
Abstract
Bone dysplasias are a group of rare hereditary diseases, with up to 436 disease types. Perinatal diagnosis is clinically important for adequate personalized management and counseling. There are no reports focused on pathogenic variants of bone dysplasias in the general population. In this study, we focused on autosomal recessive bone dysplasias. We identified pathogenic variants using whole-genome reference panel data from 3552 Japanese individuals. For the first time, we were able to estimate the carrier frequencies and the proportions of potential patients. For autosomal recessive bone dysplasias, we detected 198 pathogenic variants of 54 causative genes. We estimated the variant carrier frequencies and the proportions of potential patients with variants associated with four clinically important bone dysplasias: osteogenesis imperfecta (OI), hypophosphatasia (HPP), asphyxiating thoracic dysplasia (ATD), and Ellis–van Creveld syndrome (EvC). The proportions of potential patients with OI, ATD, and EvC based on pathogenic variants classified as “pathogenic” and “likely pathogenic” by InterVar were closer to the reported incidence rates in Japanese subjects. Furthermore, the proportions of potential patients with HPP variants classified as “pathogenic” and “likely pathogenic” in InterVar and “pathogenic” in ClinVar were closer to the reported incidence rates. For bone dysplasia, the findings of this study will provide a better understanding of the variant types and frequencies in the Japanese general population, and should be useful for clinical diagnosis, genetic counseling, and personalized medicine.
Similar content being viewed by others

Introduction
Bone dysplasias are a group of inherited disorders caused by mutations in genes affecting the development and differentiation of bones and cartilage from the fetal stage, thereby resulting in abnormalities in skeletal shape and structure1. The clinical manifestations include a wide range of prognoses, from cases diagnosed during the fetal period to mild cases in adults that are difficult to diagnose2,3. The birth incidence of bone dysplasia is estimated to be ~1/5000 births4,5. The international classification of bone diseases involves a combination of single genes or multiple related genes, specific phenotypes, and radiological findings, and is updated every 4–5 years6. The classification from 2015 comprises 42 groups of bone dysplasias and 436 specific diseases6. Thus, the actual frequency of each disease is low. In addition, new genes and genetic variants are continually being reported due to developments in next-generation sequencing (NGS)7,8, and the number of causative genes and variants are expected to increase.
Determining the incidence of bone dysplasia and its diagnosis can be difficult. First, there have been few reports on the incidence of bone dysplasia in Western countries since the 1980s. The major reports were from Italy4 and South America5. In both reports, the incidence of each bone dysplasia was calculated as the sum of live births and stillbirths in a certain region or multiple countries over a period of several years. However, no survey of bone dysplasia incidence has been reported since that time. Furthermore, the trend in disease incidence according to racial differences has not been clarified. Second, there has been no nationwide survey of the incidence of bone dysplasia in Japan. Satoh et al.9 (article in Japanese) reported the prevalence of fetal bone dysplasia in obstetrics, which included children diagnosed at a single institution for 20 years and those registered at multiple institutions for 1 year. However, there was a large bias in the target population selection and the total number of patients was not reported. There have been no subsequent reports on the frequencies of bone dysplasia in Japan. As bone dysplasia is a rare hereditary disease, it is difficult to grasp the total number of cases via nationwide efforts and the target population setting is unclear even if the patients are registered at individual institutions. Third, new causative genes and variants predicting the diagnosis of bone dysplasia are continually being identified by NGS and confirmed for their utility in diagnosis7,8. NGS is useful for the diagnosis and discovery of causative genes and variants. However, estimated causative variants and carrier frequencies of genes related to bone dysplasia have not been reported in the general population. Carriers of disease-causing genetic variants may exist in the healthy general population. Therefore, using genomic information from the general population to investigate genetic variations and determine the frequency of mutation carriers and potential patients is helpful in understanding genetic epidemiology and applying it to perinatal care, genetic counseling, and personalized medicine.
We used 3.5KJPNv210,11, a genome-wide allele-frequency reference panel, to estimate the carrier frequencies of variants associated with bone dysplasia in the Japanese population. In collaboration with the Tohoku Medical Megabank Organization and Iwate Tohoku Medical Megabank Organization, we conducted cohort studies using a biobank that integrates medical and genomic information from the general healthy population. Whole-genome sequences were analyzed in samples collected from 3552 of these healthy individuals to produce a whole-genome reference panel, 3.5KJPNv2. Allele frequency, genotype frequency, and allele count information have been made partially available to the public (https://jmorp.megabank.tohoku.ac.jp/201905).
