
Abstract
Hereditary spherocytosis is the most frequent cause of hereditary hemolytic anemia and is classified into five subtypes (SPH1-5) according to OMIM. Because the clinical and laboratory features of patients with SPH1-5 are variable, it is difficult to classify these patients into the five subtypes based only on these features. We performed target capture sequencing in 51 patients with hemolytic anemia associated with/without morphological abnormalities in red blood cells. Thirteen variants were identified in five hereditary spherocytosis-related genes (six in ANK1 [SPH1]; four in SPTB [SPH2]; and one in each of SPTA1 [SPH3], SLC4A1 [SPH4], and EPB42 [SPH5]). Among these variants, seven were novel. The distribution pattern of the variants was different from that reported previously in Japan but similar to those reported in other Asian countries. Comprehensive genomic analysis would be useful and recommended, especially for patients without a detailed family history and those receiving frequent blood transfusions due to chronic hemolytic anemia.
Similar content being viewed by others

Introduction
Hereditary spherocytosis (HS) is the most frequent cause of hereditary hemolytic anemia1. Generally, patients with HS show hemolytic anemia in association with jaundice, reticulocytosis, osmotically fragile spherocytes, gallstones, and splenomegaly2. Cholelithiasis and aplastic crises are also common complications3. The clinical severity of hemolytic anemia in patients with HS varies widely, ranging from asymptomatic to severe life-threatening hemolytic anemia. Thus, an accurate diagnosis is important to support decision-making pertaining to subsequent treatment strategies, including splenectomy. According to the Online Mendelian Inheritance in Man database (OMIM: https://www.omim.org/), HS is classified into five subtypes associated with five different genes responsible for the deficiency or dysfunction of red blood cell membrane proteins, including ankyrin 1 (ANK1; MIM #18200 [SPH1]), β-spectrin (SPTB; MIM #616649 [SPH2]), α-spectrin (SPTA1; MIM #270970 [SPH3]), band 3 protein (SLC4A1; MIM #612653 [SPH4]), and protein 4.2 (EPB42; MIM #612690 [SPH5]) (Supplemental Table S1). SPH1, SPH2, and SPH4 are associated with the autosomal dominant (AD) trait, whereas SPH3 and SPH5 are associated with autosomal recessive (AR) traits. Therefore, it is important to obtain an accurate diagnosis for proper genetic counseling. For this purpose, it is important to not only evaluate family history, the clinical course, and physical findings but to also perform laboratory examinations3. With the recent development of molecular analysis methods, it has become necessary to detect causative gene variants to obtain a final diagnosis for HS patients.
Recently, we developed an originally designed target capture sequencing (TCS) panel for the precise diagnosis of hemolytic anemia. Here, some of the results obtained with this panel are summarized to clarify the clinical and genetic features of patients with HS in association with the five well-established subtypes, SPH1-5.
Materials and methods
This study aimed to elucidate the molecular basis of HS in Japanese patients. For this purpose, we enrolled patients with hemolytic anemia, including HS, in accordance with the Declaration of Helsinki, followed by the approval of the Ethics Committee of our institution. After obtaining written informed consent, blood samples were collected from patients. From the attending doctors, we also obtained detailed clinical information, including family histories, clinical courses, and physical findings. Between 2016 and 2018, 51 patients showed clinical histories of hemolytic anemia associated with/without morphological abnormalities in red blood cells according to routine laboratory examinations and were enrolled as the subjects of this study.
In most of the patients, when possible, we first performed additional chemical tests, including the acidified glycerol hemolysis time test, the flow-cytometric osmotic fragility (FCM-OF) test, and the eosin-5′-maleimide (EMA) binding test with a negative direct antiglobulin test as per previously reported methods4,5,6,7,8.
Genomic DNA was extracted from the patients’ peripheral blood using a QIAamp DNA extraction kit according to the manufacturer’s instructions (QIAGEN, Hilden, Germany). The Haloplex HS target enrichment system (Agilent Technologies, Santa Clara, CA, USA) was used as the target panel. The target panel was designed using SureDesign (https://earray.chem.agilent.com/suredesign/home.htm) to include all coding exons and intron-exon boundaries of 74 possible candidate genes (Supplementary Table S2). Massive parallel sequencing was performed using the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). Raw data were aligned to the GRCh37/hg19 human genome. The generated FASTQ files were imported into SureCall ver3.5 (Agilent Technologies) for variant calling.
The obtained variants were filtered according to the following strategy: (1) variant frequencies were below 1% in 1000G_EAS and ALL (1000 Genomes), HGDV, and dpSNP; (2) synonymous variants were excluded (nonsynonymous variants, variants associated with a frameshift, insertion/deletion variants, and variants in splicing donor/acceptor sites were included); (3) variants with allele frequencies of less than 30% within the total read depth were excluded; and (4) the CADD_phred value was higher than 20 if obtained. Variant information obtained through wANNOVAR (http://wannovar.wglab.org/) was used for curation. The Integrative Genomics Viewer (IGV; https://software.broadinstitute.org/software/igv/) was used for visual evaluation. The final conclusion was reached following the American College of Medical Genetics and Genomics (ACMG) guidelines9.
