements
The following programs provided financial support to this research, including the study design, collection, analysis and interpretation of data, and writing of the manuscript: Grants-in-Aid from the Research Committee for Ataxic Disease (Y.T.), the Ministry of Health, Labor and Welfare, Japan, and JSPS KAKENHI Grant Numbers 16H06279 (PAGS), 17K17772 (K.K.), 19K16910 (K.K.), JP18K07495 (Y.T.), and 21K07456 (Y.T.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koh, K., Shimazaki, H., Ogawa, M. et al. A heterozygous GRID2 mutation in autosomal dominant cerebellar ataxia. Hum Genome Var 9, 27 (2022). https://doi.org/10.1038/s41439-022-00204-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00204-x