The aims of this study were to detect genetic variants responsible for autosomal recessive bone dysplasia using 3.5KJPNv2 and to estimate the frequencies of carriers and potential patients with genetic factors among Japanese individuals.
Materials and methods
This study was conducted after obtaining approval from the Ethics Committee of Tohoku Medical Megabank Organization of Tohoku University (authorization numbers: 2018-4-038) and written consent from all participants.
Causative genes of bone dysplasia
We obtained information regarding bone dysplasia registered in the Surveillance Registry for Bone Dysplasia of the Japanese Orthopedic Association (https://www.joa.or.jp/)12. A total of 7234 cases were registered between 1990 and 2016, and were classified into 217 different diseases. The top 100 diseases with the greatest number of cases in the surveillance registry were selected, among which 30 bone dysplasias showing autosomal recessive inheritance, excluding those with autosomal dominant and X-linked inheritance, were investigated. In this study, 73 causative genes of these 30 autosomal recessive bone dysplasias were selected from among the genes registered in Nosology and Classification of Genetic Skeletal Disorders: 2015 Revision6 (see Table 1 for disease–gene pairs with MIM numbers). The causative genes were unknown for mesomelic dysplasia and Pyle disease among the 30 bone dysplasias. We analyzed in detail the genetic variants associated with osteogenesis imperfecta (OI), hypophosphatasia (HPP), Ellis–van Creveld syndrome (EvC), and asphyxiating thoracic dysplasia (ATD) that were reported to be prevalent in fetal bone dysplasia in obstetrics in Japan9. The incidence rates of these 4 diseases were obtained from a report of 448,069 patients9. The 95% confidence interval for the incidence rates was calculated based on the binomial distribution.
Genetic variants and annotation
We used the 3.5KJPNv2 whole-genome reference panel10 and the variant annotations from our previous study13 with subsequent updates, in which diallelic variants (after VQSR filtering) in 3.5KJPNv2 were annotated using Annovar14, InterVar (2.0.1)15, ClinVar (March 2019)16, and the Human Gene Mutation Database professional version (HGMD) (2019.1)17. InterVar is a bioinformatics tool based on the American College of Medical Genetics and Genomics (ACMG) - the Association for Molecular Pathology (AMP) variant interpretation guidelines18. In this study, InterVar was run using the default options and the 18 criteria for variant interpretation were used for primary interpretation.
Classification of the genetic variants
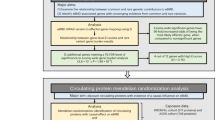
In the same way as in the previous study13, we evaluated variants in 73 genes. First, we obtained primary interpretation by InterVar and the variants were classified into five classes: pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB), and benign (B). Then, the pathogenic variants were examined by four different inclusion criteria (set 1–set 4)13 with a threshold of allele frequency (≤0.03) and correspondence to reported pathogenic variants in ClinVar and HGMD (Fig. 1).

See “Materials and Methods” and the previous report13. The variants interpreted most conservatively as P or LP by InterVar and reported as P or LP in ClinVar were categorized into set 1. Set 2 comprises all P or LP variants, including those “reported” or “predicted (unreported).” The variants interpreted as VUS, with an MAF ≤ 0.03, or reported as P or LP in ClinVar were combined with the variants in set 2 to form set 3. Finally, set 4 comprises a combination of set 3 variants and the variants reported as disease-causing mutations (DMs) in HGMD.
Estimation of the population frequency of risk alleles and expected carriers
We estimated the risk allele and carrier frequencies based on the detected pathogenic variants. First, supposing that there are n pathogenic variants of a gene, we calculated the sum of the risk allele frequencies at n sites as the estimated population frequency of pathogenic alleles of that gene (Q). It was assumed that the probability of having a risk allele for the disease in the haploid genome of a population was Q and that of not having the risk allele was 1 − Q. The estimated carrier frequency was then calculated as 2 × (1 − Q) × Q based on Hardy–Weinberg equilibrium. We also calculated Q2 as the expected frequency of individuals having pathogenic variants in both chromosomes of each gene (termed the “proportion of homozygotes” in this paper) and estimated the proportion of potential patients by summing the value of Q2 for the same disease.
Statistical analysis
A comparison of allele frequencies was performed using Excel 2016 (Microsoft, Redmond, Washington, USA). We used Fisher’s exact test to compare the allele frequencies in 3.5KJPNv2 with previously reported frequencies in the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/) for Finnish (gnomAD FIN) and non-Finnish European (gnomAD NFE) individuals. The threshold for significance was p < 0.05.