The effect of the splicing site variants was evaluated with in silico software using the Berkeley Drosophila Genome Project (https://www.fruitfly.org/seq_tools/splice.html) and DTU Bioinformatics (http://www.cbs.dtu.dk/services/NetGene2/) databases.
Results
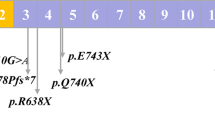
The 13 variants identified in the genes related to SPH1-5 (ANK1, SPTB, SPTA1, SLC4A1, and EPB42), together with the clinical data and laboratory test results, are summarized in Table 1. Among the 51 subjects, eight patients (patients 1, 3, 4, 7, 8, 9, 10, and 12) were primarily and clinically suspected of having HS and showed HS-related variants, indicating a 100% detection ratio. On the other hand, HS was primarily not suspected in five other patients who showed HS-related variants.
Although two variants identified in patients 6 and 10 were evaluated as “variants of uncertain significance (VUS)” in accordance with ACMG guidelines, the prediction scores for the variant in patient 6 suggested “damage”, and the variant in patient 10 was quite unique. Thus, we considered these variants to likely to be related to disease occurrence. Seven variants were novel and were not included in any database. Among these variants, the variant in patient 12 was similar to that reported by Dhermy et al.10. Three of the variants have been previously reported11,12,13. Four variants were observed in the dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), and one of them was evaluated as likely pathogenic in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
The variant identified in patient 4 (c.426 + 4_426 + 7del) has not been reported previously but was found to be similar to the Ankyrin Shiga variant (c.426 + 3_426 + 4insA). Through in silico analyses using two different websites, this insertion was predicted to cause the loss of the donor site (Supplementary Figs. S1, S2). Thus, we concluded that this variant was the likely cause underlying abnormal splicing.
Patient 13 harbored a homozygous EPB42 variant. Since the parents of patient 13 are first cousins, it is suspected that both parents are heterozygous carriers. Patient 11 also showed a homozygous splicing variant in SPTA1; however, consanguinity was not found in this patient’s family history.
Among the 38 patients who showed no pathogenic variants in the five genes, 20 patients were analyzed with the FCM-OF test, and only 5 patients showed low values (data not shown).
Discussion
Inoue et al. analyzed the genetic backgrounds of Japanese HS patients using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and reported band 3 deficiency (SPH4) in 32% of the patients, spectrin deficiency (corresponding to SPH2 and SPH3) in 15%, protein 4.2 deficiency (SPH5) in 6%, ankyrin deficiency (SPH1) in 2%, combined deficiency in 36%, and no abnormality in 9%14. In contrast, Yawata et al. analyzed 60 Japanese patients with HS using a similar method and detected protein 4.2 deficiency (SPH5) in 45% of the patients, band 3 deficiency (SPH4) in 20%, and ankyrin deficiency (SPH1) in 7%, while 28% of the patients had an unknown etiology15,16. Genetic variants in ANK1 were analyzed by Nakanishi et al.12, who identified 16 variants in 49 patients with HS, suggesting that ANK1 variants (SPH1) are not rare in the Japanese population.
Remarkable progress has been made in genomic analyses in the last decade. Next-generation sequencing (NGS) is being extensively used in this field. This has helped to expand our understanding of the genetic heterogeneity associated with HS17. Some studies have demonstrated the usefulness of the targeted NGS approach for the investigation of specific subtypes of patients with hemolytic anemia18,19,20. Exome sequencing has also been applied to hereditary hemolytic anemia21,22.
To confirm ethnic differences in HS-related variants, Choi et al. reviewed the available reports regarding HS-related mutations in comparison with the results of the present study20. In reports from the United States, SPTA1 mutation (SPH3) was found to be the most common23. A study in the Netherlands revealed that SPTA1, ANK1, and SPTB (SPH3, SPH1, and SPH2) were ranked as the top three genes with identified variants13. In Korea, SPTB mutation (SPH2) was found to be the most common, followed by ANK1 mutation (SPH1)20. Another study in Korea reported that 25 patients with HS carried mutations in ANK1 (SPH1; n = 13) or SPTB (SPH2; n = 12)24. A study from China reported that among 23 patients, 13 mutations were observed in ANK1 (SPH1), while 10 mutations were observed in SPTB (SPH2)25. Other studies have reported similar observations26,27,28. The distribution of the variants is summarized in Supplementary Table S3.