Results
Based on the annotation and interpretation of variants using InterVar, 82,818 genetic variants (from the 5 classifications [P, LP, VUS, LB, and B]) of 73 genes associated with 30 bone dysplasias were extracted (Fig. 1). Next, we selected potential pathogenic variants from sets 1–4 based on filtering those with a minor allele frequency (MAF) ≤ 0.03, evaluating the output of InterVar, and matching with classifications provided in ClinVar and HGMD. The total number of variants classified in sets 1–4 was 198 within 54 genes (Fig. 1). Thus, potentially pathogenic variants were detected in 54 of the 73 total genes associated with bone dysplasia in 3.5KJPNv2 but not in the remaining 19 genes (XYLT1, DYM, RAB33B, DMP1, SLC34A3, MMP9, SBDS, GNS, CREB3L1, PPIB, SP7, WNT1, PLEKHM1, SNX10, TNFRSF11, TNFSF11, DDR2, HES7, and MESP2) (Table 2 and Supplementary Table S1). No variant with an MAF ≤ 0.03 and classified as VUS was found (0 variant).
After the detection of pathogenic variants, we examined the status of individuals using individual genotype data from 3.5KJPNv2. Of note, we checked whether there was any compound heterozygote or homozygote for a single gene. There were two exceptional individuals with multiple pathogenic variants in single genes and we manually inspected the cases of these two individuals. One individual had seven predicted pathogenic variants of the RECQL4 gene. The seven variants are indels located in a short region (67 bp, Chr7:145737572–145737639) and all were singletons. Through the inspection of the status of mapped reads in BAM files (binary format of sequence alignment map)10 using the Integrative Genomic Viewer, we found that variant calls by HaplotypeCaller were not successful for this local region in the individual. Thus, we did not use these variants in further analysis of the estimation of frequency. Another individual had 2 predicted pathogenic variants in the BMP1 gene. These two variants are 1 bp (A) deletions at very close (3 bp) genomic sites (chr8:22034590 and chr8:22034593). They were singleton variants and were located on the same chromosome of the individual. Thus, these two variants could be a single variant. After corrections based on these two exceptional cases, the proportions (and number) of individuals having at least one pathogenic variant were 1.89% (67), 6.28% (222), 6.95% (247), and 15.0% (534) for sets 1–4, respectively. The number of individuals having 2 pathogenic variants was 1, 7, 7, and 25 for sets 1–4, respectively, and 2 individuals in set 4 had 3 pathogenic variants.
The carrier frequencies were estimated based on Hardy–Weinberg equilibrium using the allele frequencies of the variants associated with OI, EvC, ATD, and HPP (Tables 2 and 3). We calculated the expected frequency of individuals with homozygous variants from the estimated carrier frequency, estimated the proportions of potential patients, and compared them with the reported frequency in Japan (Table 4).
Osteogenesis imperfecta
BMP1 (MIM# 614856)
A stopgain variant, p.Glu547*, and a splicing variant, c.2826 + 2 T > C, were automatically classified as P by InterVar. A frameshift variant, p.His223fs (p.Glu224fs), was automatically classified as LP by InterVar. The remaining six nonsynonymous SNVs were automatically classified as LP by InterVar. One individual was heterozygous for all ten variants and the allele frequency was 0.00014 for each variant (Table 3). None had been reported in ClinVar or HGMD and, thus, these variants were included in our set 2 (Fig. 1). The carrier frequency of the BMP1 variants was estimated to be 0.00253 in set 2 (Table 2).
CRTAP (MIM# 610682)
A splicing variant, c.621 + 1 G > A, was interpreted as P by InterVar and included in set 2, although it is not reported in ClinVar or HGMD (Table 3). The allele frequency was 0.00028 and the carrier frequency of CRTAP was estimated to be 0.00056 in set 2 (Table 2).
LEPRE1 (P3H1) (MIM# 610915)
A stopgain variant, p.Gln576*, and a nonsynonymous single nucleotide variant (SNV), p.Ala614Thr, were detected in one heterozygous individual, each with an allele frequency of 0.00014, and these variants were interpreted as P by InterVar and thus included in set 2 (Table 3). A nonsynonymous SNV, p.Ala614Thr, was interpreted as LP by InterVar and thus included in set 2 (Table 3). The estimated carrier frequency of LEPRE1 was 0.00056 in set 2 (Table 2).
FKBP10 (MIM # 610968)
A nonsynonymous SNV, p.Glu575Gln, of FKBP10 was interpreted as LP by InterVar and included in our set 2, although it is not reported in ClinVar or HGMD (Table 3). The allele frequency was 0.00014 and the carrier frequency was 0.00028 in set 2 (Table 2).
SERPINH1 (MIM# 613848)
All eight variants were nonsynonymous variants and were interpreted as LP by InterVar, and thus included in set 2; none are reported in ClinVar or HGMD (Table 3). Of the eight nonsynonymous variants, all were heterozygous, with p.Thr227Ile, p.Val256Met, p.Arg339Leu, and p.Ala354Thr identified in one individual, each with an allele frequency of 0.00014; p.Ser77Leu was identified in nine individuals, with an allele frequency of 0.00127; p.Val219Met and p.Asn244Ile was identified in two individuals, each with an allele frequency of 0.00028; and p.Ala349Val was identified in five individuals, with an allele frequency of 0.0007. The carrier frequency was 0.00618 in set 2 (Table 2).
PLOD2 (MIM# 609220)
Two stopgain variants, p.Arg495* and p.Arg54*, were not reported in ClinVar or HGMD and were interpreted as P by InterVar and included in our set 2 (Table 3). These variants were found in one heterozygous individual. The allele frequency was 0.00014 and the carrier frequency was 0.00056 in set 2 (Table 2).
SERPINF1 (MIM# 613848)
A splicing variant, c.284-2 A > G, was detected in five heterozygous individuals with an allele frequency of 0.0007. This variant was interpreted as P by InterVar. However, it was not reported in ClinVar or HGMD (Table 3). A nonsynonymous SNV, p.Ala56Gly, was heterozygous in 86 individuals with an allele frequency of 0.01239, which is much higher than that reported in gnomAD in European subjects (p < 0.001, Table 3). This variant was reported in HGMD, so it was included in our set 4. The carrier frequencies were 0.00141 in set 2 and 0.02584 in set 4 (Table 2).
SEC24D (MIM# 616295)
A stopgain variant, p.Gln302*, was identified in one heterozygous individual in 3.5KJPNv2 and was interpreted as P by InterVar, and thus included in our set 2 (Table 3). The allele frequency was 0.00014 and the carrier frequency was 0.00028 in set 2 (Table 2).
Of the 12 causative genes registered in the international classification of autosomal recessive OI, 4 genes (PPIB, WNT1, CREB3L1, and SP7) were not recognized as pathogenic variants in this study (Tables 2 and 3). Except for p.Ala56Gly in SERPINF1, none of the pathogenic variants of the eight genes detected in this study was registered in ClinVar or HGMD. They were pathogenic variants detected for the first time in association with OI in this study. Most of the 12 OI-associated variant genes were included in set 2 and p.Ala56Gly was included in set 4, with a total carrier frequency of 0.01238 (set 2) and 0.03681 (set 4). The expected proportions of potential patients calculated by allele frequencies were 1/20,967 (set 2) and 1/4474 (set 4) (Table 4).
Hypophosphatasia
Thirteen pathogenic variants of ALPL (MIM# 241500) were detected in 3.5KJPNv2. Two nonsynonymous SNVs, p.Arg136His and p.Phe327Leu, were classified as LP by InterVar and reported as P in ClinVar and DM in HGMD. Thus, they were included in our set 1 (Table 3 and Fig. 1). The allele frequency of p.Arg136His was 0.00028 and this variant was identified in 2 heterozygous individuals, whereas the allele frequency of p.Phe327Leu was 0.00225 and this variant was identified in 16 individuals. Three nonsynonymous SNVs, p.Ala33Gly, p.Pro261Gln, and p.Ile395Val, were included in our set 2, because they were classified as LP by InterVar but not reported in ClinVar or HGMD (Table 3). Two variants, p.Ala33Gly and p.Ile395Val, were identified in one heterozygous individual and p.Pro261Gln was detected in two individuals. A frameshift variant, c.1559delT, was reported as P in ClinVar and DM in HGMD but classified as VUS by InterVar. This variant was classified in our set 3 and it was identified in 12 heterozygous individuals. Seven nonsynonymous SNVs, p.Met62Val, p.Ala177Thr, p.Glu191Gly, p.His341Arg, p.Gly420Ser, p.Gly426Ser, and p.Tyr436Cys, were classified as VUS by InterVar and DM in HGMD. These variants were classified in our set 4. Six variants, p.Arg136His, p.Glu191Gly, p.Phe327Leu, p.His341Arg, p.Gly426Ser, and c.1559delT, have been reported in the Japanese population19,20. Among these variants, p.Phe327Leu and c.1559delT were identified in 16 and 12 heterozygous individuals, respectively, and p.Ala177Thr was identified in 116 heterozygous individuals, with an allele frequency of 0.01689, which is much higher than that in gnomAD in European subjects (p < 0.001, Table 3). The carrier frequencies estimated from the sum of the allele frequencies were 0.00506 (set 1), 0.00618 (set 2), 0.00953 (set 3), and 0.04404 (set 4) (Table 2). The expected proportions of potential patients were 1/39,132 (set 1), 1/15,697 (set 2), 1/6473 (set 3), and 1/1511 (set 4) (Table 4).