In this study, ANK1 variants (SPH1) were found to be the most common, being found in 46% (6/13) of the patients. SPTB variants (SPH2) were identified in 31% (4/13) of the patients. Thus, the distribution of the variants was similar to those observed in other Asian countries but was different from those observed in non-Asian countries. Previously, EPB42 (SPH5) was considered the most common subtype. However, the distribution of HS-related variants observed in this study was different from that identified in a previous study on Japanese patients with HS15,16. The reason for this difference is unknown; however, the total number of samples examined in the present study is too small to be compared with this previous study. Thus, the analysis of more samples is necessary to better understand the genetic basis of HS in Japanese patients.
As mentioned above, ANK1 (SPH1), SPTB (SPH2), and SLC4A1 (SPH4) are related to AD traits, whereas SPTA1 (SPH3) and EPB42 (SPH5) are related to AR traits. In this study, variants in AD-related genes were found in 11 patients (85%). As six patients (46%) showed a positive family history, the identified variants were considered to be inherited from the affected ancestors. In comparison, five patients with variants in AD-related genes (45%) had no family history, and it remains unknown whether the identified variants occurred de novo or if they were inherited from nonsymptomatic parents. This is a limitation of this study, as parental analysis was not conducted.
We did not find any genotype-phenotype correlation. The observed severities of the clinical and laboratory findings were variable, even within the subgroups classified in accordance with the gene variants. Similar findings have been reported previously23,29. Phenotypic variabilities have been reported in a pair of twins with de novo ANK1 missense variants30. Thus, even though we found no clear genotype-phenotype correlation, our results are not contradictory to those reported previously.
Regarding laboratory testing, it is difficult to detect HS using only one method because its clinical phenotypes are widely variable. Therefore, more than one test is generally recommended31. Previously, the osmotic fragility test was considered to be the gold standard for HS diagnosis32. In this study, we found that only the results obtained from FCM-OF matched the results of TCS in this study; all patients with pathogenic HS variants who underwent the FCM-OF test showed low FCM-OF values, but only 5 of 20 patients without HS variants showed low values in the FCM-OF test. This indicates that the FCM-OF test presents high sensitivity but low specificity. Because the combination of the FCM-OF and EMA tests can correctly diagnose 100% of patients33, FCM-OF may be the best possible single test for the diagnosis of HS, followed by EMA.
In this study, 38 patients showed no pathogenic variants in the five analyzed genes related to HS. However, we cannot completely deny the possibility of those variants in the analyzed five genes may have been overlooked due to analytical limitations, even though we conducted a comprehensive genomic analysis using next-generation sequencing. Because hemolytic anemia is a heterogeneous entity and the clinical diagnosis of hereditary hemolytic anemia is often inaccurate due to overlapping phenotypes18, variable genomic backgrounds are suspected to exist in patients without pathogenic variants in known HS genes. Therefore, molecular diagnosis may help to predict the future prognosis of young patients with HS, along with genetic counseling29. Comprehensive genomic analysis would be useful and recommended, especially for patients without a detailed family history and patients with chronic hemolytic anemia receiving frequent blood transfusions.
References
Zamora E. A., Schaefer C. A. Hereditary Spherocytosis. In: StatPearls. Treasure Island (FL): StatPearls Publishing, Copyright © 2021, StatPearls Publishing LLC.; 2021.
Ciepiela, O. Old and new insights into the diagnosis of hereditary spherocytosis. Ann. Transl. Med. 6, 339 (2018).
Perrotta, S., Gallagher, P. G. & Mohandas, N. Hereditary spherocytosis. Lancet 372, 1411–1426 (2008).
Won, D. I. & Suh, J. S. Flow cytometric detection of erythrocyte osmotic fragility. Cytom. B Clin. Cytom. 76, 135–141 (2009).
Ciepiela, O., Adamowicz-Salach, A., Zgodzińska, A., Łazowska, M. & Kotuła, I. Flow cytometric osmotic fragility test: Increased assay sensitivity for clinical application in pediatric hematology. Cytom. B Clin. Cytom. 94, 189–195 (2018).
Zanella, A. et al. Acidified glycerol lysis test: a screening test for spherocytosis. Br. J. Haematol. 45, 481–486 (1980).
King, M. J. et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int. J. Lab. Hematol. 37, 304–325 (2015).
King, M. J. et al. Using the eosin-5-maleimide binding test in the differential diagnosis of hereditary spherocytosis and hereditary pyropoikilocytosis. Cytom. B Clin. Cytom. 74, 244–250 (2008).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Dhermy, D. et al. Heterogenous band 3 deficiency in hereditary spherocytosis related to different band 3 gene defects. Br. J. Haematol. 98, 32–40 (1997).
Bouhassira, E. E. et al. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood 79, 1846–1854 (1992).