Ellis–van Creveld syndrome
EVC (MIM# 225500)
One individual was heterozygous for the splicing variant c.1887-1 G > C, which was not reported in ClinVar or HGMD. This variant was interpreted as P by InterVar; thus, we included it in set 2 (Table 3 and Fig. 1). The allele frequency was 0.000141. Although two nonsynonymous variants, p.Thr295Ser and p.Leu328Phe, were considered DM in HGMD, they were interpreted as VUS by InterVar; thus, we included them in set 4. Both variants were identified in genetic testing of patients with fetal limb shortening21. We identified 2 and 7 heterozygous individuals for p.Thr295Ser and p.Leu328Phe, respectively. The allele frequencies of p.Thr295Ser and p.Leu328Phe were 0.00028 and 0.000985, respectively, which were higher than those in gnomAD in European subjects (p < 0.05). The carrier frequencies were 0.00028 in set 2 and 0.00281 in set 4 (Table 2).
EVC2 (MIM# 225500)
A stopgain variant, p.Arg399*, was heterozygous in two individuals. This variant was reported as P both by InterVar and in ClinVar but as DM in HGMD; we included it in set 1 (Table 3). This variant was detected in stillborn children with a ventricular septal defect and limb shortening with polydactyly22. A stopgain variant, p.Trp828*, was detected as heterozygous in one individual. Because it was reported as P by InterVar, as DM in HGMD and as LP in ClinVar, we included it in set 1. This was reported in genetic testing of a boy with distal limb shortening and polydactyly23. Two splicing variants, c.1230 + 1 G > C and c.906-2 A > C, were interpreted as P by InterVar and categorized in set 2, although they were not reported in ClinVar or HGMD. They were detected as heterozygous in one individual each and the allele frequencies of both were 0.00014. Two frameshift variants, p.Glu846fs and p.Asn361fs, were interpreted as LP by InterVar and included in set 2, although neither is reported in ClinVar or HGMD. Each was heterozygous in one individual, with an allele frequency of 0.00014. A stop codon, p.Arg698*, was included in set 2 according to InterVar and identified as heterozygous in one individual. A nonsynonymous variant, p.Arg950Trp, was reported as DM in HGMD but as VUS in ClinVar and InterVar by default. Therefore, p.Arg950Trp was included in set 4. This variant was identified in one heterozygous individual, with an allele frequency of 0.00014. The carrier frequencies of EVC2 were estimated to be 0.00085 in set 1, 0.00225 in set 2, and 0.00253 in set 4 (Table 2).
The sums of the estimated carrier frequencies of EVC and EVC2 were 0.00085 (set 1), 0.00254 (set 2), and 0.00535 (set 4). The proportions of potential patients calculated by allele frequencies were 1/1,398,385 (set 1), 1/193,890 (set 2), and 1/69,771 (set 4) (Table 4).
Asphyxiating thoracic dysplasia
DYNC2H1 (MIM# 613091)
A frameshift variant, p.Glu1894fs, and a stopgain variant, p.Arg3349* was classified in set 1 because they were interpreted as P by InterVar and reported as P in ClinVar (Table 3 and Fig. 1). Three individuals were identified to be heterozygous for p.Glu1894fs and 1 individual was identified to be heterozygous for p.Arg3349*. A splicing variant, c.11277 + 1 G > A, which was interpreted as P by InterVar, and three frameshift variants, p.Val1840fs, p.Lys2688fs, and p.Arg3326fs, which were interpreted as LP by InterVar, were classified in set 2. However, they were not reported in ClinVar or HGMD. Each was identified in one heterozygous individual. A nonsynonymous variant, p.Arg3004Cys, was identified in 4 heterozygous individuals. This variant was reported as DM in HGMD and was identified in a Japanese patient with prominent shortening of the ribs and extremities, evident in the fetal period24. This variant was labeled VUS by InterVar and as DM in HGMD; it was included in set 4. The carrier frequencies of DYNC2H1 were 0.00113 in set 1, 0.00225 in set 2, and 0.00337 in set 4 (Table 2).
WDR34 (MIM# 615633)
A splicing variant, c.1372 + 2 T > C, was identified as heterozygous in one individual. This variant was not reported in ClinVar or HGMD but was interpreted as P by InterVar and classified in set 2 (Table 3). The estimated carrier frequency was 0.00028 (Table 2).
IFT80 (MIM# 611263)
A stopgain variant, p.Ser134*, found in seven heterozygous individuals; two splicing variants, c.371-1 G > C, found in six heterozygous individuals and c.40-1 T > C, found in one heterozygous individual were classified as P by InterVar but were not reported in ClinVar or HGMD (Table 3). The carrier frequency was 0.00393 in set 2 (Table 2).
IFT172 (MIM# 615630)
All five variants of IFT172 result in a loss of function: p.Arg271* results in a stopgain variant; c.3229-1 G > C, c.2116-1 G > A, and c.571-1 G > A are splicing variants; and p.Thr663fs results in a frameshift variant. InterVar interpreted p.Arg271*, c.3229-1 G > C, c.2116-1 G > A, and c.571-1 G > A as P and p.Thr663fs as LP. p.Arg271* was included in set 1, and the remaining variants were included in set 2 (Table 3). One individual was heterozygous for all five variants, and the estimated carrier frequencies were 0.00028 in set 1 and 0.00141 in set 2 (Table 2).
IFT140 (MIM# 266920)
Two stopgain variants, p.Gln998* and p.Arg1072*, one frameshift variant, p.Tyr923fs, and two synonymous SNVs, p.Thr1394Thr and p.Gly163Gly, were identified. None of these variants are reported in ClinVar. In InterVar, p.Gln998* was interpreted as P and p.Arg1072* and p.Tyr923fs were interpreted as LP, and thus included in set 2. Two variants, p.Thr1394Thr and p.Gly163Gly, were reported as DM in HGMD but not in InterVar and ClinVar; thus, they were included in set 4 (Table 3). All variants were found in one heterozygous individual each and the carrier frequencies were 0.00085 in set 2 and 0.00141 in set 4 (Table 2).
WDR19 (MIM# 614376)
One frameshift variant, p.Leu211fs, and two splicing variants, c.2165 + 1 G > T and c.2782-2 A > G, were interpreted as P, and one nonsynonymous SNV, p.Gly789Arg, was identified as LP by InterVar. Only p.Leu211fs was included in set 1 and the remaining three pathogenic variants were included in set 2 (Table 3). The carrier frequencies were 0.00056 in set 1 and 0.00169 in set 2 (Table 2).
TTC21B (MIM# 613819)
A stopgain variant, p.Glu371*, and a splicing variant, c.430-2 A > C, were interpreted as P, and two frameshift variants, p.Tyr1282fs and p.Val1075fs, were interpreted as LP by InterVar. None of these four variants are reported in ClinVar or HGMD and all were included in set 2. Two nonsynonymous SNVs, p.Arg867Cys and pHis566Arg, were interpreted as VUS by InterVar but were included in set 4 because of their DM in HGMD (Table 3). The estimated carrier frequencies were 0.00141 in set 2 and 0.00197 in set 4 (Table 2).
The carrier frequencies, estimated from the sum of the allele frequencies, of the variants of the six ATD-associated genes were 0.00197 (set 1), 0.01182 (set 2), and 0.01408 (set 4). The proportions of potential patients calculated were 1/601,010 (set 1), 1/35,498 (set 2), and 1/26,553 (set 4) (Table 4).
Discussion
We investigated variants of genes related to bone dysplasia using 3.5KJPNv2, which contains genomic information from a large general population. This is the first investigation of this type. We tried to estimate the allele frequency, carrier frequency, and proportion of potential patients in the general population by evaluating the pathogenic significance by original variant interpretation13. However, an automatic and efficient method for variant interpretation has not yet been established. Although many studies refer to existing databases such as ClinVar and HGMD for variant interpretation, the assessment of pathogenic significance in these databases is heterogeneous, with some reports misclassified as false-positive variants25,26,27. By combining useful bioinformatic tools such as InterVar with databases such as ClinVar and HGMD, we detected and evaluated not only previously reported but also novel variants and further classified the detected variants from sets 1–4 based on the reliability of pathogenic significance (Fig. 1).