Nakanishi, H., Kanzaki, A., Yawata, A., Yamada, O. & Yawata, Y. Ankyrin gene mutations in japanese patients with hereditary spherocytosis. Int. J. Hematol. 73, 54–63 (2001).
van Vuren, A. et al. The complexity of genotype-phenotype correlations in hereditary spherocytosis: a cohort of 95 patients: genotype-phenotype correlation in hereditary spherocytosis. Hemasphere 3, e276 (2019).
Inoue, T. et al. Uniquely higher incidence of isolated or combined deficiency of band 3 and/or band 4.2 as the pathogenesis of autosomal dominantly inherited hereditary spherocytosis in the Japanese population. Int. J. Hematol. 60, 227–238 (1994).
Yawata, Y. et al. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int. J. Hematol. 71, 118–135 (2000).
Yawata, Y., Kanzaki, A., Yawata, A., Nakanishi, H. & Kaku, M. Hereditary red cell membrane disorders in Japan: their genotypic and phenotypic features in 1014 cases studied. Hematology 6, 399–422 (2001).
Andolfo, I., Russo, R., Gambale, A. & Iolascon, A. New insights on hereditary erythrocyte membrane defects. Haematologica 101, 1284–1294 (2016).
Russo, R. et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol. 93, 672–682 (2018).
Del Orbe Barreto, R. et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int. J. Lab. Hematol. 38, 629–638 (2016).
Choi, H. S. et al. Molecular diagnosis of hereditary spherocytosis by multi-gene target sequencing in Korea: matching with osmotic fragility test and presence of spherocyte. Orphanet. J. Rare Dis. 14, 114 (2019).
Wang, R. et al. Exome sequencing confirms molecular diagnoses in 38 Chinese families with hereditary spherocytosis. Sci. China Life Sci. 61, 947–953 (2018).
Shen, H., Huang, H., Luo, K., Yi, Y. & Shi, X. Two different pathogenic gene mutations coexisted in the same hereditary spherocytosis family manifested with heterogeneous phenotypes. BMC Med. Genet. 20, 90 (2019).
Agarwal, A. M. et al. Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. Br. J. Haematol. 174, 806–814 (2016).
Park, J. et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin. Genet. 90, 69–78 (2016).
Wang, X. et al. Genetic and clinical characteristics of patients with hereditary spherocytosis in Hubei Province of China. Front. Genet. 11, 953 (2020).
Lin, P. C. et al. Whole-exome sequencing for the genetic diagnosis of congenital red blood cell membrane disorders in Taiwan. Clin. Chim. Acta 487, 311–317 (2018).
Mansour-Hendili, L. et al. Exome sequencing for diagnosis of congenital hemolytic anemia. Orphanet J. Rare Dis. 15, 180 (2020).
Xie, F. et al. Clinical manifestation and phenotypic analysis of novel gene mutation in 28 Chinese children with hereditary spherocytosis. Mol. Genet. Genom. Med. 9, e1577 (2021).
Tole, S. et al. Genotype-phenotype correlation in children with hereditary spherocytosis. Br. J. Haematol. 191, 486–496 (2020).
Smith, A. R. et al. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol. Aging 74, 70–76 (2019).
Farias, M. G. Advances in laboratory diagnosis of hereditary spherocytosis. Clin. Chem. Lab. Med. 55, 944–948 (2017).
Xue, J., He, Q., Xie, X., Su, A. & Cao, S. Clinical utility of targeted gene enrichment and sequencing technique in the diagnosis of adult hereditary spherocytosis. Ann. Transl. Med. 7, 527 (2019).
Arora, R. D. et al. Flow cytometric osmotic fragility test and eosin-5′-maleimide dye-binding tests are better than conventional osmotic fragility tests for the diagnosis of hereditary spherocytosis. Int. J. Lab. Hematol. 40, 335–342 (2018).
Acknowledgements
We appreciate the cooperation of the patients and their families. This work was supported by grants received from the Ministry of Health, Labor, and Welfare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, K.S., Utshigisawa, T., Ogura, H. et al. Clinical and genetic diagnosis of thirteen Japanese patients with hereditary spherocytosis. Hum Genome Var 9, 1 (2022). https://doi.org/10.1038/s41439-021-00179-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00179-1
This article is cited by
-
De novo variations of ANK1 gene caused hereditary spherocytosis in two Chinese children by affecting pre-mRNA splicing
BMC Pediatrics (2023)
-
Genetic mutation analysis of hereditary spherocytosis in Guangxi Zhuang Autonomous Region
Journal of Hematopathology (2023)
-
Targeted next-generation sequencing identifies novel deleterious variants in ANK1 gene causing severe hereditary spherocytosis in Indian patients: expanding the molecular and clinical spectrum
Molecular Genetics and Genomics (2023)
-
Variant spectrum of PIEZO1 and KCNN4 in Japanese patients with dehydrated hereditary stomatocytosis
Human Genome Variation (2023)