We used a similar method as a previous study13, to estimate carrier frequency for diseases subject to neonatal screening. The proportions of potential patients with OI, ATD, and EvC based on pathogenic variants in set 2 were close to the reported incidence rates in Japanese patients9. Furthermore, the proportions of potential patients with HPP based on set 1 variants were close to the reported incidence rates in Japanese patients (Table 4).
Several factors are considered to be the reason for the difference between the estimated proportion of potential patients and the frequency in the previous report9. They might include variants with penetrance of <100%, those with false-positive results and those that are not present or have a mild effect on onset because allele frequencies are estimated from genomic information. It is possible that the carrier frequencies and the proportions of potential patients were overestimated, because they were considered to be pathogenically significant despite not being reported previously by the automatic determination of InterVar. On the other hand, some of the pathogenic variants identified as VUS by InterVar may actually be highly pathogenically significant. Furthermore, genetic testing is rarely performed in clinical practice, because many cases of bone dysplasia show severe clinical findings from the fetal stage28, resulting in abortion and fetal death29. Thus, the number of reported variants may be less than the actual number. For these reasons, the carrier frequencies and the proportions of potential patients may have been underestimated. In addition, we assume that the following factors could alter variant assessments and affect estimates of the carrier frequency and proportion of potential patients: (1) increasing the sample size of the whole-genome reference panel beyond the 3552 individuals and expanding the number of subjects analyzed in this study; (2) revising the ACMG-AMP variant interpretation guidelines; and (3) increasing the reported disease-associated variants and the number of causative variants enrolled in ClinVar and HGMD.
Few studies have compared bone dysplasia incidence between Japanese and other ethnic groups or reported bone dysplasia-associated variants unique to the Japanese population. Two pathogenic variants, p.Phe327Leu and c.1559delT, are frequent mutations in the ALPL gene in Japanese patients with HPP19,30. On the other hand, in Caucasians, there are two common variants: c.571 G > A, which is observed in 50% of mild HPP cases, and c.1133 A > T, which is the most common variant in perinatal benign HPP cases31. These variants were not detected in 3.5KJPNv2, suggesting ethnic differences in bone dysplasia-associated variants. In the future, with the expansion of whole-genome reference panels, variants with a high frequency in Caucasians and those that are potentially pathogenic will be detected.
This study has some limitations. First, the genomic information used here was based on the whole-genome reference panel data from 3552 individuals and the number of samples may not have been sufficient to obtain reliable estimates of the variant frequencies for rare diseases. However, it is possible to estimate the proportion of potential patients with autosomal recessive inheritance, making this the first study meaningful for autosomal recessive bone dysplasia in the Japanese population. Second, this study did not use the ten criteria in the ACMG-AMP variant interpretation guidelines that are not subject to the automatic determinations of InterVar. The reliability of pathogenic significance in the variants obtained in this study may be changed by adding evidence and evaluating them by custom analysis of InterVar. Third, large insertions/deletions and structural polymorphisms were not included in the public version of 3.5KJPNv2. That is, 3.5KJPNv2 was constructed by sequence analysis of short reads, but large insertions/deletions and structural polymorphisms may be detected by reanalysis using long-read NGS. Finally, the study looked at genes associated with bone dysplasia that are inherited in an autosomal recessive mode, including those with autosomal dominant or X-linked inheritance (OI and HPP). Investigating genomic information, including these modes of inheritance, may lead to more accurate estimates of carrier frequency and the proportion of potential patients.
It is difficult to appropriately estimate the carrier frequency and the proportion of potential patients based on pathogenic variants detected from genomic information. Furthermore, the establishment of an ideal and plausible method remains challenging. However, this is an unprecedented study of rare autosomal recessive bone dysplasias and the first study to attempt to estimate the carrier frequency and the proportion of potential patients from the allele frequency of pathogenic variants. From a genetic epidemiologic perspective, the findings from this study will help in the understanding of what types of pathogenic variants of bone dysplasia exist in the general population and the differences between reported frequencies observed in clinical practices in Japan and the proportions of potential patients calculated from allele frequencies. In terms of perinatal care, the findings of this study are expected to be useful for clinical diagnosis in cases where differential diagnosis is difficult, for accurate risk calculation such as the risk of recurrence for the next child, for information for parents who may be carriers, for genetic counseling such as prenatal diagnosis, and for personalized medicine such as neonatal treatment by utilizing variant information.
References
Krakow, D. & Rimoin, D. L. The skeletal dysplasias. Genet. Med. 12, 327–341 (2010).
Forlino, A. & Marini, J. C. Osteogenesis imperfecta. Lancet 387, 1657–1671 (2016).
Ozono, K. et al. Identification of novel missense mutations (Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. J. Clin. Endocrinol. Metab. 81, 4458–4461 (1996).
Camera, G. & Mastroiacovo, P. Birth prevalence of skeletal dysplasias in the Italian Multicentric Monitoring System for Birth Defects. Prog. Clin. Biol. Res. 104, 441–449 (1982).
Orioli, I. M., Castilla, E. E. & Barbosa-Neto, J. G. The birth prevalence rates for the skeletal dysplasias. J. Med. Genet. 23, 328–332 (1986).
Bonafe, L. et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A 167A, 2869–2892 (2015).
Bae, J. S. et al. Comprehensive genetic exploration of skeletal dysplasia using targeted exome sequencing. Genet. Med. 18, 563–569 (2016).
Taillandier, A. et al. Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted next generation sequencing. Mol. Genet. Metab. 116, 215–220 (2015).
Satoh, N., Murotsuki, J. & Sawai, H. Incidence rates of fetal bone dysplasias in Japanese (in Japanese). J. Jpn. Soc. Perin. Neon. Med. 45, 1005–1007 (2009).
Tadaka, S. et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 6, 28 (2019).
Kuriyama, S. et al. The Tohoku Medical Megabank Project: design and mission. J. Epidemiol. 26, 493–511 (2016).
Kato, H., Kitano, T. & Iba, K. (eds). Surveillance registry for bone dysplasias of Japanese Orthopedic Association. p 20–26. (The Japanese Orthopaedic Association, Japan, 2016).
Yamaguchi-Kabata, Y. et al. Estimating carrier frequencies of newborn screening disorders using a whole-genome reference panel of 3552 Japanese individuals. Hum. Genet. 138, 389–409 (2019).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Li, Q. & Wang, K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280 (2017).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Stenson, P. D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Michigami, T. et al. Common mutations F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur. J. Pediatr. 164, 277–282 (2005).
Taketani, T. et al. Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch. Dis. Child 99, 211–215 (2014).
Ren, Y. et al. [Clinical analysis of 21 cases with short fetal femur in the third trimester]. Zhonghua Fu Chan Ke Za Zhi 52, 86–92 (2017).
Ruiz-Perez, V. L. et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am. J. Hum. Genet. 72, 728–732 (2003).
Zhang, Z. et al. L. Identification of one novel mutation in the EVC2 gene in a Chinese family with Ellis-van Creveld syndrome. Gene 511, 380–382 (2012).
Okamoto, T. et al. Novel compound heterozygous mutations in DYNC2H1 in a patient with severe short-rib polydactyly syndrome type III phenotype. Congenit. Anom. 55, 155–157 (2015).
Dorschner, M. O. et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am. J. Hum. Genet. 93, 631–640 (2013).
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014).
Shi, L. et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 7, 12065 (2016).
Ikenoue, S. et al. Discordant fetal phenotype of hypophosphatasia in two siblings. Am. J. Med. Genet. A 176, 171–174 (2018).
Oberklaid, F., Danks, D. M., Mayne, V. & Campbell, P. Asphyxiating thoracic dysplasia. Clinical, radiological, and pathological information on 10 patients. Arch. Dis. Child 52, 758–765 (1977).
Watanabe, A. et al. Prevalence of c.1559delT in ALPL, a common mutation resulting in the perinatal (lethal) form of hypophosphatasia in Japanese and effects of the mutation on heterozygous carriers. J. Hum. Genet. 56, 166–168 (2011).
Mornet, E. Hypophosphatasia. Best. Pr. Res Clin. Rheumatol. 22, 113–127 (2008).
Acknowledgements
We thank all past and present members of the Tohoku Medical Megabank Organization at Tohoku University (present members are listed at https://www.megabank.tohoku.ac.jp/english/a200601/). The Tohoku Medical Megabank is supported by grants from the Reconstruction Agency; the Ministry of Education, Culture, Sports, Science and Technology (MEXT); and the Japan Agency for Medical Research and Development (AMED). This study was supported by AMED under grant numbers JP19km0105001 and JP19km0105002.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagaoka, S., Yamaguchi-Kabata, Y., Shiga, N. et al. Estimation of the carrier frequencies and proportions of potential patients by detecting causative gene variants associated with autosomal recessive bone dysplasia using a whole-genome reference panel of Japanese individuals. Hum Genome Var 8, 2 (2021). https://doi.org/10.1038/s41439-020-00133-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-00133-7
